
Rational Design of Covalent Organic Frameworks-Based Single Atom Catalysts for Oxygen Evolution Reaction and Oxygen Reduction Reaction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Computational Details
3. Results and Discussion
3.1. Structural Stability and Electronic Properties of TM/TQBQ
3.2. Catalytic Performance for OER and ORR
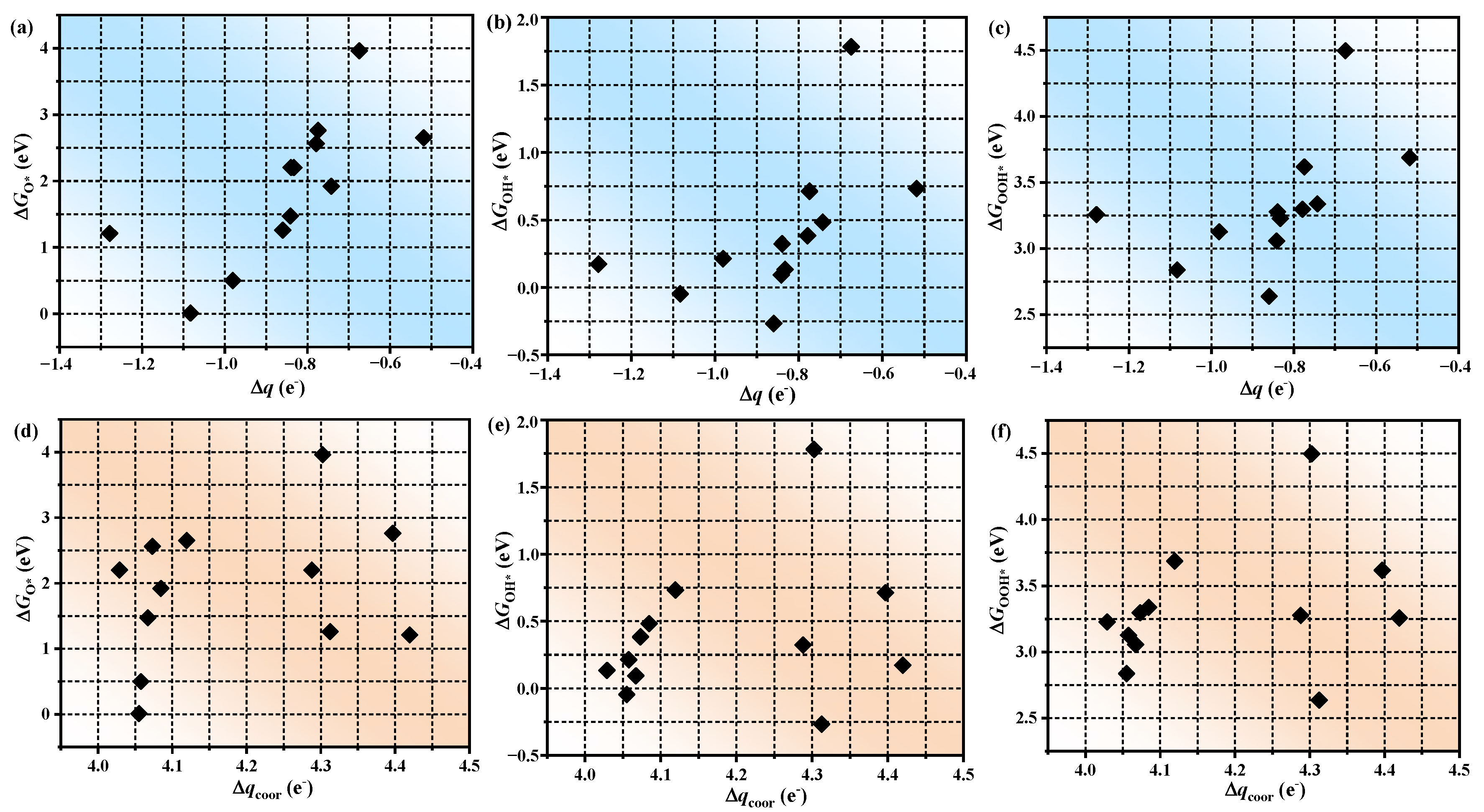
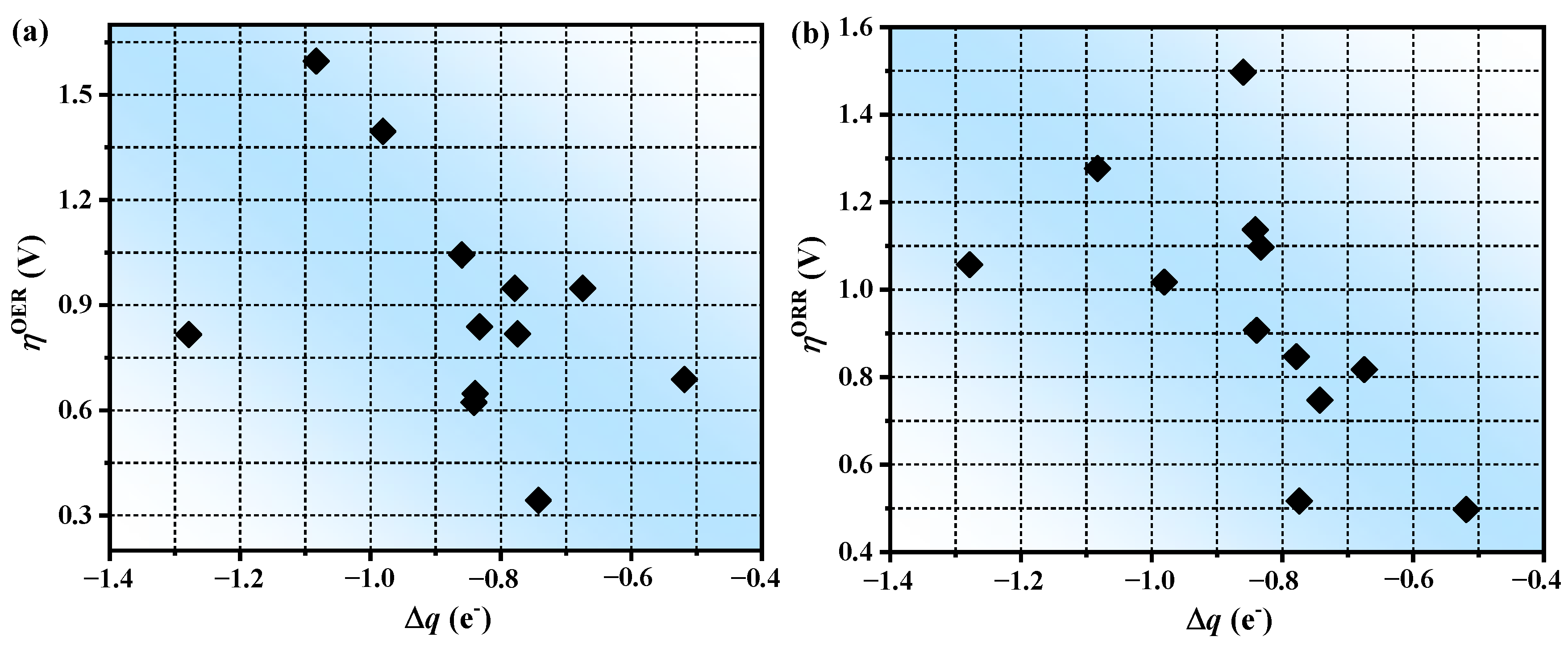
3.3. The Origin of Catalytic Activity
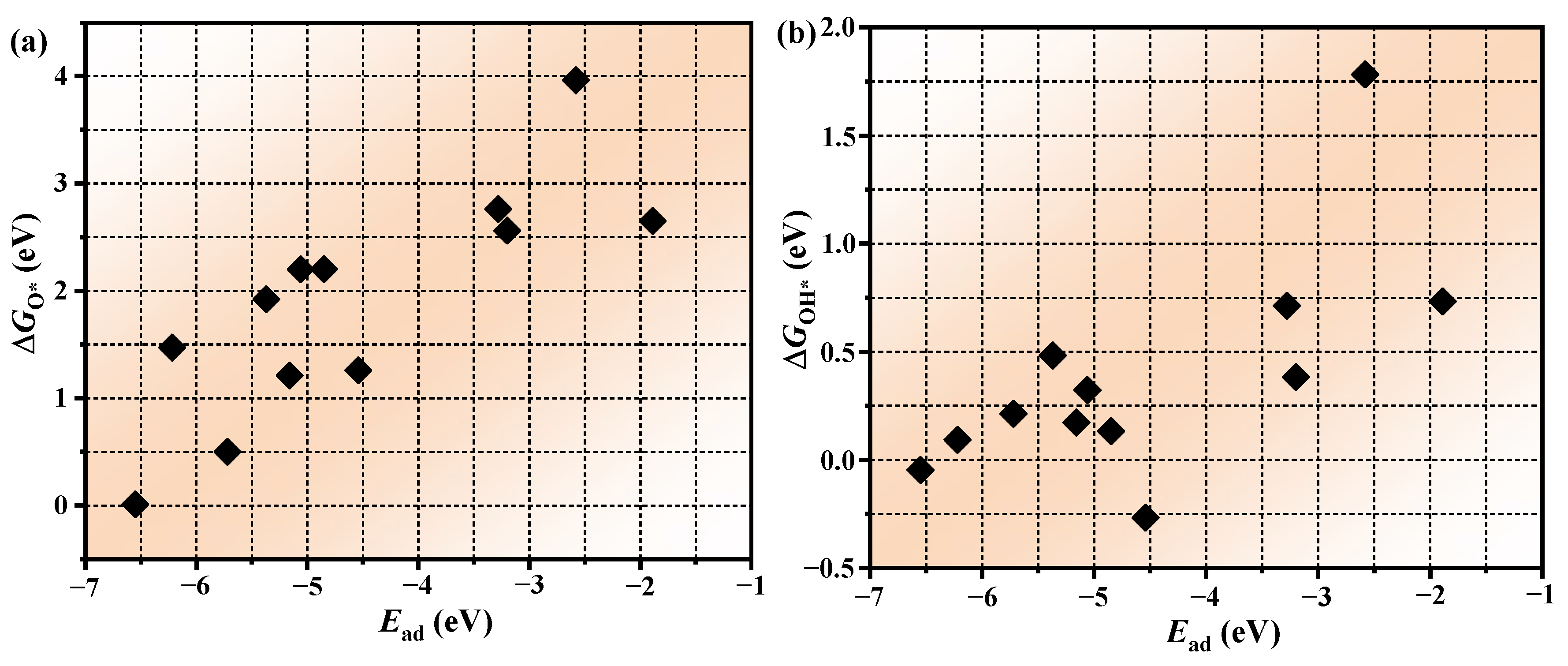
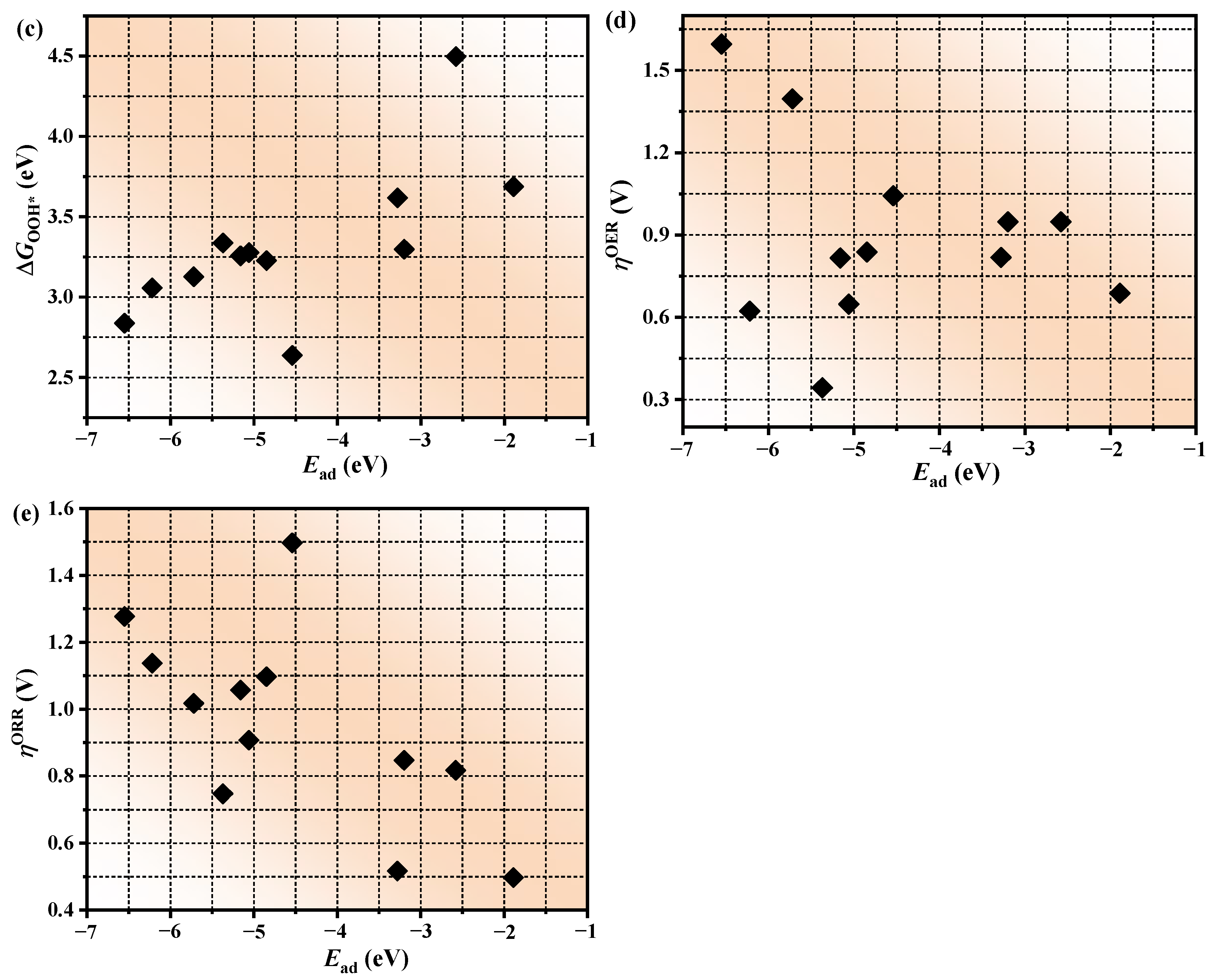
3.4. The Prediction of Catalytic Performance
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhu, Z.X.; Jiang, T.L.; Ali, M.; Meng, Y.H.; Jin, Y.; Cui, Y.; Chen, W. Rechargeable batteries for grid scale energy storage. Chem. Rev. 2022, 122, 16610–16751. [Google Scholar] [PubMed]
- Ma, S.C.; Yu, L.Z.; Jiang, H.X.; Yang, P.W.; Li, Q.; Du, M.Z.; Ma, J.X.; Zhu, H.T. Coal-fired power plants using ammonia for flexibility enhancement under carbon control strategies: Status, development, and perspectives. Energy Fuels 2024, 38, 4946–4965. [Google Scholar]
- Li, C.; Wang, F.; Cui, B.; Pan, Z.; Jia, Y. Localized magnetic moment induced by boron adatoms chemisorbed on graphene. J. Phys. Condens. Matter 2023, 35, 29. [Google Scholar]
- Li, C.; Sun, X.Y.; Yuan, P.F.; Wang, F.; Niu, C.Y.; Cui, B.; Jia, Y. Design of atomically localized magnetic moment by adatoms chemisorbed on graphene. Phys. Lett. A 2024, 504, 129435. [Google Scholar]
- Deng, Y.P.; Jiang, Y.; Liang, R.L.; Chen, N.; Chen, W.W.; Yin, Z.W.; King, G.; Su, D.; Wang, X.; Chen, Z.W. Reconstructing 3d-metal electrocatalysts through anionic evolution in zinc-air batteries. J. Am. Chem. Soc. 2023, 145, 20248–20260. [Google Scholar] [PubMed]
- Xu, W.; Zeng, R.; Rebarchik, M.; Posada-Borbón, A.; Li, H.; Pollock, C.J.; Mavrikakis, M.; Abruña, H.D. Atomically dispersed Zn/Co-N-C as ORR electrocatalysts for alkaline fuel cells. J. Am. Chem. Soc. 2024, 146, 2593–2603. [Google Scholar]
- Niu, X.D.; Wei, J.; Xu, D.Y.; Pei, J.J.; Sui, R. Charge-asymmetry Fe1Cu single-atom alloy catalyst for efficient oxygen reduction reaction. Nano Res. 2024, 17, 4702–4710. [Google Scholar]
- Huang, H.C.; Wang, T.T.; Li, J.; Chen, J.; Bu, Y.X.; Cheng, S.B. A strain-engineered self-intercalation Ta9Se12 based bifunctional single atom catalyst for oxygen evolution and reduction reactions. Appl. Surf. Sci. 2022, 602, 154378. [Google Scholar]
- Qiao, B.T.; Wang, A.Q.; Yang, X.F.; Allard, L.F.; Jiang, Z.; Cui, Y.T.; Liu, J.Y.; Li, J.; Zhang, T. Single-atom catalysis of CO oxidation using Pt1/FeOx. Nat. Chem. 2011, 3, 634–641. [Google Scholar]
- Fan, W.; Duan, Z.; Liu, W.; Mehmood, R.; Qu, J.; Cao, Y.; Guo, X.; Zhong, J.; Zhang, F. Rational design of heterogenized molecular phthalocyanine hybrid single-atom electrocatalyst towards two-electron oxygen reduction. Nat. Commun. 2023, 14, 1426. [Google Scholar]
- Peng, Y.; Lu, B.Z.; Chen, S.W. Carbon-supported single atom catalysts for electrochemical energy conversion and storage. Adv. Mater. 2018, 30, 1801995. [Google Scholar] [CrossRef]
- Zhang, Q.Q.; Guan, J.Q. Single-atom catalysts for electrocatalytic applications. Adv. Funct. Mater. 2020, 30, 2000768. [Google Scholar] [CrossRef]
- Cao, D.; Zhang, Z.R.; Cui, Y.H.; Zhang, R.H.; Zhang, L.P.; Zeng, J.; Cheng, D.J. One-step approach for constructing high-density single-atom catalysts toward overall water splitting at industrial current densities. Angew. Chem. Int. Edit. 2023, 135, e202214259. [Google Scholar]
- Zhang, S.B.; Jin, M.; Shi, T.F.; Han, M.M.; Sun, Q.; Lin, Y.; Ding, Z.H.; Zheng, L.R.; Wang, G.Z.; Zhang, Y.X.; et al. Electrocatalytically active Fe-(O-C2)4 single-atom sites for efficient reduction of nitrogen to ammonia. Angew. Chem. Int. Edit. 2020, 59, 13423–13429. [Google Scholar]
- Wang, M.H.; Kong, L.Y.; Lu, X.Q.; Wu, C.M.L. Transition metal doped pyrrole-NC for high-performance CO2 reduction reaction to C1 products. Appl. Surf. Sci. 2023, 618, 156678. [Google Scholar]
- Zhou, S.Q.; Shang, L.; Zhao, Y.X.; Shi, R.; Waterhouse, G.I.N.; Huang, Y.C.; Zheng, L.R.; Zhang, T.R. Pd single-atom catalysts on nitrogen-doped graphene for the highly selective photothermal hydrogenation of acetylene to ethylene. Adv. Mater. 2019, 31, 1900509. [Google Scholar] [CrossRef]
- Liu, G.M.; Huang, Y.; Lv, H.Q.; Wang, H.; Zeng, Y.B.; Yuan, M.Z.; Meng, Q.G.; Wang, C.Y. Confining single-atom Pd on g-C3N4 with carbon vacancies towards enhanced photocatalytic NO conversion. Appl. Catal. B Energy 2020, 284, 119683. [Google Scholar]
- Zhang, J.Q.; Zhao, Y.F.; Guo, X.; Chen, C.; Dong, C.L.; Liu, R.S.; Han, C.P.; Li, Y.D.; Gogotsi, Y.; Wang, G.X. Single platinum atoms immobilized on an MXene as an efficient catalyst for the hydrogen evolution reaction. Nat. Catal. 2018, 1, 985–992. [Google Scholar]
- Jiang, K.; Luo, M.; Liu, Z.; Peng, M.; Chen, D.; Lu, Y.-R.; Chan, T.-S.; de Groot, F.M.F.; Tan, Y. Rational strain engineering of single-atom ruthenium on nanoporous MoS2 for highly efficient hydrogen evolution. Nat. Commun. 2021, 12, 1687. [Google Scholar] [CrossRef]
- Wang, C.D.; Humayun, M.; Debecker, D.P.; Wu, Y. Electrocatalytic water oxidation with layered double hydroxides confining single atomss. Coord. Chem. Rev. 2022, 478, 214973. [Google Scholar] [CrossRef]
- Zhang, R.; Liu, W.S.; Zhang, F.M.; Yang, Z.D.; Zhang, G.L.; Zeng, Z.C. COF-C4N Nanosheets with uniformly anchored single metal sites for electrocatalytic OER: From theoretical screening to target synthesis. Appl. Catal. B Environ. 2023, 325, 122366. [Google Scholar]
- Dong, P.Y.; Wang, Y.; Zhang, A.C.J.; Cheng, T.; Xi, X.G.; Zhang, J.L. Platinum single atoms anchored on a covalent organic framework: Boosting active sites for photocatalytic hydrogen evolution. ACS Catal. 2021, 11, 13266–13279. [Google Scholar]
- Chen, Y.J.; Zhuo, H.Y.; Pan, Y.; Liang, J.X.; Liu, C.G.; Li, J. Triazine COF-supported single-atom catalyst (Pd1/trzn-COF) for CO oxidation. Sci. China Mater. 2021, 64, 1939–1951. [Google Scholar]
- Wang, J.; Zhang, Z.H.; Li, Y.Y.; Qu, Y.Y.; Li, Y.Q.; Li, W.F.; Zhao, M.W. Screening of transition-metal single-atom catalysts anchored on covalent-organic frameworks for efficient nitrogen fixation. ACS Appl. Mater. Interfaces 2021, 14, 1024–1033. [Google Scholar]
- Shi, R.J.; Liu, L.J.; Lu, Y.; Wang, C.C.; Li, Y.X.; Li, L.; Yan, Z.H.; Chen, J. Nitrogen-rich covalent organic frameworks with multiple carbonyls for high-performance sodium batteries. Nat. Commun. 2020, 11, 178. [Google Scholar] [PubMed]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953. [Google Scholar] [CrossRef]
- Perdew, J.P.; Chevary, J.A.; Vosko, S.H.; Jackson, K.A.; Pederson, M.R.; Singh, D.J.; Fiolhais, C. Atoms, molecules; solids, and surfaces: Applications of the generalized gradient approximation for exchange and correlation. Phys. Rev. B 1992, 46, 6671–6687. [Google Scholar]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar]
- Momma, K.; Izumi, F. VESTA: A three-dimensional visualization system for electronic and structural analysis. J. Appl. Crystallogr. 2008, 41, 653–658. [Google Scholar]
- Wang, V.; Xu, N.; Liu, J.C.; Tang, G.; Geng, W.T. VASPKIT: A user-friendly interface facilitating high-throughput computing and analysis using VASP code. Comput. Phys. Commun. 2021, 267, 108033. [Google Scholar]
- Zhou, W.Y.; Shen, H.M.; Xie, H.H.; Shen, Y.H.; Kang, W.; Wang, Q.; Kawazoe, Y.; Jena, P. Boron-functionalized organic framework as a high-performance metal-free catalyst for N2 fixation. J. Phys. Chem. Lett. 2021, 12, 12142–12149. [Google Scholar] [PubMed]
- Talib, S.H.; Lu, Z.S.; Yu, X.H.; Ahmad, K.; Bashir, B.; Yang, Z.X.; Li, J. Theoretical inspection of M1/PMA single-atom electrocatalyst: Ultra-high performance for water splitting (HER/OER) and oxygen reduction reactions (OER). ACS Catal. 2021, 11, 8929–8941. [Google Scholar]
- Li, L.L.; Chang, X.; Lin, X.Y.; Zhao, Z.J.; Gong, J.L. Theoretical insights into single-atom catalysts. Chem. Soc. Rev. 2020, 49, 8156–8178. [Google Scholar]
- Seh, Z.W.; Kibsgaard, J.; Dickens, C.F.; Chorkendorff, I.; Nørskov, J.K.; Jaramillo, T.F. Combining theory and experiment in electrocatalysis: Insights into materials design. Science 2017, 355, eaad4998. [Google Scholar]
- Greeley, J.; Stephens, I.E.L.; Bondarenko, A.S.; Johansson, T.P.; Hansen, H.A.; Jaramillo, T.F.; Rossmeisl, J.; Chorkendorff, I.; Nørskov, J.K. Alloys of platinum and early transition metals as oxygen reduction electrocatalysts. Nat. Chem. 2009, 1, 552–556. [Google Scholar]
- Shi, X.C.; Li, Y.C.; Zhang, S.; Hu, R.M.; Gao, S.; Jin, P.P.; Shang, J.X.; Shui, J.L. Precious trimetallic single-cluster catalysts for oxygen and hydrogen electrocatalytic reactions: Theoretical considerations. Nano Res. 2023, 16, 8042–8050. [Google Scholar]
- Li, Z.R.; Li, X.R.; Gu, Y.K.; Hu, X.H.; Wang, L.B.; Li, P. Theoretical screening of transition metal atoms doped two-dimensional VSe2 monolayers as single-atom catalysts for HER, OER and ORR. Appl. Surf. Sci. 2023, 646, 158959. [Google Scholar]
- Song, Z.J.; Zhao, B.; Wang, Q.; Cheng, P. Steering reduction and decomposition of peroxide compounds by interface interactions between MgO thin film and transition-metal support. Appl. Surf. Sci. 2018, 459, 812–821. [Google Scholar]
- Song, Z.J.; Shao, X.J.; Wang, Q.; Ma, C.X.; Wang, K.D.; Han, D.M. Generation of molybdenum hydride species via addition of molecular hydrogen across metal-oxygen bond at monolayer oxide/metal composite interface. Int. J. Hydrogen Energy 2020, 45, 2975–2988. [Google Scholar]
- Huang, H.C.; Li, J.; Zhao, Y.; Chen, J.; Bu, Y.X.; Cheng, S.B. Adsorption energy as a promising single-parameter descriptor for single atom catalysis in the oxygen evolution reaction. J. Mater. Chem. A 2021, 9, 6442–6450. [Google Scholar]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xie, W.; Cui, B.; Liu, D.; Huang, H.; Yang, C. Rational Design of Covalent Organic Frameworks-Based Single Atom Catalysts for Oxygen Evolution Reaction and Oxygen Reduction Reaction. Molecules 2025, 30, 1505. https://doi.org/10.3390/molecules30071505
Xie W, Cui B, Liu D, Huang H, Yang C. Rational Design of Covalent Organic Frameworks-Based Single Atom Catalysts for Oxygen Evolution Reaction and Oxygen Reduction Reaction. Molecules. 2025; 30(7):1505. https://doi.org/10.3390/molecules30071505
Chicago/Turabian StyleXie, Wenli, Bin Cui, Desheng Liu, Haicai Huang, and Chuanlu Yang. 2025. "Rational Design of Covalent Organic Frameworks-Based Single Atom Catalysts for Oxygen Evolution Reaction and Oxygen Reduction Reaction" Molecules 30, no. 7: 1505. https://doi.org/10.3390/molecules30071505
APA StyleXie, W., Cui, B., Liu, D., Huang, H., & Yang, C. (2025). Rational Design of Covalent Organic Frameworks-Based Single Atom Catalysts for Oxygen Evolution Reaction and Oxygen Reduction Reaction. Molecules, 30(7), 1505. https://doi.org/10.3390/molecules30071505