

Synthesis of Functionalized 1H-Imidazoles via Denitrogenative Transformation of 5-Amino-1,2,3-Triazoles
 , , and
, , and
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
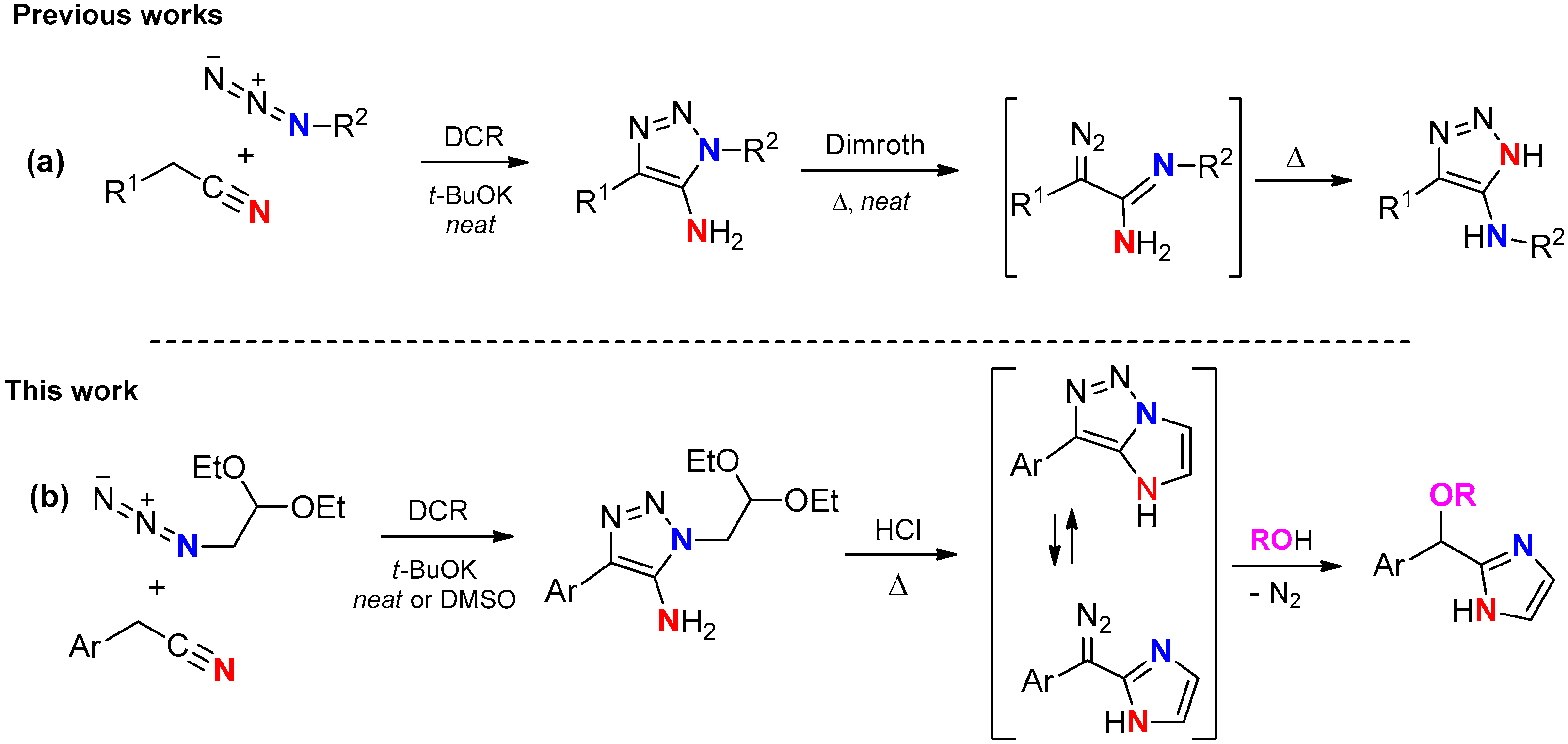
1. Introduction
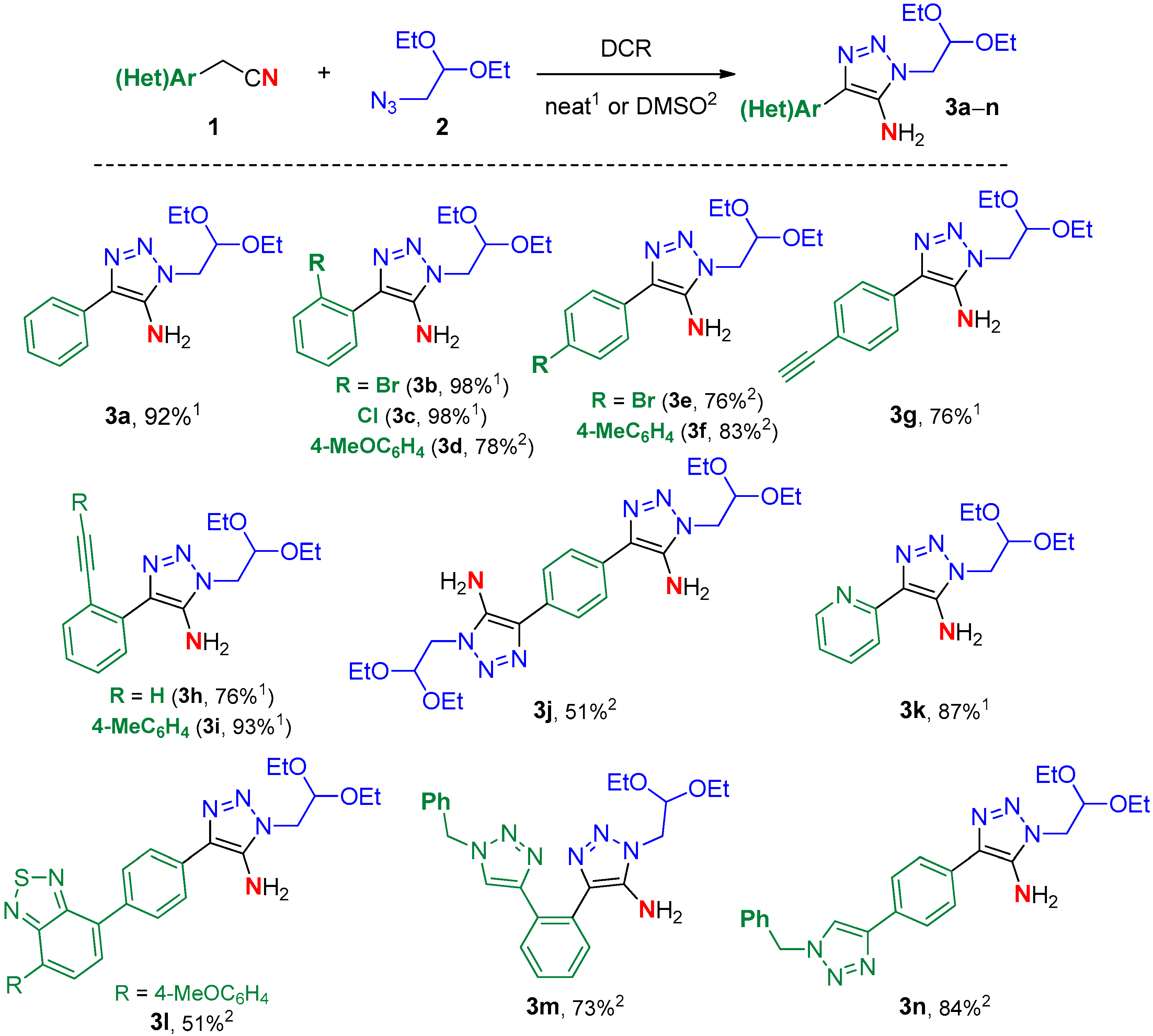
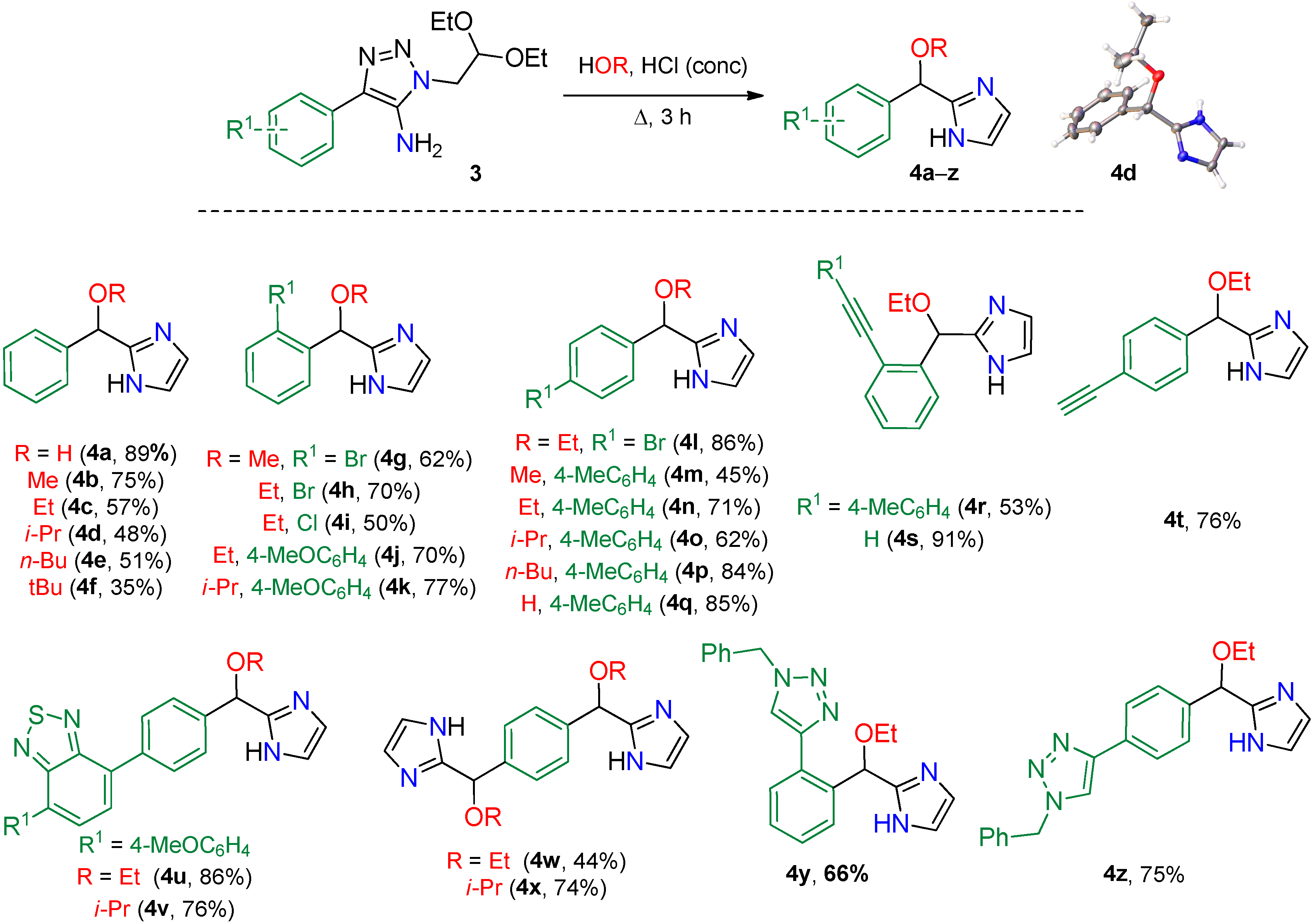
2. Results
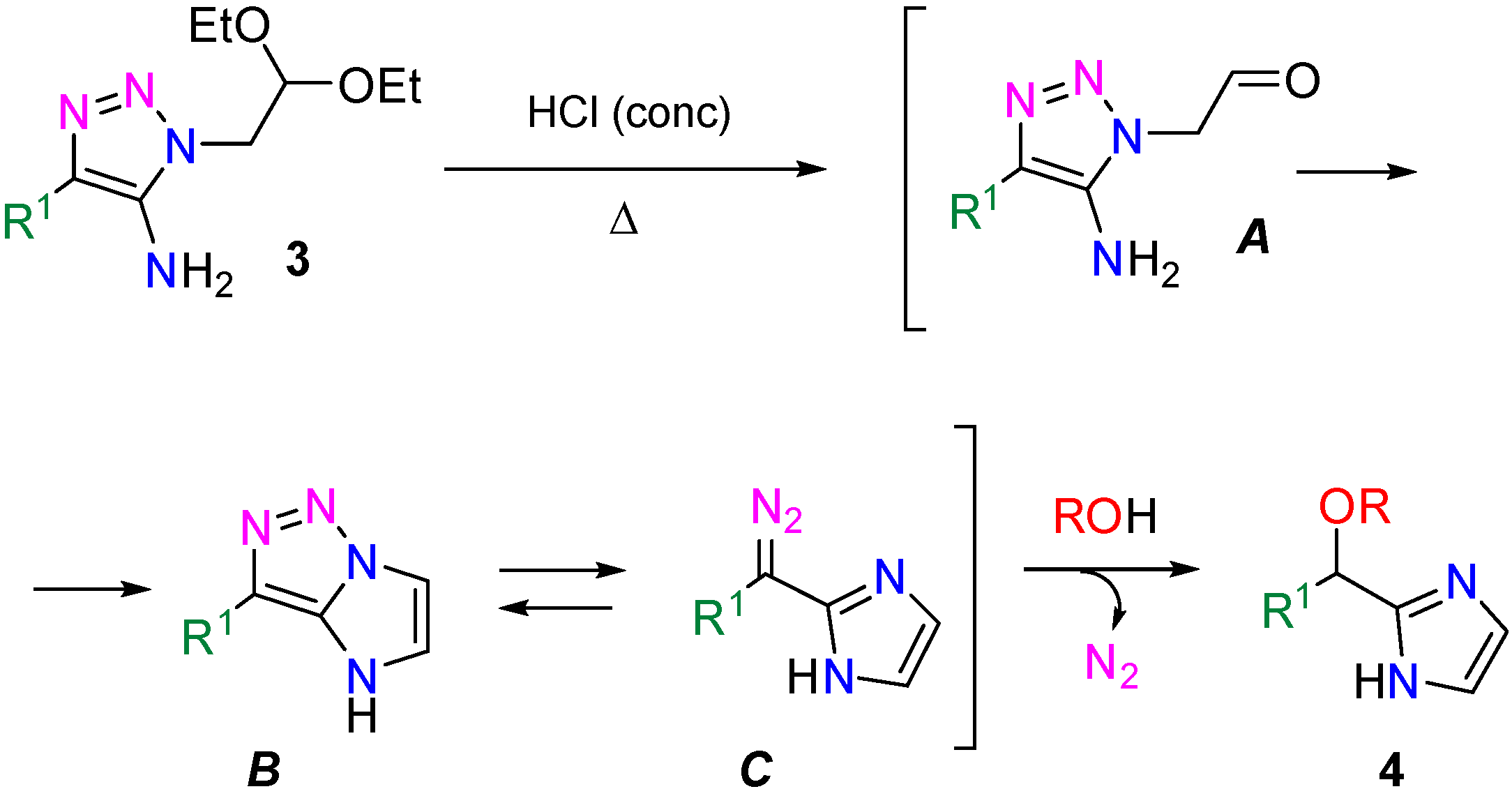
Synthesis
3. Materials and Methods
3.1. General Information
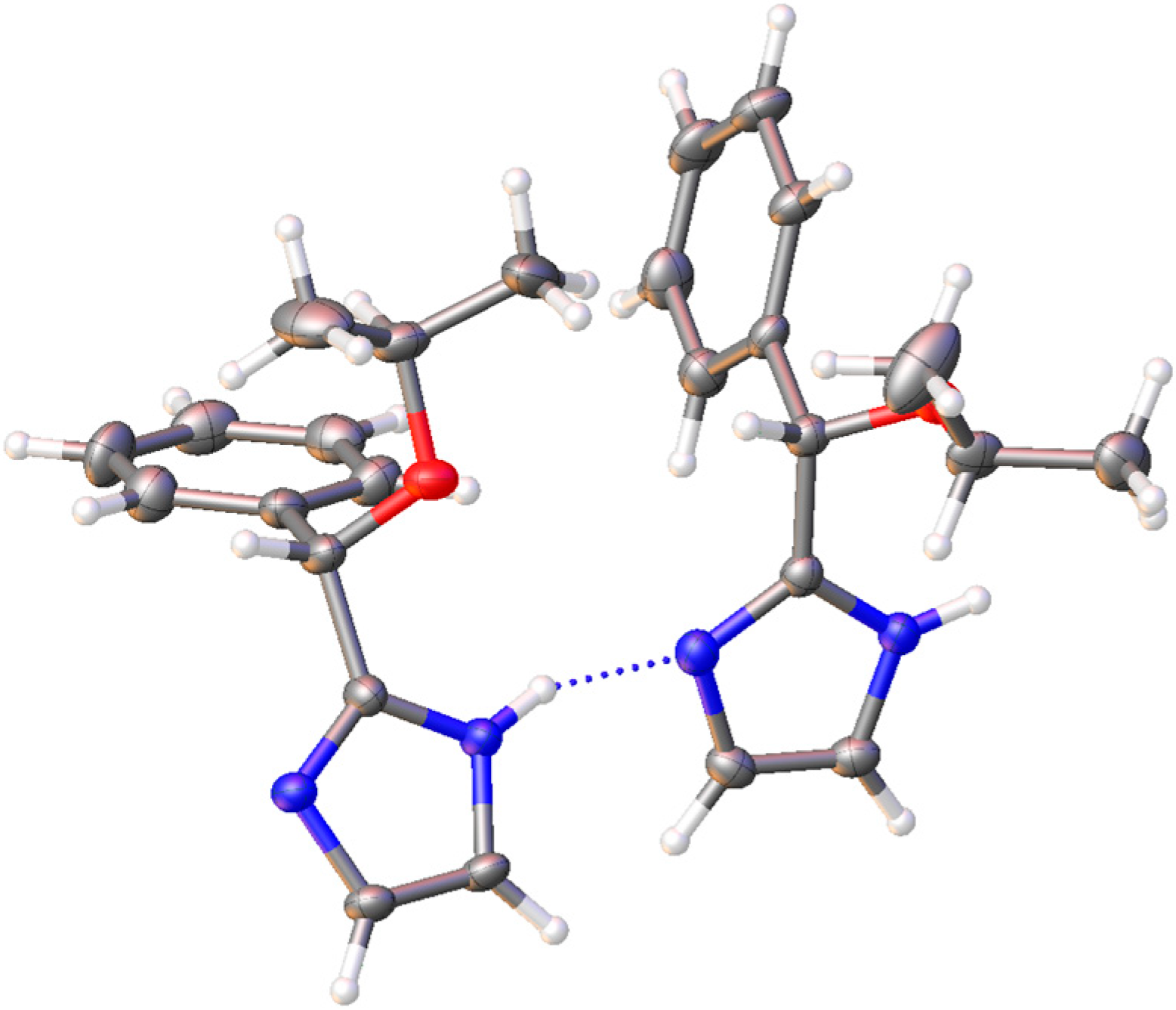
3.2. Crystal Structure Determination
3.3. Preparation and Characterization of Starting Acetonitrile Derivatives
3.4. Preparation and Characterization of Novel Compounds
3.4.1. General Procedure A for Solvent-Free Preparation of 5-Amino-1-(2,2-Diethoxyethyl)-1,2,3-Triazoles 3 via Dipolar Azide−Nitrile Cycloaddition (DCR)
3.4.2. General Procedure B for Preparation of 5-Amino-1-(2,2-Diethoxyethyl)-1,2,3-Triazoles 3 via Dipolar Azide−Nitrile Cycloaddition (DCR) in DMSO
3.4.3. General Procedure C for Preparation of 1H-Imidazole Derivatives 4
3.4.4. General Procedure D for CuAAC Synthesis of 4y and 4z
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Patel, A.; Sharp, S.Y.; Hall, K.; Lewis, W.; Stevens, M.F.; Workman, P.; Moody, C.J. Fused imidazoles as potential chemical scaffolds for inhibition of heat shock protein 70 and induction of apoptosis. Synthesis and biological evaluation of phenanthro [9,10-d]imidazoles and imidazo[4,5-f][1,10]phenanthrolines. Org. Biomol. Chem. 2016, 14, 3889–3905. [Google Scholar] [PubMed]
- Shabalin, D.A.; Camp, J.E. Recent advances in the synthesis of imidazoles. Org. Biomol. Chem. 2020, 18, 3950–3964. [Google Scholar] [PubMed]
- Tagare, J.; Dubey, D.K.; Jou, J.-H.; Vaidyanathan, S. Synthesis, photophysical, theoretical and electroluminescence study of triphenylamine-imidazole based blue fluorophores for solution-processed organic light emitting diodes. Dyes Pigment 2019, 160, 944–956. [Google Scholar]
- Dong, Y.; Qian, J.; Liu, Y.; Zhu, N.; Xu, B.; Ho, C.-L.; Tian, W.; Wong, W.-Y. Imidazole-containing cyanostilbene-based molecules with aggregation-induced emission characteristics: Photophysical and electroluminescent properties. New J. Chem. 2019, 43, 1844–1850. [Google Scholar]
- Mohagheghnezhad, M.; Rafiee, Z. Synthesis and characterization of two novel poly(amide-imide)s containing pendent (5-(4,5-diphenyl-1H-imidazol-2-yl)furan-2-yl)phenyl moieties and natural amino acids linkages for adsorption of Cu(II). Polym. Bull. 2019, 77, 4739–4757. [Google Scholar]
- Martínez, R.; Pastor, I.M.; Yus, M. 1,2-Functionalized Imidazoles as Palladium Ligands: An Efficient and Robust Catalytic System for the Fluorine-Free Hiyama Reaction. Eur. J. Org. Chem. 2013, 2014, 872–877. [Google Scholar]
- Földesi, T.; Dancsó, A.; Simig, G.; Volk, B.; Milen, M. Efficient synthesis of a new compound family, 9-aryl-5H-imidazo[2,1-d][1,2,5]triazepin-6(7 H)-ones. Tetrahedron 2016, 72, 5427–5432. [Google Scholar]
- Perl, N.R.; Leighton, J.L. Enantioselective imidazole-directed allylation of aldimines and ketimines. Org. Lett. 2007, 9, 3699–3701. [Google Scholar]
- Sinha, N.; Mistry, P.; Das, S.; Datta, T.; Roy, B. Intramolecular Hydroarylation of Arenes via Imidazole-Directed C-H Activation in Aqueous Methanol Using Rhodium(III) as the Catalyst and Mechanistic Study. J. Org. Chem. 2023, 88, 8969–8983. [Google Scholar]
- Dutta, P.K.; Sen, S. (Benz)Imidazole-Directed Cobalt(III)-Catalyzed C–H Activation of Arenes: A Facile Strategy to Access Polyheteroarenes by Oxidative Annulation. Eur. J. Org. Chem. 2018, 2018, 5512–5519. [Google Scholar]
- Zhao, Q.; Meng, G.; Nolan, S.P.; Szostak, M. N-Heterocyclic Carbene Complexes in C-H Activation Reactions. Chem. Rev. 2020, 120, 1981–2048. [Google Scholar]
- Liang, Q.; Song, D. Iron N-heterocyclic carbene complexes in homogeneous catalysis. Chem. Soc. Rev. 2020, 49, 1209–1232. [Google Scholar] [CrossRef]
- Fortman, G.C.; Nolan, S.P. N-Heterocyclic carbene (NHC) ligands and palladium in homogeneous cross-coupling catalysis: A perfect union. Chem. Soc. Rev. 2011, 40, 5151–5169. [Google Scholar] [CrossRef] [PubMed]
- Joshi, A.; Mohan, D.C.; Adimurthy, S. Lewis Acid-Catalyzed Denitrogenative Transannulation of Pyridotriazoles with Nitriles: Synthesis of Imidazopyridines. J. Org. Chem. 2016, 81, 9461–9469. [Google Scholar] [PubMed]
- Clarke, P.; Reeder, A.; Winn, J. Transannulation Reactions in the Synthesis of Natural Products. Synthesis 2009, 2009, 691–709. [Google Scholar] [CrossRef]
- Chuprakov, S.; Hwang, F.W.; Gevorgyan, V. Rh-catalyzed transannulation of pyridotriazoles with alkynes and nitriles. Angew. Chem. Int. Ed. 2007, 46, 4757–4759. [Google Scholar] [CrossRef]
- Zhang, Z.; Gevorgyan, V. Co-Catalyzed Transannulation of Pyridotriazoles with Isothiocyanates and Xanthate Esters. Org. Lett. 2020, 22, 8500–8504. [Google Scholar] [CrossRef]
- Rawat, D.; Adimurthy, S. Transannulation of Pyridotriazoles with Naphthoquinones and Indoles: Synthesis of Benzo[f]Pyrido[1,2-a]Indoles and Indolizino[3,2-b]indoles. Adv. Synth. Catal. 2021, 364, 71–76. [Google Scholar] [CrossRef]
- Yang, Y.; Zhou, M.B.; Ouyang, X.H.; Pi, R.; Song, R.J.; Li, J.H. Rhodium(III)-Catalyzed [3+2]/[5+2] Annulation of 4-Aryl 1,2,3-Triazoles with Internal Alkynes through Dual C(sp2)—H Functionalization. Angew. Chem. Int. Ed. 2015, 127, 6695–6699. [Google Scholar] [CrossRef]
- Barashkova, X.A.; Gevondian, A.G.; Latyshev, G.V.; Kotovshchikov, Y.N.; Bezzubov, S.I.; Lukashev, N.V.; Beletskaya, I.P. Access to Bicyclo[3.1.0]hexane and Cyclopenta[c]pyrazole Scaffolds via Solvent-Directed Divergent Reactivity of 5-Iodotriazoles. Org. Lett. 2024, 26, 9625–9630. [Google Scholar] [CrossRef]
- Motornov, V.; Markos, A.; Beier, P. A rhodium-catalyzed transannulation of N-(per)fluoroalkyl-1,2,3-triazoles under microwave conditions—A general route to N-(per)fluoroalkyl-substituted five-membered heterocycles. Chem. Commun. 2018, 54, 3258–3261. [Google Scholar]
- Horneff, T.; Chuprakov, S.; Chernyak, N.; Gevorgyan, V.; Fokin, V.V. Rhodium-catalyzed transannulation of 1,2,3-triazoles with nitriles. J. Am. Chem. Soc. 2008, 130, 14972–14974. [Google Scholar] [PubMed]
- Akter, M.; Rupa, K.; Anbarasan, P. 1,2,3-Triazole and Its Analogues: New Surrogates for Diazo Compounds. Chem. Rev. 2022, 122, 13108–13205. [Google Scholar]
- Lieber, E.; Chao, T.S.; Rao, C.N.R. Synthesis and Isomerization of Substituted 5-Amino-1,2,3-triazoles1. J. Org. Chem. 1957, 22, 654–662. [Google Scholar]
- Chattopadhyay, B.; Gevorgyan, V. Transition-metal-catalyzed denitrogenative transannulation: Converting triazoles into other heterocyclic systems. Angew. Chem. Int. Ed. 2012, 51, 862–872. [Google Scholar]
- Li, Y.; Yang, H.; Zhai, H. The Expanding Utility of Rhodium-Iminocarbenes: Recent Advances in the Synthesis of Natural Products and Related Scaffolds. Chem.—Eur. J. 2018, 24, 12757–12766. [Google Scholar]
- Tichy, D.; Kost Al, V.; Motornov, V.; Klimankova, I.; Beier, P. Preparation of 1-Azido-2-Bromo-1,1,2,2-Tetrafluoroethane and Its Use in the Synthesis of N-Fluoroalkylated Nitrogen Heterocycles. J. Org. Chem. 2020, 85, 11482–11489. [Google Scholar] [PubMed]
- Rostovskii, N.V.; Titov, G.D.; Filippov, I.P. Recent Advances in Denitrogenative Reactions of Pyridotriazoles. Synthesis 2020, 52, 3564–3576. [Google Scholar]
- Yadagiri, D.; Rivas, M.; Gevorgyan, V. Denitrogenative Transformations of Pyridotriazoles and Related Compounds: Synthesis of N-Containing Heterocyclic Compounds and Beyond. J. Org. Chem. 2020, 85, 11030–11046. [Google Scholar]
- Gevondian, A.G.; Kotovshchikov, Y.N.; Latyshev, G.V.; Lukashev, N.V.; Beletskaya, I.P. Metal-Free Reductive C–C-Coupling between Arylboronic Acids and 2-(5-Iodo-1,2,3-triazol-1-yl)phenols. Russ. J. Org. Chem. 2023, 59, 1465–1472. [Google Scholar]
- Kotovshchikov, Y.N.; Voloshkin, V.A.; Latyshev, G.V.; Lukashev, N.V.; Beletskaya, I.P. Cascade Transformations of [1,2,3]Triazolo[1,5-a]pyridines as Convenient Precursors of Diazo Compounds and Metal Carbenes. Russ. J. Org. Chem. 2021, 57, 1212–1244. [Google Scholar]
- Kotovshchikov, Y.N.; Latyshev, G.V.; Navasardyan, M.A.; Erzunov, D.A.; Beletskaya, I.P.; Lukashev, N.V. Annulation-Induced Cascade Transformation of 5-Iodo-1,2,3-triazoles to 2-(1-Aminoalkyl)benzoxazoles. Org. Lett. 2018, 20, 4467–4470. [Google Scholar]
- Chuprakov, S.; Gevorgyan, V. Regiodivergent metal-catalyzed rearrangement of 3-iminocyclopropenes into N-fused heterocycles. Org. Lett. 2007, 9, 4463–4466. [Google Scholar] [PubMed]
- Gribanov, P.S.; Atoian, E.M.; Philippova, A.N.; Topchiy, M.A.; Asachenko, A.F.; Osipov, S.N. One-Pot Synthesis of 5-Amino-1,2,3-triazole Derivatives via Dipolar Azide−Nitrile Cycloaddition and Dimroth Rearrangement under Solvent-Free Conditions. Eur. J. Org. Chem. 2021, 2021, 1378–1384. [Google Scholar]
- Gribanov, P.S.; Philippova, A.N.; Topchiy, M.A.; Minaeva, L.I.; Asachenko, A.F.; Osipov, S.N. General Method of Synthesis of 5-(Het)arylamino-1,2,3-triazoles via Buchwald-Hartwig Reaction of 5-Amino- or 5-Halo-1,2,3-triazoles. Molecules 2022, 27, 1999. [Google Scholar] [CrossRef] [PubMed]
- Gribanov, P.S.; Philippova, A.N.; Topchiy, M.A.; Lypenko, D.A.; Dmitriev, A.V.; Tokarev, S.D.; Smol’yakov, A.F.; Rodionov, A.N.; Asachenko, A.F.; Osipov, S.N. Synthesis of 5-(Aryl)amino-1,2,3-triazole-containing 2,1,3-Benzothiadiazoles via Azide-Nitrile Cycloaddition Followed by Buchwald-Hartwig Reaction. Molecules 2024, 29, 2151. [Google Scholar] [CrossRef]
- Tischler, A.N.; Lanza, T.J. 6-substituted indoles from o-halonitrobenzenes. Tetrahedron Lett. 1986, 27, 1653–1656. [Google Scholar]
- Arai, E.; Tokuyama, H.; Linsell, M.S.; Fukuyama, T. 2-(2-Aminophenyl)-acetaldehyde Dimethyl Acetal: A Novel Reagent for the Protection of Carboxylic Acids. Tetrahedron Lett. 1998, 39, 71–74. [Google Scholar]
- Gilley, C.B.; Buller, M.J.; Kobayashi, Y. New entry to convertible isocyanides for the Ugi reaction and its application to the stereocontrolled formal total synthesis of the proteasome inhibitor omuralide. Org. Lett. 2007, 9, 3631–3634. [Google Scholar]
- Maisto, S.K.; Leersnyder, A.P.; Pudner, G.L.; Scheerer, J.R. Synthesis of Pyrrolopyrazinones by Construction of the Pyrrole Ring onto an Intact Diketopiperazine. J. Org. Chem. 2020, 85, 9264–9271. [Google Scholar]
- Artyushin, O.I.; Genkina, G.K.; Moiseeva, A.A.; Vinogradova, N.M.; Sharova, E.V.; Nelyubina, Y.V.; Klemenkova, Z.S.; Brel, V.K. Synthesis and Structure of New gem-Diols with 1,2,3-Triazole Fragment. Russ. J. Gen. Chem. 2018, 88, 1108–1113. [Google Scholar]
- Torregrosa, R.; Pastor, I.M.; Yus, M. Isoprene-catalysed lithiation: Deprotection and functionalisation of imidazole derivatives. Tetrahedron 2007, 63, 947–952. [Google Scholar]
- Gribanov, P.S.; Loginov, D.A.; Lypenko, D.A.; Dmitriev, A.V.; Pozin, S.I.; Aleksandrov, A.E.; Tameev, A.R.; Martynov, I.L.; Chernyadyev, A.Y.; Osipov, S.N. New Unsymmetrically Substituted Benzothiadiazole-Based Luminophores: Synthesis, Optical, Electrochemical Studies, Charge Transport, and Electroluminescent Characteristics. Molecules 2021, 26, 7596. [Google Scholar] [CrossRef] [PubMed]
- Charushin, V.N.; Verbitskiy, E.V.; Chupakhin, O.N.; Vorobyeva, D.V.; Gribanov, P.S.; Osipov, S.N.; Ivanov, A.V.; Martynovskaya, S.V.; Sagitova, E.F.; Dyachenko, V.D.; et al. The chemistry of heterocycles in the 21st century. Russ. Chem. Rev. 2024, 93, RCR5125. [Google Scholar]
- Hirayama, T.; Ueda, S.; Okada, T.; Tsurue, N.; Okuda, K.; Nagasawa, H. Facile one-pot synthesis of [1, 2, 3]triazolo[1, 5-a]pyridines from 2-acylpyridines by copper(II)-catalyzed oxidative N-N bond formation. Chem.—Eur. J. 2014, 20, 4156–4162. [Google Scholar]
- Lapier, M.; Ballesteros-Garrido, R.; Guzman-Rivera, D.; Gonzalez-Herrera, F.; Aguilera-Venegas, B.; Moncada-Basualto, M.; Ballesteros, R.; Abarca, B.; Pesce, B.; Kemmerling, U.; et al. Novel [1,2,3]triazolo[1,5-a]pyridine derivatives are trypanocidal by sterol biosynthesis pathway alteration. Future Med. Chem. 2019, 11, 1137–1155. [Google Scholar]
- Ezawa, M.; Togo, H. One-Pot Preparation of C1-Homologated Aliphatic Nitriles from Aldehydes through a Wittig Reaction under Metal-Cyanide-Free Conditions. Eur. J. Org. Chem. 2017, 2017, 2379–2384. [Google Scholar]
- Wilde, F.; Lemmerhirt, H.; Emmrich, T.; Bednarski, P.J.; Link, A. Microwave-assisted synthesis and evaluation of acylhydrazones as potential inhibitors of bovine glutathione peroxidase. Mol. Divers. 2014, 18, 307–322. [Google Scholar]
- Arun Kumar, G.; Gomathi Priya, P.; Alagar, M. Functional phenylethynylene side arm poly(arylene ethynylene) conjugated polymers: Optical and electrochemical behavior for enrichment of electronic applications. New J. Chem. 2018, 42, 5767–5773. [Google Scholar]
- An, B.K.; Kwon, S.K.; Jung, S.D.; Park, S.Y. Enhanced emission and its switching in fluorescent organic nanoparticles. J. Am. Chem. Soc. 2002, 124, 14410–14415. [Google Scholar]
- Chen, X.; He, Q.; Xie, Y.; Yang, C. Palladium(II)-catalyzed synthesis of functionalized indenones via oxidation and cyclization of 2-(2-arylethynylphenyl)acetonitriles. Org. Biomol. Chem. 2013, 11, 2582–2585. [Google Scholar]
- Thompson, B.C.; Kim, Y.G.; McCarley, T.D.; Reynolds, J.R. Soluble narrow band gap and blue propylenedioxythiophene-cyanovinylene polymers as multifunctional materials for photovoltaic and electrochromic applications. J. Am. Chem. Soc. 2006, 128, 12714–12725. [Google Scholar]
- Bidal, Y.D.; Lesieur, M.; Melaimi, M.; Nahra, F.; Cordes, D.B.; Athukorala Arachchige, K.S.; Slawin, A.M.Z.; Bertrand, G.; Cazin, C.S.J. Copper(I) Complexes Bearing Carbenes Beyond Classical N-Heterocyclic Carbenes: Synthesis and Catalytic Activity in “Click Chemistry”. Adv. Synth. Catal. 2015, 357, 3155–3161. [Google Scholar]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A 2015, 71, 3–8. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. A 2008, 64, 112–122. [Google Scholar]
- Gribanov, P.S.; Vorobyeva, D.V.; Tokarev, S.D.; Petropavlovskikh, D.A.; Loginov, D.A.; Nefedov, S.E.; Dolgushin, F.M.; Osipov, S.N. Rhodium-Catalyzed C−H Activation/Annulation of Aryl Hydroxamates with Benzothiadiazol-Containing Acetylenes: Access to Isoquinoline-Bridged Donor-Acceptor Luminophores. Eur. J. Org. Chem. 2022, 2022, e202101572. [Google Scholar]
- Moro, A.V.; Ferreira, P.C.; Migowski, P.; Rodembusch, F.S.; Dupont, J.; Lüdtke, D.S. Synthesis and photophysical properties of fluorescent 2,1,3-benzothiadiazole-triazole-linked glycoconjugates: Selective chemosensors for Ni(II). Tetrahedron 2013, 69, 201–206. [Google Scholar]
- Li, H.; Huang, Q.; Chen, C.; Xu, B.; Wang, H.Y.; Long, Y.Q. Discovery of Potent and Orally Bioavailable GPR40 Full Agonists Bearing Thiophen-2-ylpropanoic Acid Scaffold. J. Med. Chem. 2017, 60, 2697–2717. [Google Scholar]
- Topchiy, M.A.; Ageshina, A.A.; Chesnokov, G.A.; Sterligov, G.K.; Rzhevskiy, S.A.; Gribanov, P.S.; Osipov, S.N.; Nechaev, M.S.; Asachenko, A.F. Alkynyl- or Azido-Functionalized 1,2,3-Triazoles: Selective MonoCuAAC Promoted by Physical Factors. ChemistrySelect 2019, 4, 7470–7475. [Google Scholar]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gribanov, P.S.; Philippova, A.N.; Smol’yakov, A.F.; Tukhvatullina, D.N.; Vlasova, V.A.; Topchiy, M.A.; Asachenko, A.F.; Osipov, S.N. Synthesis of Functionalized 1H-Imidazoles via Denitrogenative Transformation of 5-Amino-1,2,3-Triazoles. Molecules 2025, 30, 1401. https://doi.org/10.3390/molecules30071401
Gribanov PS, Philippova AN, Smol’yakov AF, Tukhvatullina DN, Vlasova VA, Topchiy MA, Asachenko AF, Osipov SN. Synthesis of Functionalized 1H-Imidazoles via Denitrogenative Transformation of 5-Amino-1,2,3-Triazoles. Molecules. 2025; 30(7):1401. https://doi.org/10.3390/molecules30071401
Chicago/Turabian StyleGribanov, Pavel S., Anna N. Philippova, Alexander F. Smol’yakov, Diana N. Tukhvatullina, Viktoria A. Vlasova, Maxim A. Topchiy, Andrey F. Asachenko, and Sergey N. Osipov. 2025. "Synthesis of Functionalized 1H-Imidazoles via Denitrogenative Transformation of 5-Amino-1,2,3-Triazoles" Molecules 30, no. 7: 1401. https://doi.org/10.3390/molecules30071401
APA StyleGribanov, P. S., Philippova, A. N., Smol’yakov, A. F., Tukhvatullina, D. N., Vlasova, V. A., Topchiy, M. A., Asachenko, A. F., & Osipov, S. N. (2025). Synthesis of Functionalized 1H-Imidazoles via Denitrogenative Transformation of 5-Amino-1,2,3-Triazoles. Molecules, 30(7), 1401. https://doi.org/10.3390/molecules30071401