The Adsorption of Ru-Based Dyes on the TiO2 Surface to Enhance the Photovoltaic Efficiency of Dye-Sensitized Solar Cell Devices

 , , ,
, , ,  and
and
Abstract
1. Introduction
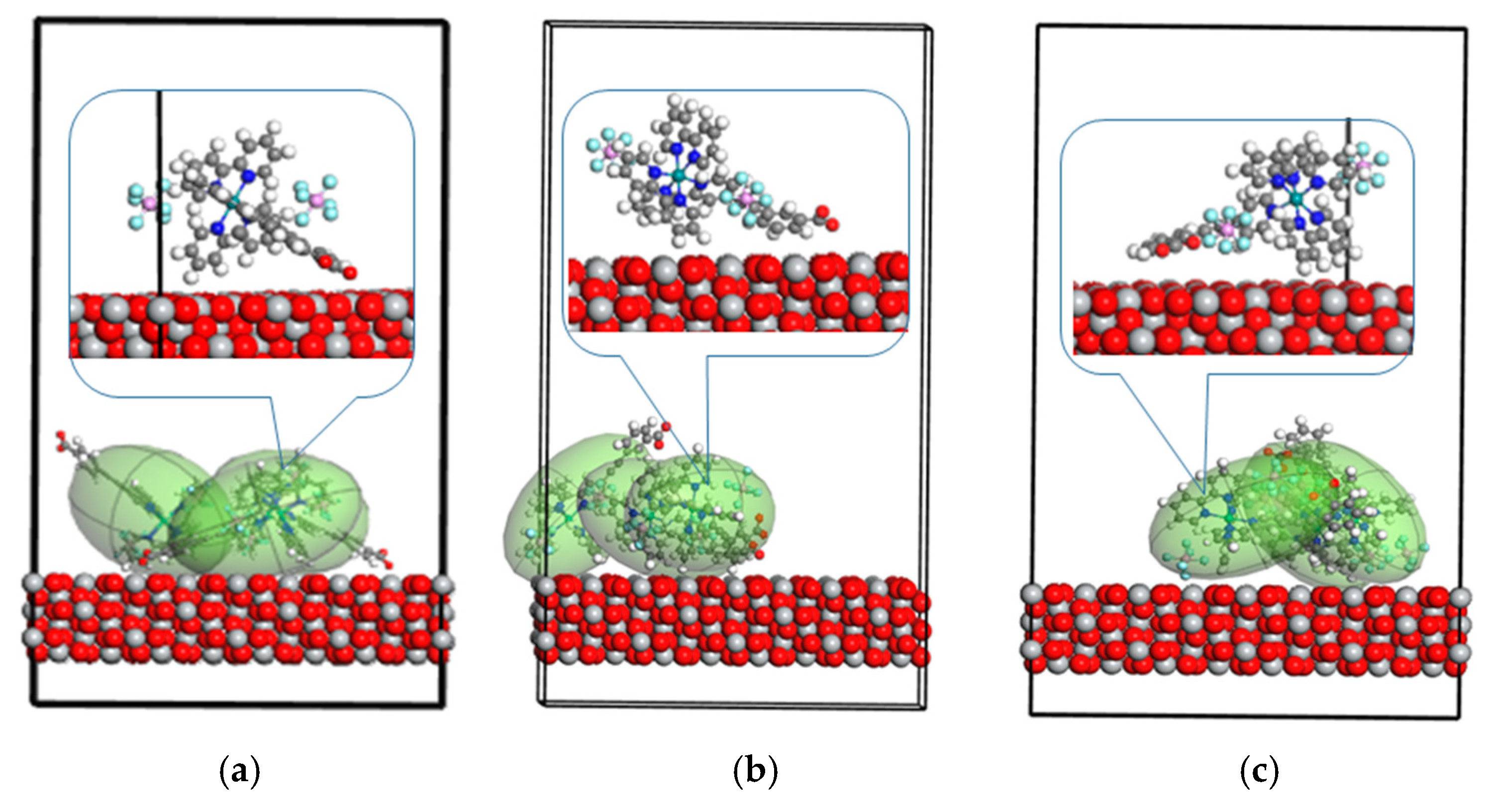
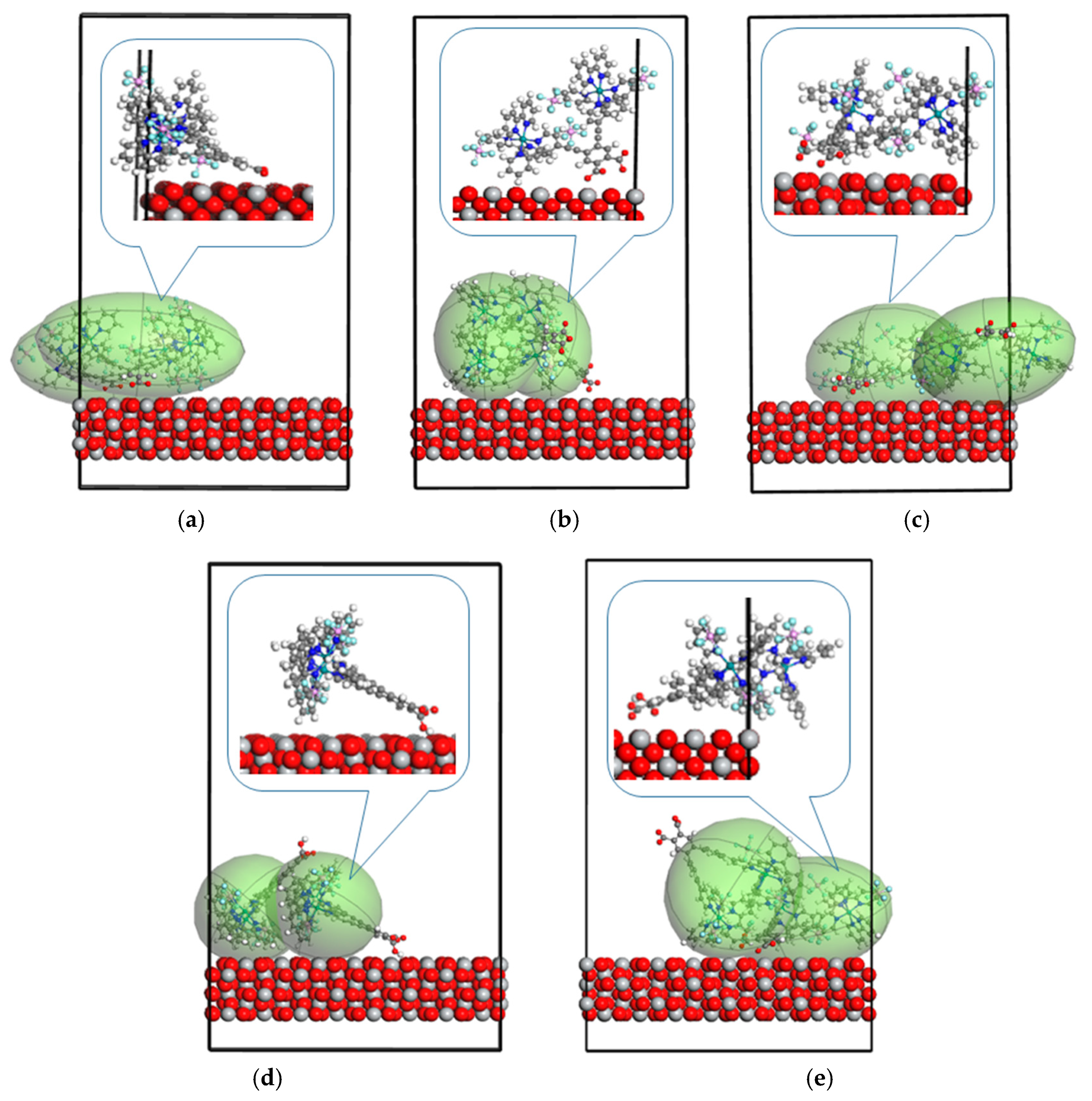
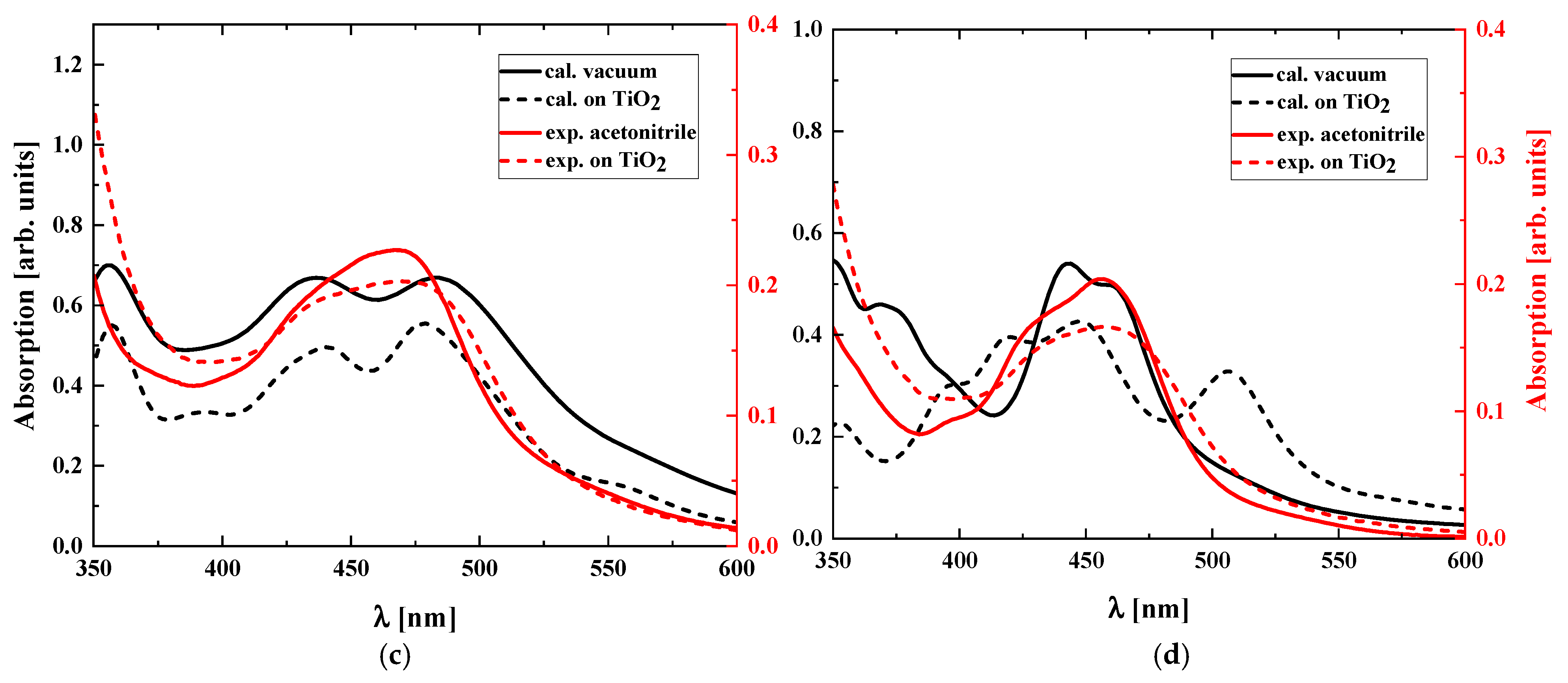
2. Results
3. Discussion
4. Computer Simulation Methodology
4.1. Computer Simulations
4.2. Experimental Setup and Sample Preparation
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Carella, A.; Borbone, F.; Centore, R. Research Progress on Photosensitizers for DSSC. Front. Chem. 2018, 6, 481. [Google Scholar] [CrossRef] [PubMed]
- Sasikumar, R.; Thirumalaisamy, S.; Kim, B.; Hwang, B. Dye-sensitized solar cells: Insights and research divergence towards alternatives. Renew. Sustain. Energy Rev. 2024, 199, 114549. [Google Scholar] [CrossRef]
- Gong, J.; Sumathy, K.; Qiao, Q.; Zhou, Z. Review on dye-sensitized solar cells (DSSCs): Advanced techniques and research trends. Renew. Sustain. Energy Rev. 2017, 68, 234–246. [Google Scholar] [CrossRef]
- Baby, R.; Nixon, P.D.; Kumar, N.M.; Subathra, M.S.P.; Ananthi, N. A comprehensive review of dye-sensitized solar cell optimal fabrication conditions, natural dye selection, and application-based future perspectives. Environ. Sci. Pollut. Res. 2022, 29, 371–404. [Google Scholar] [CrossRef]
- O’Regan, B.; Gratzel, M. A low-cost, high-efficiency solar cell based on dye-sensitized colloidal TiO2 films. Nature 1991, 353, 737–740. [Google Scholar] [CrossRef]
- Ren, Y.; Zhang, D.; Suo, J.; Cao, Y.; Eickemeyer, F.T.; Vlachopoulos, N.; Zakeeruddin, S.M.; Hagfeldt, A.; Gratzel, M. Hydroxamic acid pre-adsorption raises the efficiency of cosensitized solar cells. Nature 2022, 613, 60–65. [Google Scholar] [CrossRef]
- Srivastava, K.V.; Srivastava, P.; Srivastava, A.; Maurya, R.K.; Singha, Y.P.; Srivastava, A. 1D TiO2 photoanodes: A game-changer for high efficiency dye-sensitized solar cells. RSC Adv. 2025, 15, 4789–4819. [Google Scholar] [CrossRef]
- Vallejo, W.; Lerma, M.; Díaz-Uribe, C. Dye sensitized solar cells: Meta-analysis of effect sensitizer-type on photovoltaic efficiency. Heliyon 2025, 11, e41092. [Google Scholar] [CrossRef]
- Brewster, T.P.; Konezny, S.J.; Sheehan, S.W.; Martini, L.A.; Schmuttenmaer, C.A.; Batista, V.S.; Crabtree, R.H. Hydroxamate Anchors for Improved Photoconversion in Dye-Sensitized Solar Cells. Inorg. Chem. 2013, 52, 6752–6764. [Google Scholar] [CrossRef]
- Hirva, P.; Haukka, M. Effect of Different Anchoring Groups on the Adsorption of Photoactive Compounds on the Anatase (101) surface. Langmuir 2010, 26, 17075–17081. [Google Scholar] [CrossRef]
- Baik, C.; Kim, D.; Kang, M.-S.; Kang, S.O.; Ko, J.; Nazeeruddin, M.K.; Grätzel, M. Organic Dyes with a Novel Anchoring Group for Dye-Sensitized Solar Cell Applications. J. Photochem. Photobiol. A 2009, 201, 168–174. [Google Scholar] [CrossRef]
- Mao, J.; He, N.; Ning, Z.; Zhang, Q.; Guo, F.; Chen, L.; Wu, W.; Hua, J.; Tian, H. Stable Dyes Containing Double Acceptors without COOH as Anchors for Highly Efficient Dye-Sensitized Solar Cells. Angew. Chem. Int. Ed. 2012, 51, 9873–9876. [Google Scholar] [CrossRef] [PubMed]
- Qu, Q.; Geng, H.; Peng, R.; Cui, Q.; Gu, X.; Li, F.; Wang, M. Chemically Binding Carboxylic Acids onto TiO2 Nanoparticles with Adjustable Coverage by Solvothermal Strategy. Langmuir 2010, 26, 9161–10396. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.-Q.; Ding, D.; Zhang, M.; Chen, S.; Qiu, Z.; Yan, J.-W.; Yang, Z.-L.; Chen, M.-S.; Mao, B.-W.; Tian, Z.-Q. Adsorption of Dye Molecules on Single Crystalline SemiconductorSurfaces: An Electrochemical Shell-Isolated Nanoparticle EnhancedRaman Spectroscopy Study. J. Phys. Chem. C 2016, 120, 22500–22507. [Google Scholar] [CrossRef]
- Murakoshi, K.; Kano, G.; Wada, Y.; Yanagida, S.; Miyazaki, H.; Matsumoto, M.; Murasawa, S. Importance of Binding States between Photosensitizing Molecules and the TiO2 Surface for Efficiency in aDye-Sensitized Solar Cell. J. Electroanal. Chem. 1995, 396, 27–34. [Google Scholar] [CrossRef]
- De Angelis, F.; Fantacci, S.; Selloni, A.; Nazeeruddin, M.K.; Grätzel, M. First-Principles Modeling of the Adsorption Geometry and Electronic Structure of Ru(II) Dyes on Extended TiO2 Substrates for Dye-Sensitized Solar Cell Applications. J. Phys. Chem. C 2010, 114, 6054–6061. [Google Scholar] [CrossRef]
- Anselmi, C.; Mosconi, E.; Pastore, M.; Ronca, E.; De Angelis, F. Adsorption of organic dyes on TiO2 surfaces in dye-sensitized solar cells: Interplay of theory and experiment. Phys. Chem. Chem. Phys. 2012, 14, 15963–15974. [Google Scholar] [CrossRef]
- Manaa, M.B.; Issaoui, N.; Bouaziz, N.; Lamine, A.B. Combined statistical physics models and DFT theory to study the adsorption process of paprika dye On TiO2 for dye sensitized solar cells. J. Mater. Res. Technol. 2020, 9, 1175–1188. [Google Scholar] [CrossRef]
- Narayanaswamy, V.; Alaabed, S.; AL-Akhras, M.-A.; Obaidat, I.M. Molecular simulation of adsorption of methylene blue and rhodamine B on graphene and graphene oxide for water purification. Mater. Today 2020, 28, 1078–1083. [Google Scholar] [CrossRef]
- Pastore, M.; De Angelis, F. Computational modelling of TiO2 surfaces sensitized by organic dyes with different anchoring groups: Adsorption modes, electronic structure and implication for electron injection/recombination. Phys. Chem. Chem. Phys. 2012, 14, 920–928. [Google Scholar] [CrossRef]
- Vittadini, A.; Selloni, A.; Rotzinger, F.P.; Grätzel, M. Formic Acid Adsorption on Dry and Hydrated TiO2 Anatase (101) Surfaces by DFT Calculations. J. Phys. Chem. B 2000, 104, 1300–1306. [Google Scholar] [CrossRef]
- Zalas, M.; Gierczyk, B.; Bossi, A.; Mussini, P.R.; Kleine, M.; Pankiewicz, R.; Makowska-Janusik, M.; Popenda, Ł.; Stampor, W. The influence of anchoring group position in ruthenium dye molecule on performance of dye-sensitized solar cells. Dyes Pigments 2018, 150, 335–346. [Google Scholar] [CrossRef]
- Bartkowiak, A.; Korolevych, O.; Gierczyk, B.; Pelczarski, D.; Bossi, A.; Klein, M.; Popenda, Ł.; Stampor, W.; Makowska-Janusik, M.; Zalas, M. The importance of anchoring ligands of binuclear sensitizers on electron transfer processes and photovoltaic action in dye-sensitized solar cells. Sci. Rep. 2023, 13, 16808. [Google Scholar] [CrossRef]
- Brogdon, P.; McNamara, L.E.; Peddapuram, A.; Hammer, N.I.; Delcamp, J.H. Toward tightly bound carboxylic acid-based organic dyes for DSCs: Relative TiO2 binding strengths of benzoic acid, cyanoacrylic acid, and conjugated double carboxylic acid anchoring dyes. Synth. Met. 2016, 222, 66–75. [Google Scholar] [CrossRef]
- Amoli, V.; Bhat, S.; Maurya, A.; Banerjee, B.; Bhaumik, A.; Sinha, A.K. Tailored Synthesis of Porous TiO2 Nanocubes and Nanoparallelepipeds with Exposed {111} Facets and Mesoscopic Void Space: A Superior Candidate for Efficient Dye-Sensitized Solar Cells. ACS Appl. Mater. Interfaces 2015, 7, 26022–26035. [Google Scholar] [CrossRef]
- Adhikari, S.G.; Gascooke, J.R.; Alotabi, A.S.; Andersson, G.G. Anchoring Modes of Ru-Based N719 Dye onto Titania Substrates. J. Phys. Chem. C 2024, 128, 3136–3147. [Google Scholar] [CrossRef]
- Nilsing, M.; Persson, P.; Lunell, S.; Ojamäe, L. Dye-Sensitization of the TiO2 Rutile (110) Surface by Perylene Dyes: Quantum-Chemical Periodic B3LYP Computations. Phys. Chem. C 2007, 111, 12116–12123. [Google Scholar] [CrossRef]
- Mydlova, L.; Sahraoui, B.; El-Ghayoury, A.; Berdowski, J.; Migalska-Zalas, A.; Makowska-Janusik, M. Hierarchical Modeling of the Nonlinear Optical Response of Composite Materials Based on Tetrathiafulvalene Derivatives. Molecules 2024, 29, 3720. [Google Scholar] [CrossRef]
- Funaki, T.; Yanagida, M.; Onozawa-Komatsuzaki, N.; Kawanishi, Y.; Kasuga, K.; Sugihara, H. Ruthenium (II) complexes with π expanded ligand having phenylene–ethynylene moiety as sensitizers for dye-sensitized solar cells. Sol. Energy Mater. Sol. Cells 2009, 93, 729–732. [Google Scholar] [CrossRef]
- Wu, M.; Zhang, Z.; Yong, J.; Schenk, P.M.; Tian, D.; Xu, Z.P.; Zhang, R. Determination and Imaging of Small Biomolecules and Ions Using Ruthenium(II) Complex-Based Chemosensors; Springer International Publishing: Berlin/Heidelberg, Germany, 2022; Volume 380, pp. 1–45. [Google Scholar] [CrossRef]
- Filevich, O.; Zayat, L.; Baraldo, L.M.; Etchenique, R. Long Wavelength Phototriggering: Ruthenium-Based Caged Compounds. In Luminescent and Photoactive Transition Metal Complexes as Biomolecular Probes and Cellular Reagents. Structure and Bonding; Springer: Berlin/Heidelberg, Germany, 2014; Volume 165, pp. 47–68. [Google Scholar] [CrossRef]
- Shi, W.; Peng, B.; Lin, L.; Li, R.; Zhang, J.; Peng, T. Effect of carboxyl anchoring groups in asymmetric zinc phthalocyanine with large steric hindrance on the dye-sensitized solar cell performance. Mater. Chem. Phys. 2015, 163, 348–354. [Google Scholar] [CrossRef]
- Korolevych, O.; Zalas, M.; Stampor, W.; Kassiba, A.H.; Makowska-Janusik, M. Impact of dyes isomerization effect on the charge transfer phenomenon occurring on the dye/nanosemiconductor interface. Sol. Energy Mater. Sol. Cells 2021, 219, 110771. [Google Scholar] [CrossRef]
- Douglas, M.; Kroll, N.M. Quantum electrodynamical corrections to the fine structure of helium. Ann. Phys. 1974, 82, 89–155. [Google Scholar] [CrossRef]
- Hess, B.A. Relativistic electronic-structure calculations employing a two-component no-pair formalism with external-field projection operators. Phys. Rev. A 1986, 33, 3742–3748. [Google Scholar] [CrossRef] [PubMed]
- Jansen, G.; Hess, B.A. Revision of the Douglas-Kroll transformation. Phys. Rev. A 1989, 39, 6016–6017. [Google Scholar] [CrossRef]
- Jorge, F.E.; Canal Neto, A.; Camiletti, G.G.; MacHado, S.F. Contracted Gaussian basis sets for Douglas–Kroll–Hess calculations: Estimating scalar relativistic effects of some atomic and molecular properties. J. Chem. Phys. 2009, 130, 064108. [Google Scholar] [CrossRef]
- Pelczarski, D.; Korolevych, O.; Gierczyk, B.; Zalas, M.; Makowska-Janusik, M.; Stampor, W. Electronic States of Tris(bipyridine) Ruthenium(II) Complexes in Neat Solid Films Investigated by Electroabsorption Spectroscopy. Materials 2022, 15, 2278. [Google Scholar] [CrossRef]
- Rappé, A.K.; Casewit, C.J.; Colwell, K.S.; Goddard, W.A.; Skiff, W.M. UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations. J. Am. Chem. Soc. 1992, 114, 10024–10035. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Perdew, J.P.; Chevary, J.A.; Vosko, S.H.; Jackson, K.A.; Pederson, M.R.; Singh, D.J.; Fiolhais, C. Atoms, molecules, solids, and surfaces: Applications of the generalized gradient approximation for exchange and correlation. Phys. Rev. B 1992, 46, 6671–6687. [Google Scholar] [CrossRef] [PubMed]
- Hüfner, S. Electronic structure of NiO and related 3d-transition-metal compounds. Adv. Phys. 1994, 43, 183–356. [Google Scholar] [CrossRef]
- Liechtenstein, A.I.; Anisimov, V.I.; Zaanen, J. Density-functional theory and strong interactions: Orbital ordering in Mott-Hubbard insulators. Phys. Rev. B 1995, 52, R5467–R5470. [Google Scholar] [CrossRef]
- Bartkowiak, A.; Korolevych, O.; Chiarello, G.L.; Makowska-Janusik, M.; Zalas, M. Experimental and theoretical insight into DSSCs mechanism influenced by different doping metal ions. Appl. Surf. Sci. 2022, 597, 153607. [Google Scholar] [CrossRef]
- Pelczarski, D.; Gierczyk, B.; Zalas, M.; Makowska-Janusik, M.; Stampor, W. Excited states of mono- and biruthenium(II) complexes adsorbed on nanocrystalline titanium dioxide studied by electroabsorption spectroscopy. Sci. Rep. 2025, 15, 4562. [Google Scholar] [CrossRef]
- Zalas, M.; Gierczyk, B.; Klein, M.; Siuzdak, K.; Pȩdziński, T.; Łuczak, T. Synthesis of a novel dinuclear ruthenium polypyridine dye for dye-sensitized solar cells application. Polyhedron 2014, 67, 381–387. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule | Total Energy [kcal/mol] | Total Energy/Atom [kcal/mol] | Adsorption Energy [kcal/mol] | [kcal/mol] |
|---|---|---|---|---|
| RuLp | 3.98 × 103 | 0.45 × 103 | −113.55 | −52.24 |
| RuLm | 6.95 × 103 | 0.78 × 103 | −109.56 | −42.50 |
| RuLo | 6.90 × 103 | 0.77 × 103 | −83.04 | −33.47 |
| B1 | 5.29 × 103 | 0.55 × 103 | −72.72 | −43.64 |
| B2 | 5.05 × 103 | 0.53 × 103 | −68.77 | −53.71 |
| B2H | 5.02 × 103 | 0.52 × 103 | −83.36 | −53.94 |
| B3 | 5.45 × 103 | 0.55 × 103 | −84.11 | −52.61 |
| B3H | 5.44 × 103 | 0.55 × 103 | −81.34 | −46.84 |
| Molecule | RuLp | RuLm | RuLo | B1 | B2 | B2H | B3 | B3H |
|---|---|---|---|---|---|---|---|---|
| Distance [Å] | 1.05 | 2.13 | 2.42 | 1.35 | 1.15 | 2.39 | 1.08 | 1.47 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Makowska-Janusik, M.; Filipecka-Szymczyk, K.; Pelczarski, D.; Stampor, W.; Zalas, M. The Adsorption of Ru-Based Dyes on the TiO2 Surface to Enhance the Photovoltaic Efficiency of Dye-Sensitized Solar Cell Devices. Molecules 2025, 30, 1312. https://doi.org/10.3390/molecules30061312
Makowska-Janusik M, Filipecka-Szymczyk K, Pelczarski D, Stampor W, Zalas M. The Adsorption of Ru-Based Dyes on the TiO2 Surface to Enhance the Photovoltaic Efficiency of Dye-Sensitized Solar Cell Devices. Molecules. 2025; 30(6):1312. https://doi.org/10.3390/molecules30061312
Chicago/Turabian StyleMakowska-Janusik, Malgorzata, Katarzyna Filipecka-Szymczyk, Daniel Pelczarski, Waldemar Stampor, and Maciej Zalas. 2025. "The Adsorption of Ru-Based Dyes on the TiO2 Surface to Enhance the Photovoltaic Efficiency of Dye-Sensitized Solar Cell Devices" Molecules 30, no. 6: 1312. https://doi.org/10.3390/molecules30061312
APA StyleMakowska-Janusik, M., Filipecka-Szymczyk, K., Pelczarski, D., Stampor, W., & Zalas, M. (2025). The Adsorption of Ru-Based Dyes on the TiO2 Surface to Enhance the Photovoltaic Efficiency of Dye-Sensitized Solar Cell Devices. Molecules, 30(6), 1312. https://doi.org/10.3390/molecules30061312