
Synthesis and Structural Analysis of New (−)-Cytisine Squaramides



Abstract
1. Introduction

2. Results and Discussion
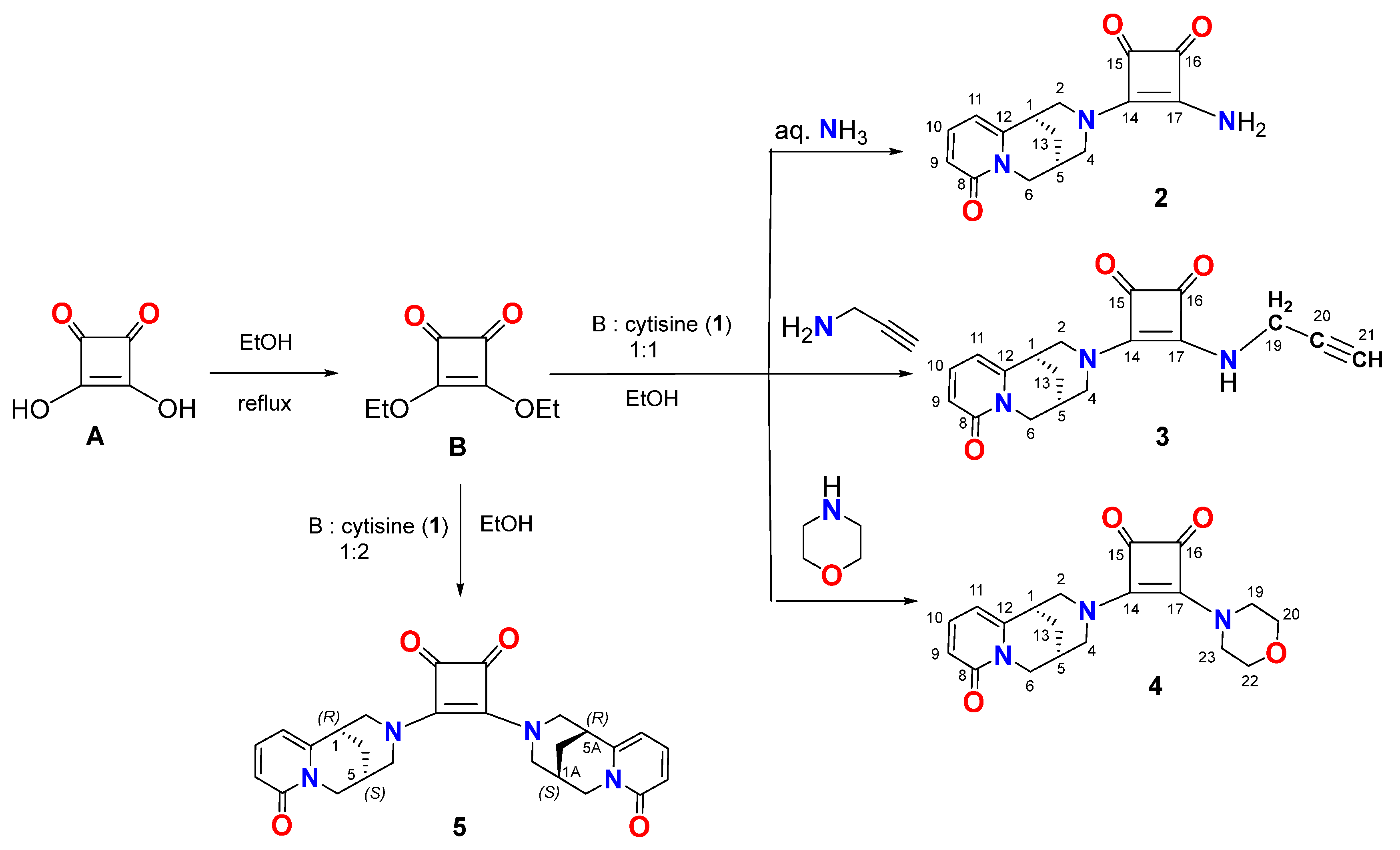
2.1. Chemistry
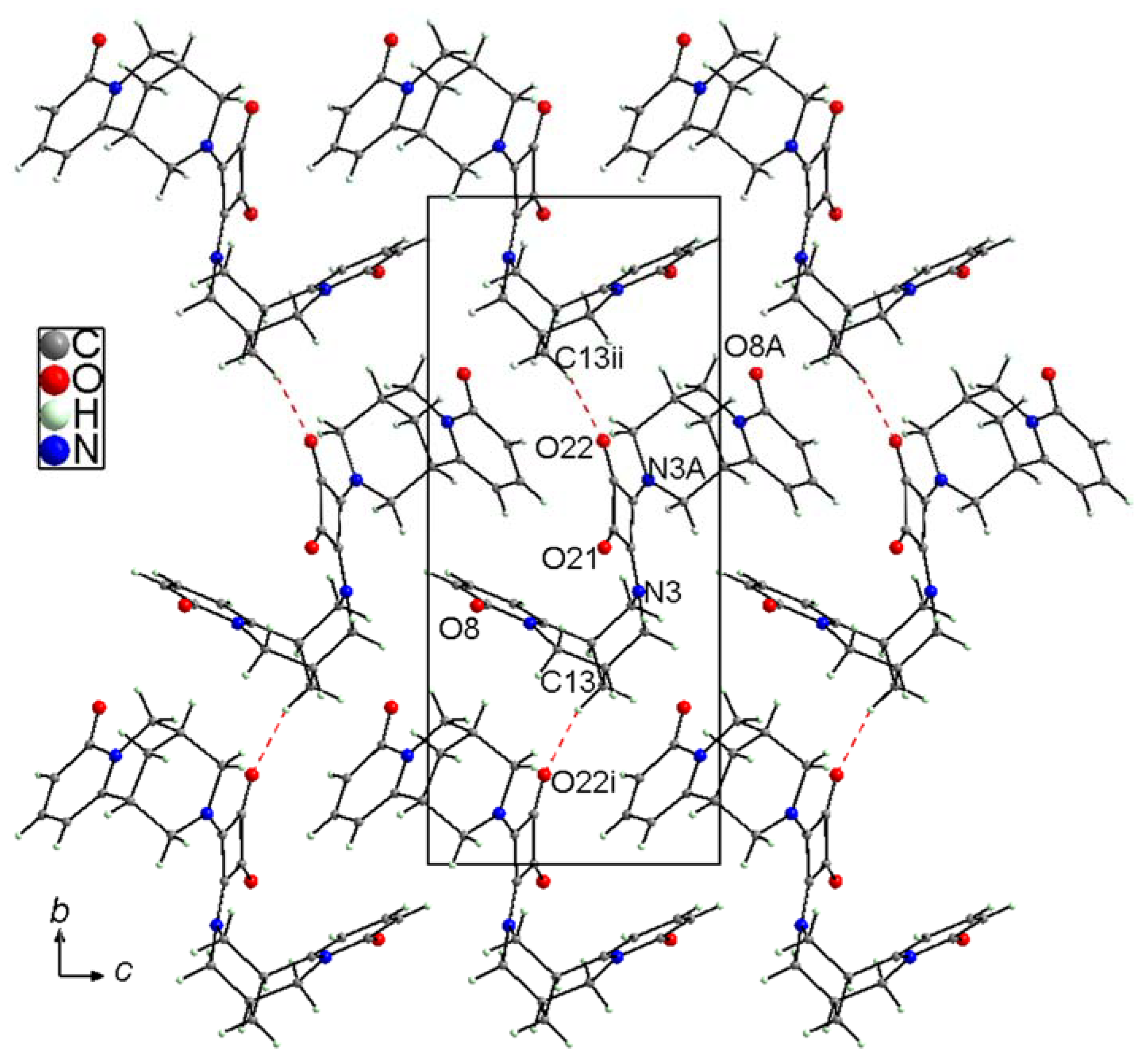
2.2. X-Ray Structure Characterization
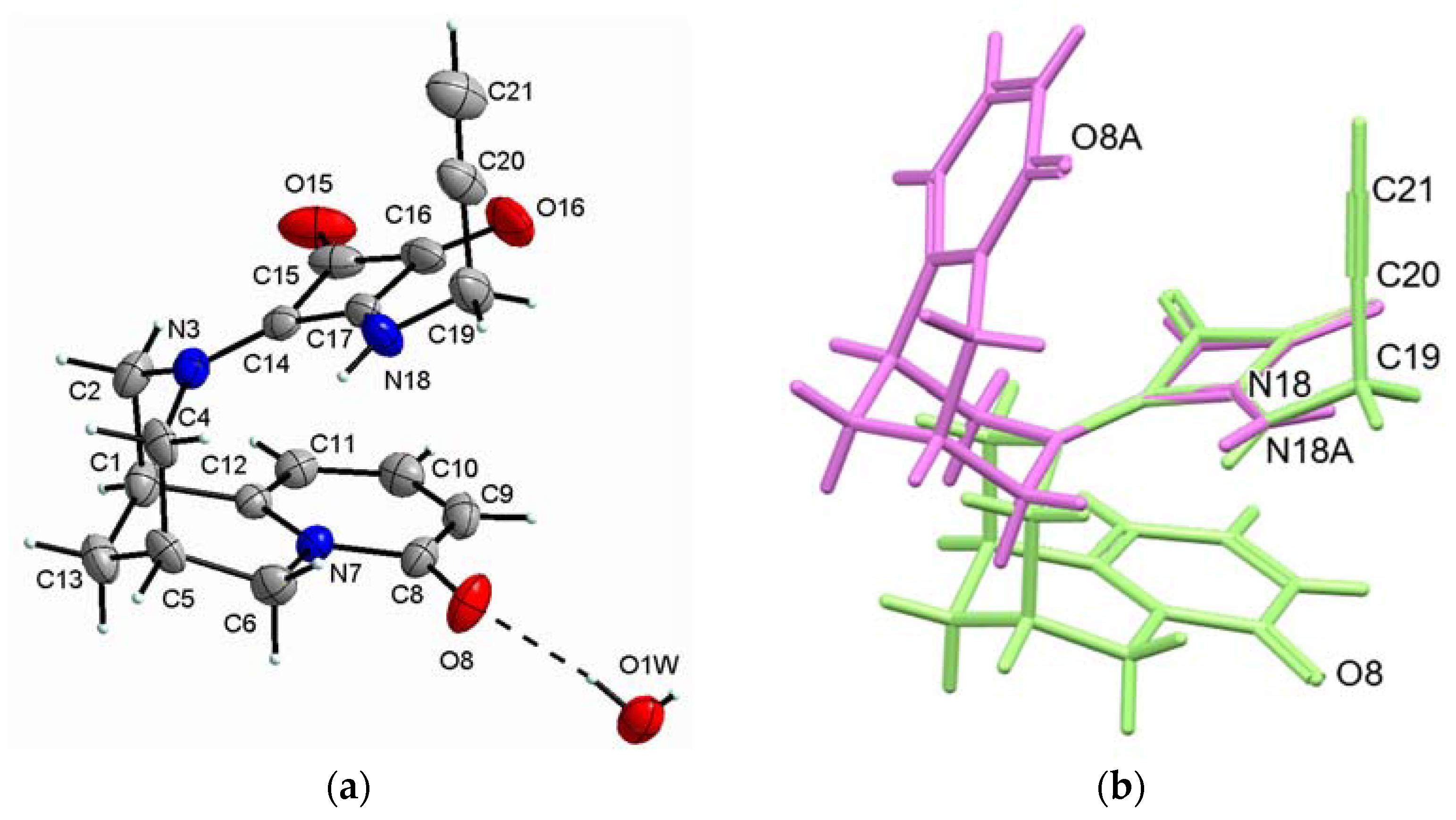
2.2.1. (−)-Cytisine Squaramide 2
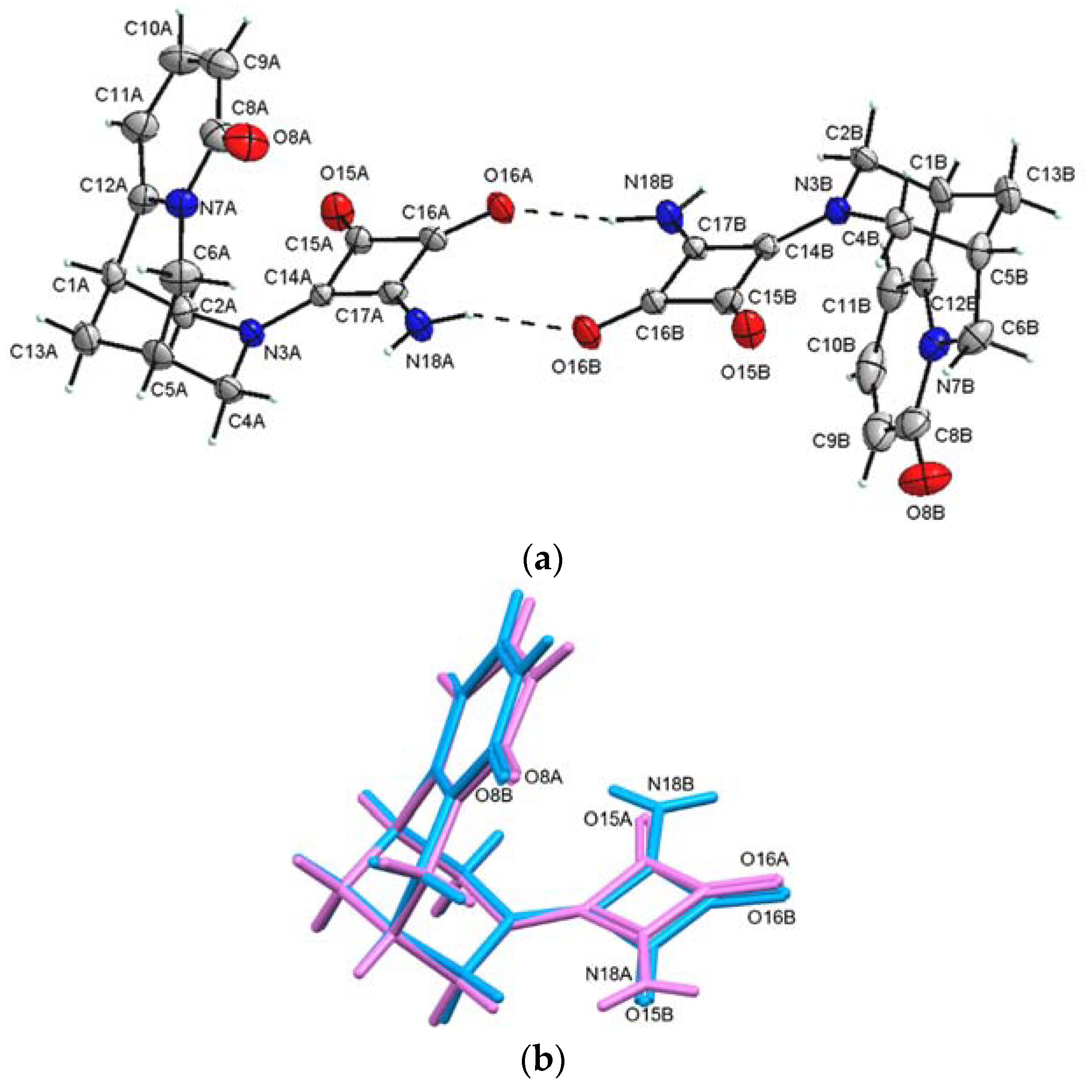
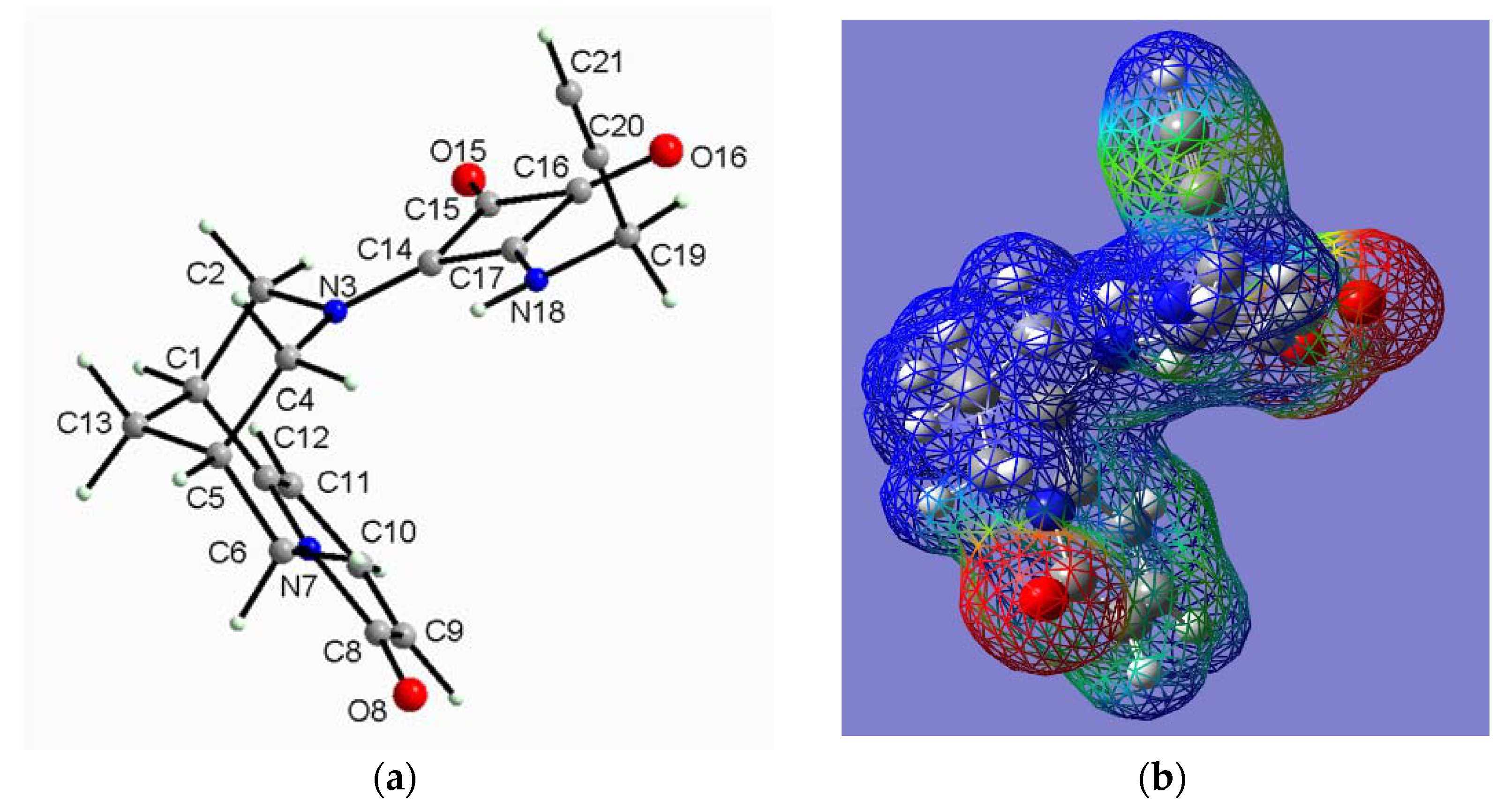
2.2.2. (−)-Cytisine Squaramide 3‧H2O
2.2.3. (−)-Cytisine Squaramide 4
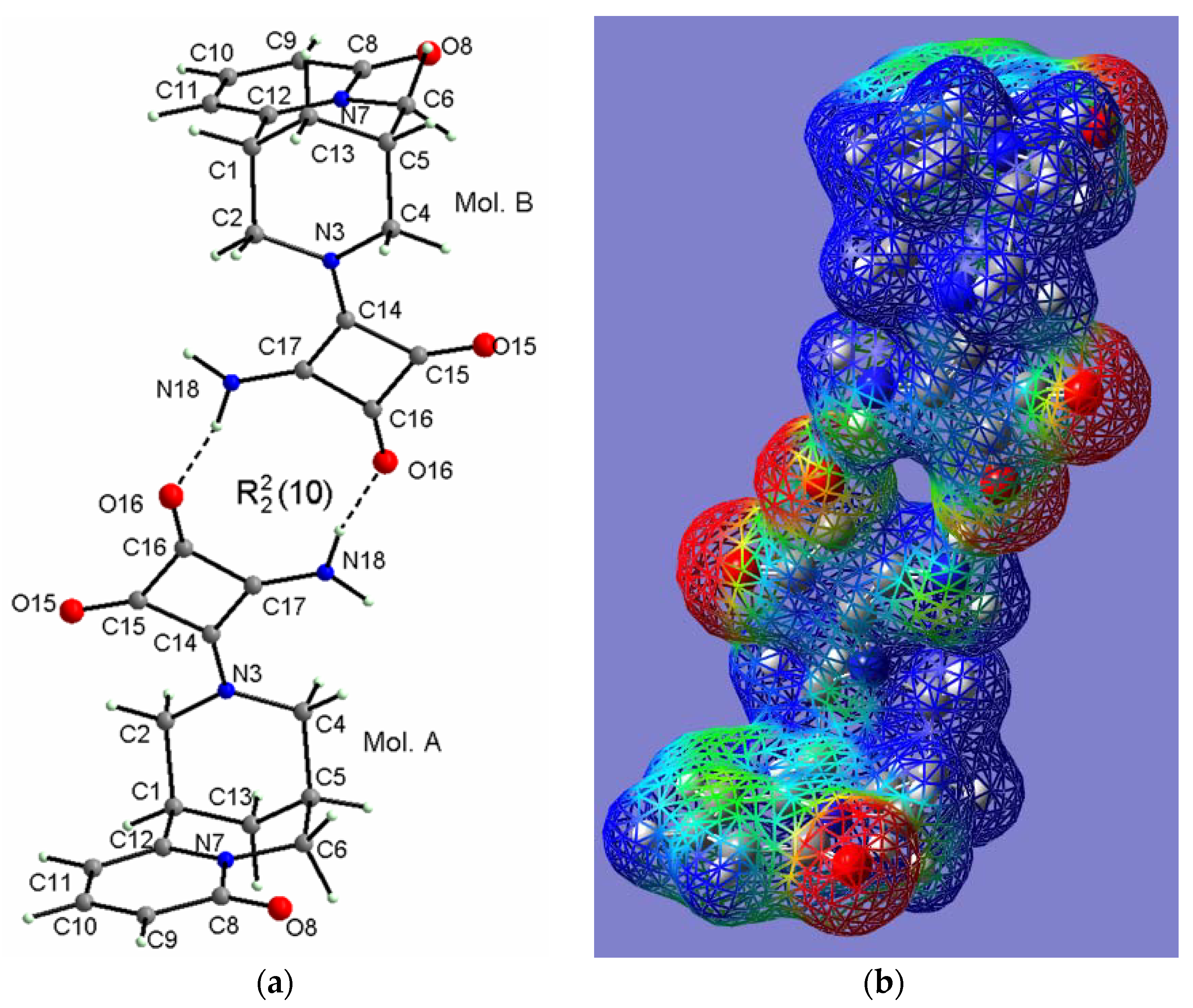
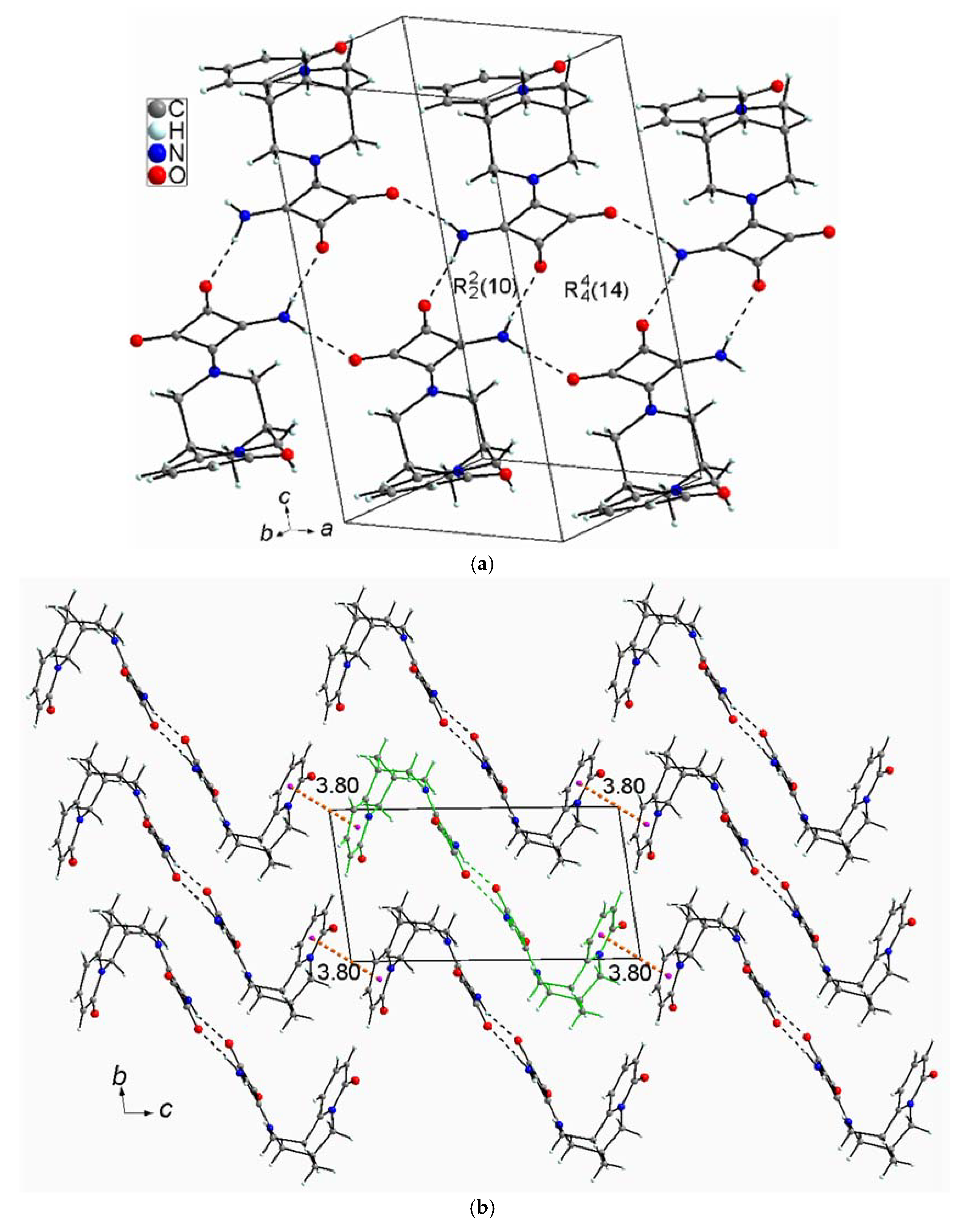
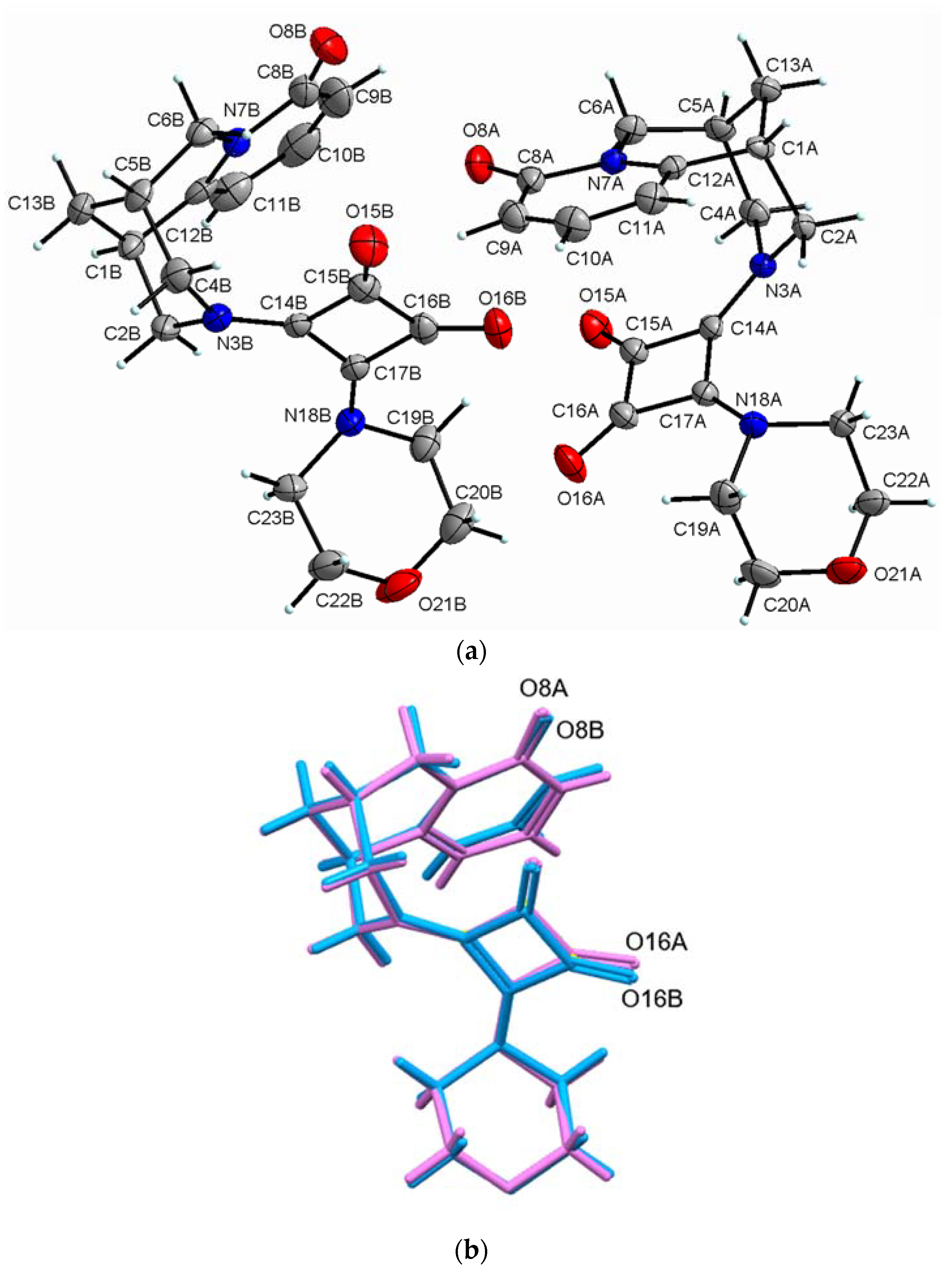
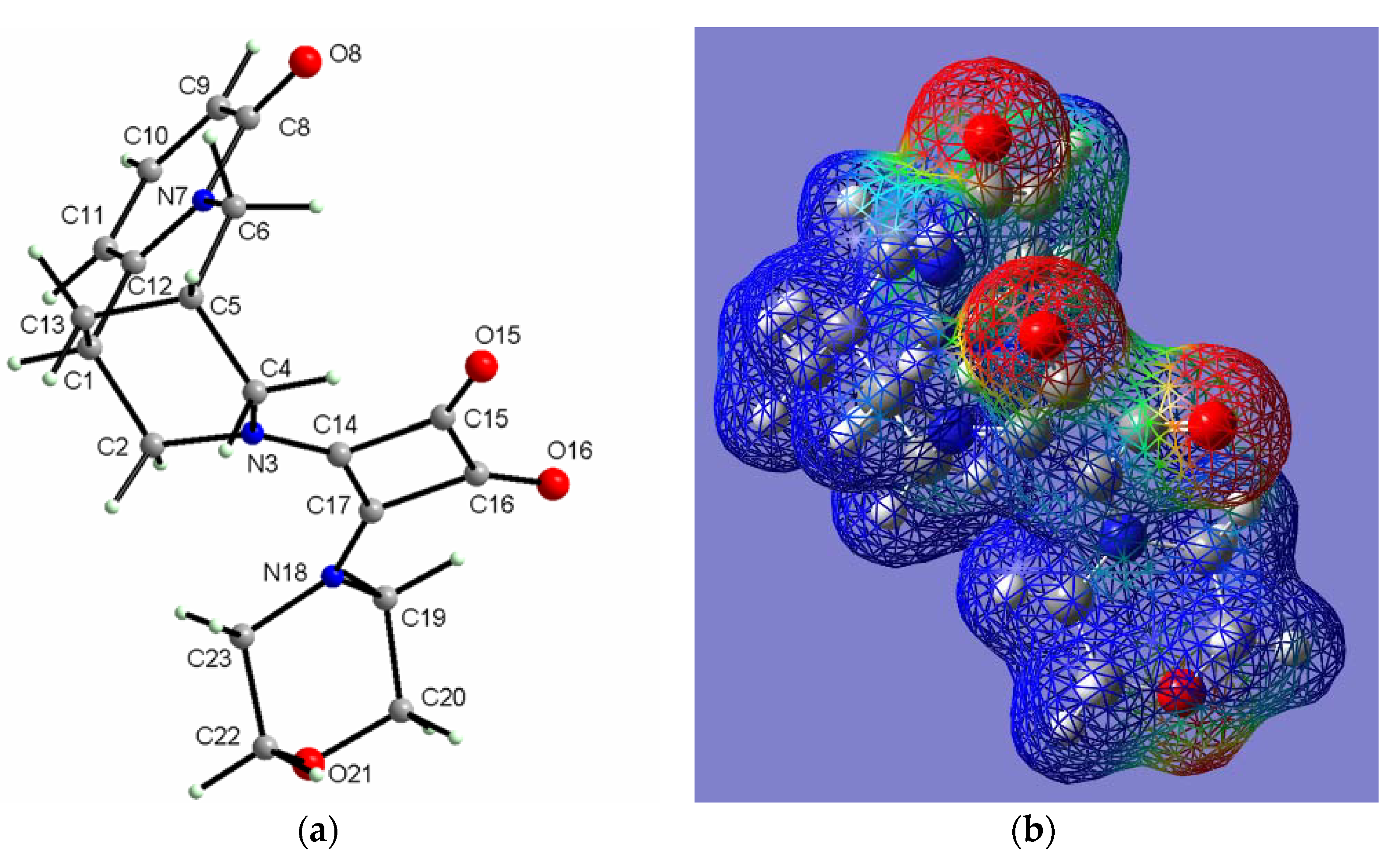
2.2.4. (−)-Cytisine Squaramide 5
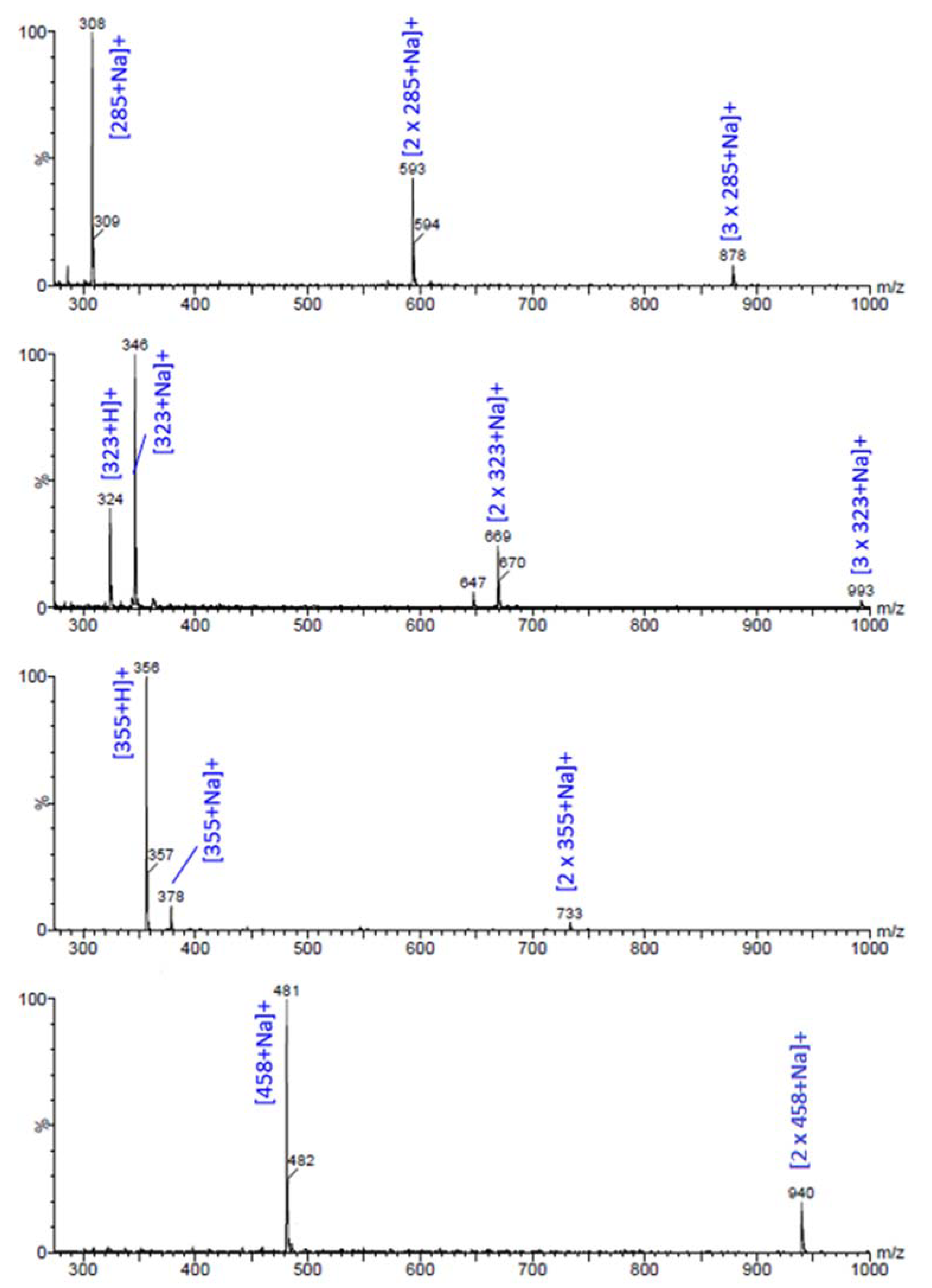
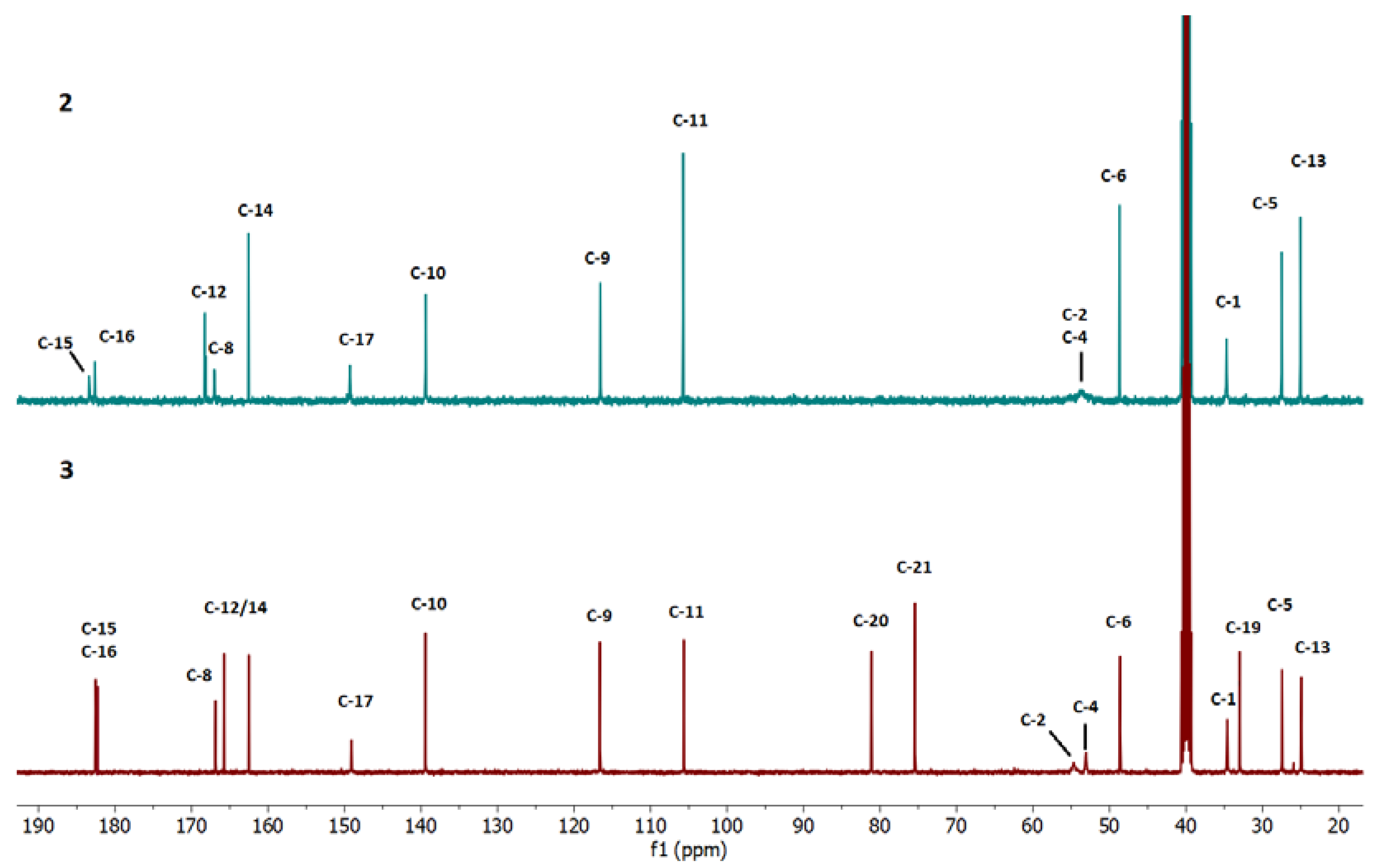
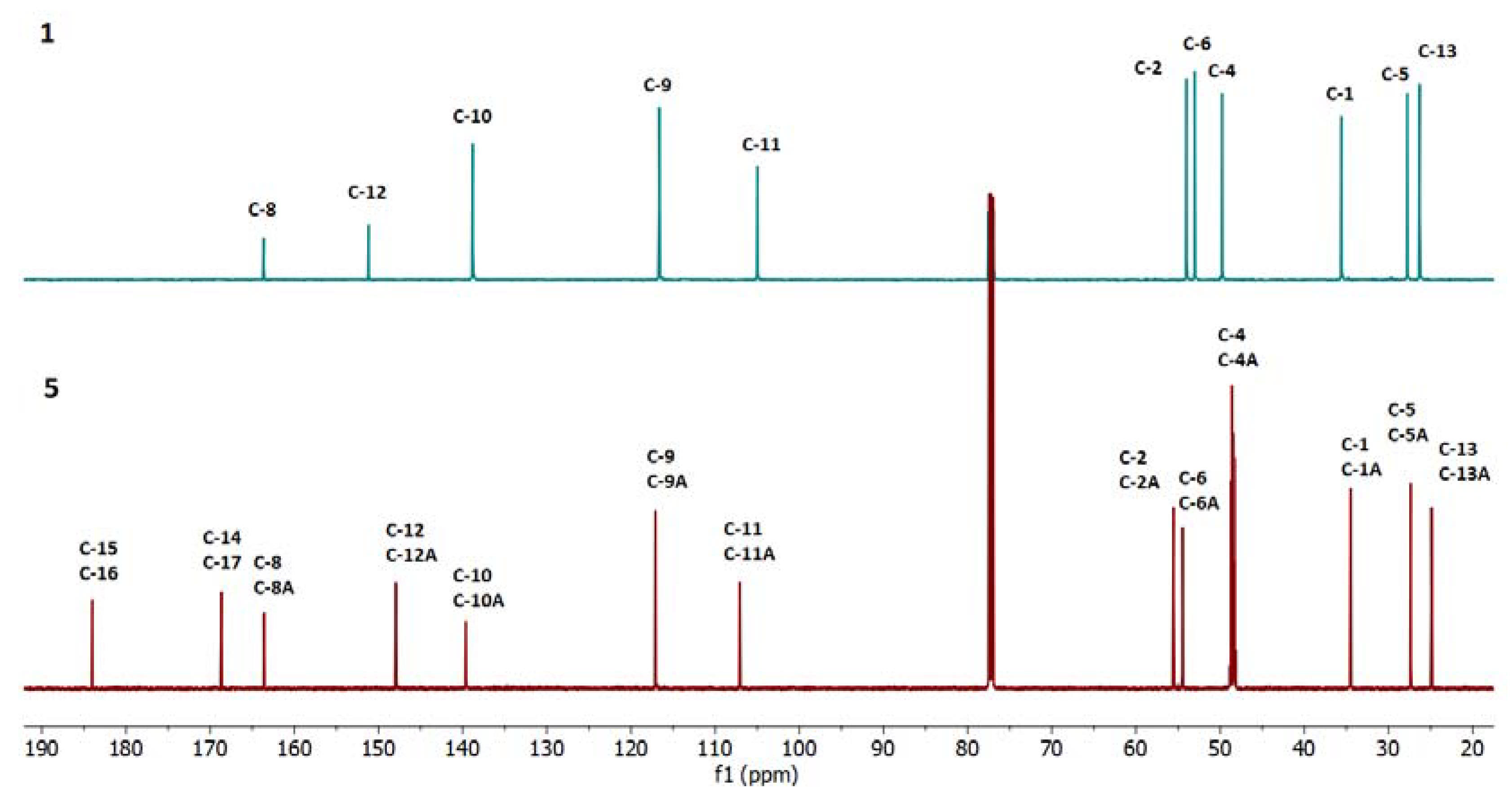
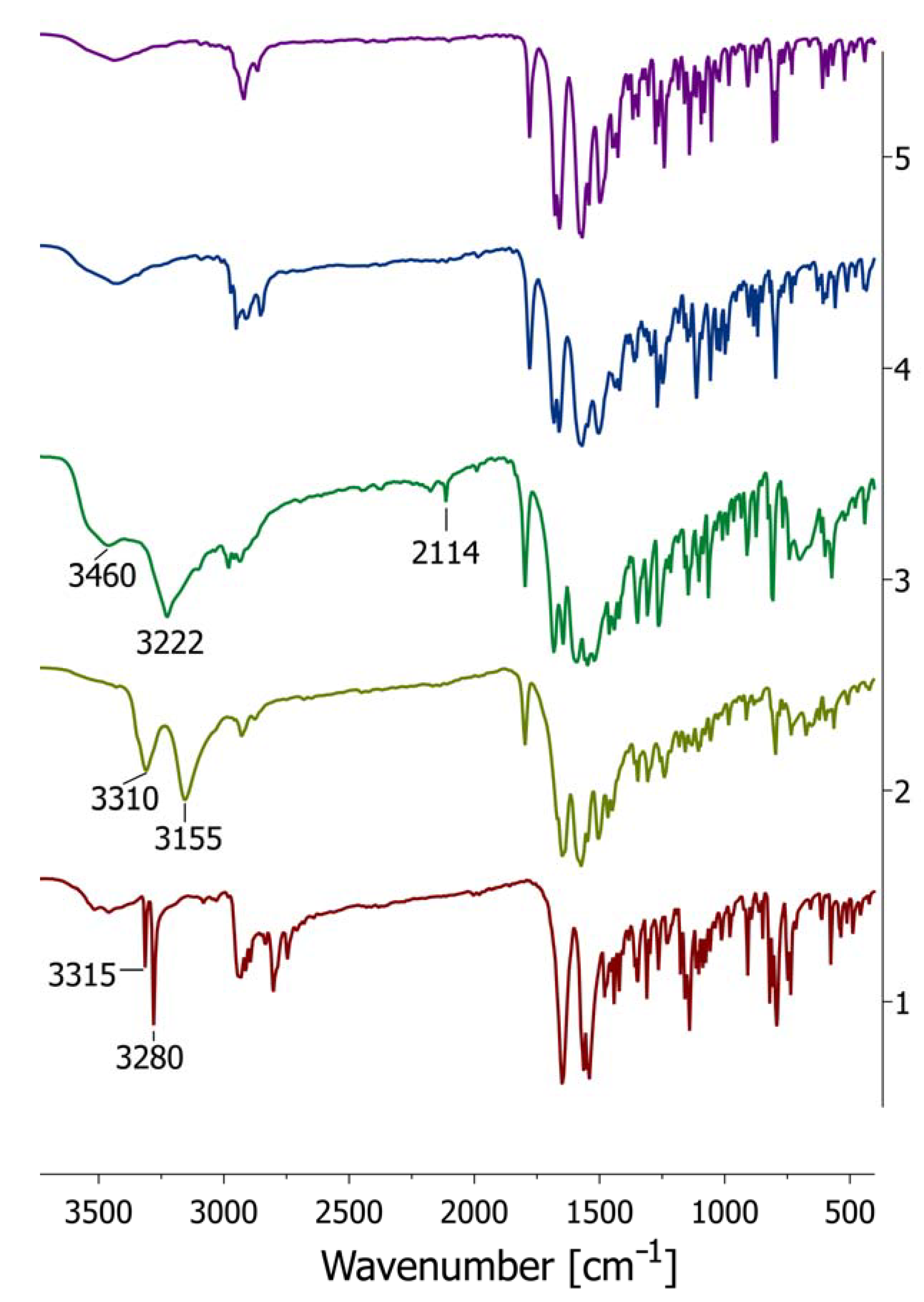
2.3. Spectroscopic Analysis
3. Materials and Methods
3.1. General
3.2. Synthesis of New (−)-Cytisine Squaramides (2-5)
3.3. X-Ray Single Crystal Data Collection
3.4. DFT Calculation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Agnew-Francis, K.A.; Williams, C.M. Squaramides as Bioisosteres in Contemporary Drug Design. Chem. Rev. 2020, 120, 11616–11650. [Google Scholar] [CrossRef] [PubMed]
- Chasák, J.; Šlachtová, V.; Urban, M.; Brulíková, L. Squaric acid analogues in medicinal chemistry. Eur. J. Med. Chem. 2021, 209, 112872–112894. [Google Scholar] [CrossRef]
- Bakalova, A.G.; Ruseva, N.K.; Cherneva, E.D. A Concise Review on Application of Squaric Acid Derivatives and Their Metal Complexes in Medicine. Arkivoc 2022, 1, 285–303. [Google Scholar] [CrossRef]
- Tremblay, T.; Bergeron, C.; Gagnon, D.; Bérubé, C.; Voyer, N.; Richard, D.; Giguè, D. Squaramide Tethered Clindamycin, Chloroquine, and Mortiamide Hybrids: Design, Synthesis, and Antimalarial Activity. ACS Med. Chem. Lett. 2023, 14, 217–222. [Google Scholar] [CrossRef]
- Wurm, F.R.; Klok, H.-A. Be Squared: Expanding the Horizon of Squaric Acid-Mediated Conjugations. Chem. Soc. Rev. 2013, 42, 8220–8236. [Google Scholar] [CrossRef]
- Long, N.; Gresley, A.L.; Solomonsz, A.; Wozniak, A.; Brough, S.; Wren, S.P. Synthesis of Squaric Acid Monoamides as Building Blocks for Drug Discovery. SynOpen 2023, 7, 401–407. [Google Scholar] [CrossRef]
- De Oliveira, V.; Da Cunha, A.R.; De Almeida Rizzutto, M.; Lamy, M.T.; De Oliveira, L.F.C.; Milán-Garcés, E.A. Vibrational Spectroscopic Analysis of 1,3-Dianiline Squarate: Infrared, Normal Raman, Surface-Enhanced Raman Scattering, and Density Functional Theory Calculations. J. Phys. Chem. C 2023, 127, 421–428. [Google Scholar] [CrossRef]
- He, J.; Jo, Y.J.; Sun, X.; Qiao, W.; Ok, J.; Kim, T.; Li, Z. Squaraine Dyes for Photovoltaic and Biomedical Applications. Adv. Funct. Mater. 2021, 31, 2008201. [Google Scholar] [CrossRef]
- Kelly, M.; Mandlik, A.; Charles, R.C.; Verma, S.; Calderwood, S.B.; Leung, D.T.; Biswas, R.; Islam, K.; Kamruzzaman, M.; Chowdhury, F.; et al. Development of Shigella conjugate vaccines targeting Shigella flexneri 2a and S. flexneri 3a using a simple platform-approach conjugation by squaric acid chemistry. Vaccine 2023, 41, 4967–4977. [Google Scholar] [CrossRef] [PubMed]
- Grus, T.; Lahnif, H.; Klasen, B.; Moon, E.S.; Greifenstein, L.; Roesch, F. Squaric Acid-Based Radiopharmaceuticals for Tumor Imaging and Therapy. Bioconjug. Chem. 2021, 32, 1223–1231. [Google Scholar] [CrossRef] [PubMed]
- Greifenstein, L.; Engelbogen, N.; Lahnif, H.; Sinnes, J.P.; Bergmann, R.; Bachmann, M.; Rösch, F. Synthesis, Labeling and Preclinical Evaluation of a Squaric Acid Containing PSMA Inhibitor Labeled with 68 Ga: A Comparison with PSMA-11 and PSMA-617. ChemMedChem 2020, 15, 695–704. [Google Scholar] [CrossRef] [PubMed]
- Lach, P.; Garcia-Cruz, A.; Canfarotta, F.; Groves, A.; Kalecki, J.; Korol, D.; Borowicz, P.; Nikiforow, K.; Cieplak, M.; Kutner, W.; et al. Electroactive molecularly imprinted polymer nanoparticles for selective glyphosate determination. Biosens. Bioelectron. 2023, 236, 115381. [Google Scholar] [CrossRef] [PubMed]
- Picci, G.; Farotto, S.; Milia, J.; Caltagirone, C.; Lippolis, V.; Aragoni, M.C.; Di Natale, C.; Paolesse, R.; Lvova, L. Potentiometric Sensing of Nonsteroidal Painkillers by Acyclic Squaramide Ionophores. ACS Sens. 2023, 8, 3225–3239. [Google Scholar] [CrossRef]
- Mamidi, N.; Delgadillo, R.M.V. Squaramide-Immobilized Carbon Nanoparticles for Rapid and High-Efficiency Elimination of Anthropogenic Mercury Ions from Aquatic Systems. ACS Appl. Mater. Interfaces 2022, 14, 35789–35801. [Google Scholar] [CrossRef] [PubMed]
- Ruseva, N.; Sbirkova-Dimitrova, H.; Atanasova, M.; Marković, A.; Šmelcerović, Ž.; Šmelcerović, A.; Bakalova, A.; Cherneva, E. Synthesis and DNase I inhibitory properties of new squaramides. Molecules 2023, 28, 538. [Google Scholar] [CrossRef]
- Dattatray Shinde, S.; Kumar Behera, S.; Kulkarni, N.; Dewangan, B.; Sahu, B. Bifunctional backbone modified squaramide dipeptides as amyloid beta (Aβ) aggregation inhibitors. Bioorg. Med. Chem. 2024, 97, 117538. [Google Scholar] [CrossRef]
- Perez, E.G.; Mendez-Galvez, C.; Cassels, B.K. Cytisine: A Natural Product Lead for the Development of Drugs Acting at Nicotinic Acetylcholine Receptors. Nat. Prod. Rep. (NPR) 2012, 29, 555–567. [Google Scholar] [CrossRef]
- Rouden, J.; Lasne, M.-C.; Blanchet, J.; Baudoux, J. (−)-Cytisine and Derivatives: Synthesis, Reactivity, and Applications. Chem. Rev. 2014, 114, 712–778. [Google Scholar] [CrossRef] [PubMed]
- Bartusik-Aebisher, D.; Aebisher, D.; Courtney, R.; Sobczak, A.; Bober, Z.; Podgorski, R.; Kolodziejczyk, P.; Tutka, P. Applications of Cytisine Extraction and Detection in Biological Materials for Clinical Medicine. Acta Pol. Pharm.-Drug Res. 2019, 76, 797–804. [Google Scholar] [CrossRef]
- Ofori, S.; Lu, C.; Olasupo, O.O.; Dennis, B.B.; Fairbairn, N.; Devereaux, P.J.; Mbuagbaw, L. Cytisine for Smoking Cessation: A Systematic Review and Meta-Analysis. Drug Alcohol Depend. 2023, 251, 110936. [Google Scholar] [CrossRef]
- Wang, X.; Yang, J.; Huang, P.; Wang, D.; Zhang, Z.; Zhou, Z.; Liang, L.; Yao, R.; Yang, L. Cytisine: State of the Art in Pharmacological Activities and Pharmacokinetics. Biomed. Pharmacother. 2024, 171, 116210. [Google Scholar] [CrossRef] [PubMed]
- Przybył, A.K.; Kubicki, M.; Jastrzab, R. Complexing ability of (−)-cytisine—Synthesis, spectroscopy and crystal structures of the new copper and zinc complexes. J. Inorg. Biochem. 2014, 138, 47–55. [Google Scholar] [CrossRef]
- Li, X.-S.; Zhao, J.; Jiao, Z.-H.; Zhao, X.-Y.; Hou, S.-L.; Zhao, B. Portably and Visually Sensing Cytisine through Smartphone Scanning Based on a Post-Modified Luminescence Center Strategy in Zinc-Organic Frameworks. Angew. Chem. Int. Ed. 2024, 63, e202401880. [Google Scholar] [CrossRef]
- Wijata, A.; Grajewska, A.; Huczyński, A. Synthesis and structural characterization of a new colchicine monosquarate-amide derivative. J. Mol. Struct. 2025, 1325, 140913. [Google Scholar] [CrossRef]
- Rigaku Oxford Diffraction, CrysAlisPro 1.171.42.93a Software System, Oxford, UK. 2023. Available online: https://rigaku.com/products/crystallography/x-ray-diffraction/crysalispro (accessed on 28 February 2025).
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Putz, H.; Brandenburg, K. DIAMOND, Version 3.0; Crystal Impact GbR: Bonn, Germany, 2014. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian09; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Becke, A.D. Density Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. PhysRev B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Dennington, R.; Keith, T.; Millam, J. GaussView, Version 5.0.8; Semichem Inc.: Shawnee Mission, KS, USA, 2009. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| N18A—H18A⋯O16B | 0.88 (4) | 2.03 (4) | 2.870 (5) | 160 (4) |
| N18A—H18B⋯O15A i | 0.93 (6) | 2.03 (6) | 2.955 (5) | 178 (5) |
| N18B—H18C⋯O16A | 0.96 (4) | 1.94 (5) | 2.890 (5) | 169 (4) |
| N18B—H18D⋯O15B ii | 0.88 (5) | 2.09 (5) | 2.935 (5) | 161 (4) |
| X-Ray | DFT | |||
|---|---|---|---|---|
| Mol A | Mol B | Mol A | Mol B | |
| C1—C2—N3—C4 | 55.9 (5) | 56.1 (5) | 53.69 | 54.19 |
| C1—C2—N3—C14 | −113.4 (4) | −121.3 (4) | −140.66 | −129.52 |
| C2—N3—C14—C15 | −15.9 (7) | −179.7 (4) | 1.79 | −166.54 |
| C2—C1—C12—C11 | 86.3 (5) | 84.6 (5) | 87.46 | 88.41 |
| X-Ray | DFT | |
|---|---|---|
| C1—C2—N3—C4 | −56.9 (3) | −51.80 |
| C1—C2—N3—C14 | 102.8 (3) | 144.73 |
| C2—N3—C14—C15 | 9.1 (4) | −13.59 |
| C2—C1—C12—C11 | −82.4 (3) | −87.29 |
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| O1W—H1WB⋯O8 | 0.85 | 1.93 | 2.775 (3) | 170 |
| N18—H18⋯O1W i | 0.86 | 2.11 | 2.935 (3) | 160 |
| X-Ray | DFT | ||
|---|---|---|---|
| Mol A | Mol B | ||
| C1—C2—N3—C4 | 54.1 (5) | 53.8 (5) | 49.73 |
| C1—C2—N3—C14 | −101.1 (4) | −108.6 (5) | −128.14 |
| C2—N3—C14—C15 | 143.3 (5) | 150.9 (5) | 158.30 |
| C2—C1—C12—C11 | 78.0 (5) | 86.1 (5) | 88.49 |
| X-Ray | DFT | |
|---|---|---|
| C1—C2—N3—C4 | 51.2 (3) | 49.50 |
| C1A—C2A—N3A—C4A | 51.5 (3) | 49.50 |
| C1—C2—N3—C14 | −114.8 (2) | −134.76 |
| C1A—C2A—N3A—C17 | −125.1 (2) | −134.76 |
| C2—N3—C14—C15 | 146.3 (3) | 161.00 |
| C2A—N3A—C17—C16 | 153.7 (3) | 161.00 |
| C2—C1—C12—C11 | 93.1 (3) | 88.41 |
| C2A—C1A—C12A—C11A | 87.2 (3) | 88.41 |
| C Atoms | 2 DMSO | 3 DMSO | 4 D2O | 5 CDCl3/MeOD (1:0.5) | 1 CDCl3 |
|---|---|---|---|---|---|
| 1 | 27.5 | 27.5 | 35.1 | 33.5 (1 and 1A) | 35.4 |
| 2 | 53.9 a | 54.7 a # | 57.0 | 55.2 (2 and 2A) | 53.8 |
| 4 | 53.9 a | 53.1 a # | 49.1 | 48.7 (4 and 4A) | 49.5 |
| 5 | 34.7 | 34.6 | 27.7 | 27.4 (5 and 5A) | 27.5 |
| 6 | 48.7 | 48.7 | 54.2 | 54.5 (6 and 6A) | 52.8 |
| 8 | 167.0 | 166.9 | 164.5 | 163.6 (8 and 8A) | 163.4 |
| 9 | 116.6 | 116.7 | 116.2 | 117.1 (9 and 9A) | 116.4 |
| 10 | 139.4 | 139.5 | 141.2 | 139.6 (10 and 10A) | 138.6 |
| 11 | 105.8 | 105.7 | 109.7 | 107.1 (11 and 11A) | 104.7 |
| 12 | 168.3 | 165.8 | 149.0 | 147.9 (12 and 12A) | 150.9 |
| 13 | 25.1 | 25.0 | 24.1 | 24.9 (13 and 13A) | 26.1 |
| 14 | 162.6 | 162.5 | 168.0 (or 17) | 168.7 (14 and 17) | |
| 15 | 183.4 | 182.6 * | 182.8 (or 16) | 184.0 (15 and 16) | |
| 16 | 182.7 | 182.5 * | 181.6 (or 15) | 184.0 (16 and 15) | |
| 17 | 149.3 | 149.1 | 166.6 (or 14) | 168.7 (17 and 14) | |
| 19 | 33.0 | 48.7 (19/23) | |||
| 20 | 81.1 | 66.3 (20/22) | |||
| 21 | 75.5 | - |
| 2 | 3 | 4 | 5 | |
|---|---|---|---|---|
| CCDC No. | 2421174 | 2421175 | 2421176 | 2421177 |
| Formula | C15H15N3O3 | C18H17N3O3•H2O | C19H21N3O4 | C26H26N4O4 |
| Molecular weight | 285.30 | 341.36 | 355.39 | 458.51 |
| Temperature (K) | 295 (2) | 295 (2) | 295 (2) | 100 (2) |
| Crystal system | triclinic | orthorhombic | monoclinic | monoclinic |
| Space group | P1 | P 212121 | P 21 | P 21 |
| a (Å) | 6.9269 (5) | 8.2405 (3) | 10.7027 (5) | 6.9626 (2) |
| b (Å) | 7.7099 (6) | 12.6825 (5) | 7.4702 (3) | 18.6664 (6) |
| c (Å) | 13.9934 (9) | 16.3143 (7) | 22.0755 (11) | 8.6303 (3) |
| α (o) | 92.495 (5) | |||
| β (o) | 103.055 (6) | 102.381 (4) | 109.095 (4) | |
| γ (o) | 110.026 (5) | |||
| V (Å3) | 678.00 (9) | 1705.01 (12) | 1723.92 (14) | 1059.94 (6) |
| Z | 2 | 4 | 4 | 2 |
| F(000) | 300 | 720 | 752 | 484 |
| Dcal (g.cm−1) | 1.397 | 1.330 | 1.369 | 1.437 |
| θ range(o) | 2.837–29.288 | 2.769–29.312 | 2.886–29.448 | 2.497–29.380 |
| μ (mm−1) | 0.100 | 0.096 | 0.098 | 0.099 |
| Crystal size (mm) | 0.26 × 0.21 × 0.13 | 0.28 × 0.26 × 0.15 | 0.29 × 0.26 × 0.19 | 0.29 × 0.25 × 0.14 |
| Tmin/Tmax | 0.965/1.00 | 0.980/1.00 | 0.974/1.00 | 0.977/1.00 |
| Total/unique/obs. refls (F2 > 2σ(F2)) | 10596/6186/3886 | 12022/4143/2642 | 28225/8269/5037 | 23797/5253/4470 |
| Rint | 0.0273 | 0.0331 | 0.0493 | 0.0473 |
| R [F2 > 2σ(F2)] (*) | 0.0543 | 0.0481 | 0.0673 | 0.0422 |
| wR [F2 all refls] (*) | 0.1114 | 0.1067 | 0.1475 | 0.0899 |
| S | 1.020 | 1.033 | 1.002 | 1.002 |
| Flack parameter | −1.7 (10) | −0.2(3) | −0.5(7) | −0.1(3) |
| Δρmax, Δρmin (eÅ−3) | +0.196, −0.172 | +0.157, −0.138 | +0.216, −0.180 | +0.264, −0.227 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Przybył, A.K.; Janczak, J.; Huczyński, A. Synthesis and Structural Analysis of New (−)-Cytisine Squaramides. Molecules 2025, 30, 1135. https://doi.org/10.3390/molecules30051135
Przybył AK, Janczak J, Huczyński A. Synthesis and Structural Analysis of New (−)-Cytisine Squaramides. Molecules. 2025; 30(5):1135. https://doi.org/10.3390/molecules30051135
Chicago/Turabian StylePrzybył, Anna K., Jan Janczak, and Adam Huczyński. 2025. "Synthesis and Structural Analysis of New (−)-Cytisine Squaramides" Molecules 30, no. 5: 1135. https://doi.org/10.3390/molecules30051135
APA StylePrzybył, A. K., Janczak, J., & Huczyński, A. (2025). Synthesis and Structural Analysis of New (−)-Cytisine Squaramides. Molecules, 30(5), 1135. https://doi.org/10.3390/molecules30051135