
CoCeOx-PVP Catalyst for Effective CO-SCR in the Presence of O2


Abstract
1. Introduction
2. Results and Discussion
2.1. Physical Properties (N2 Physisorption, SEM, XRD, and Raman)
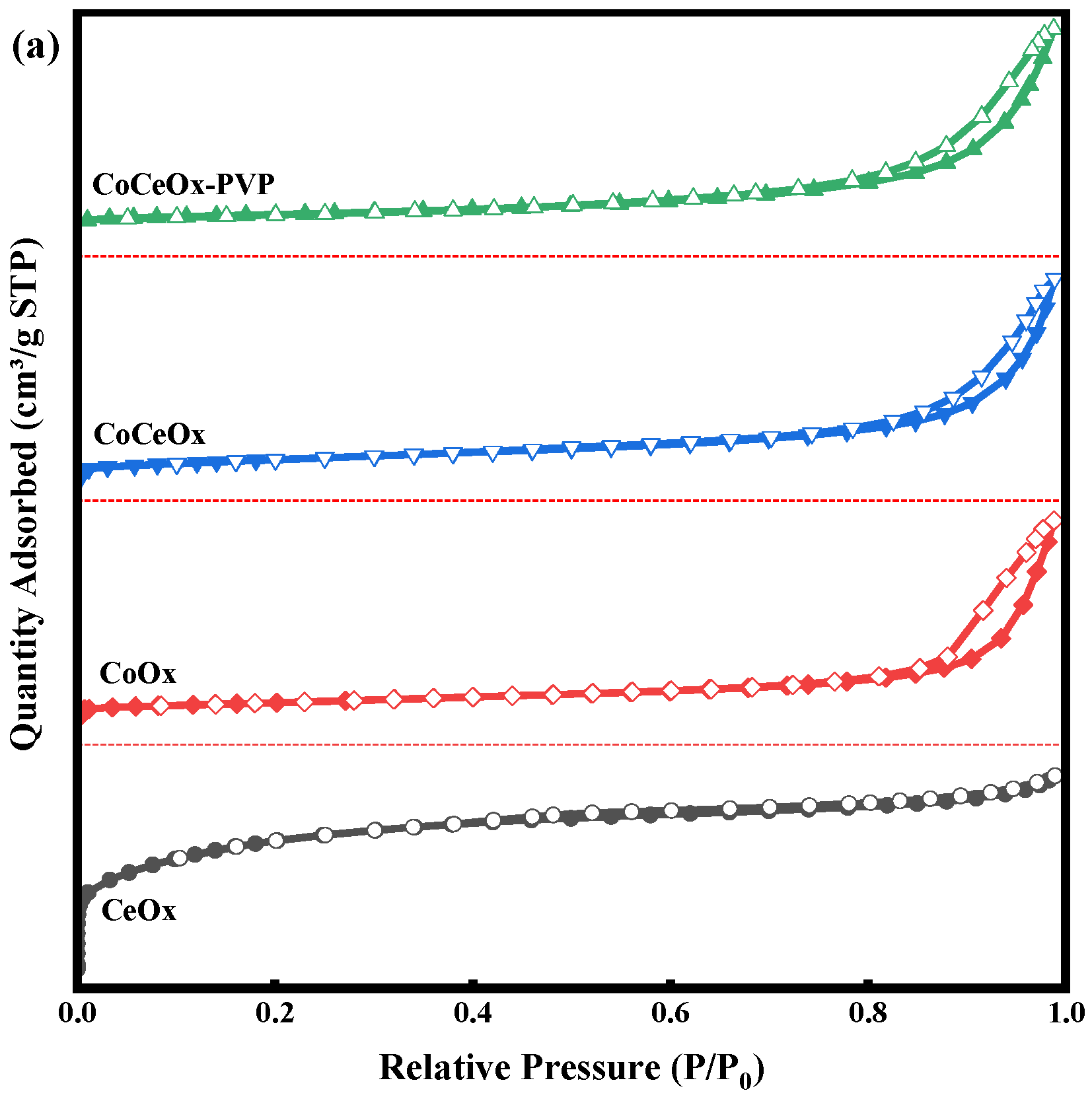
2.1.1. N2 Physisorption
2.1.2. SEM Observation
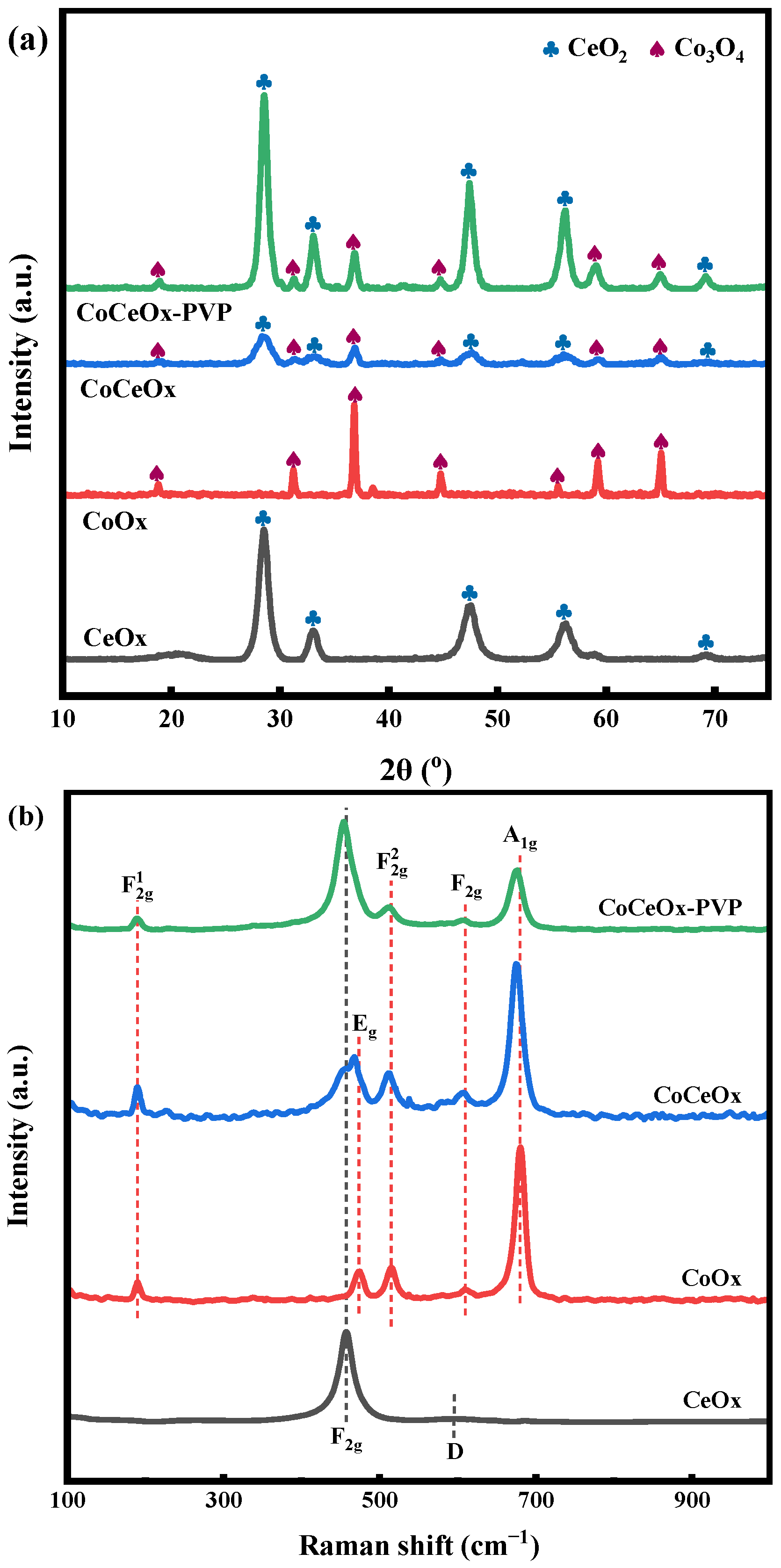
2.1.3. XRD and Raman Analysis
2.2. Chemical Properties (XPS, H2-TPR, and O2-TPD)
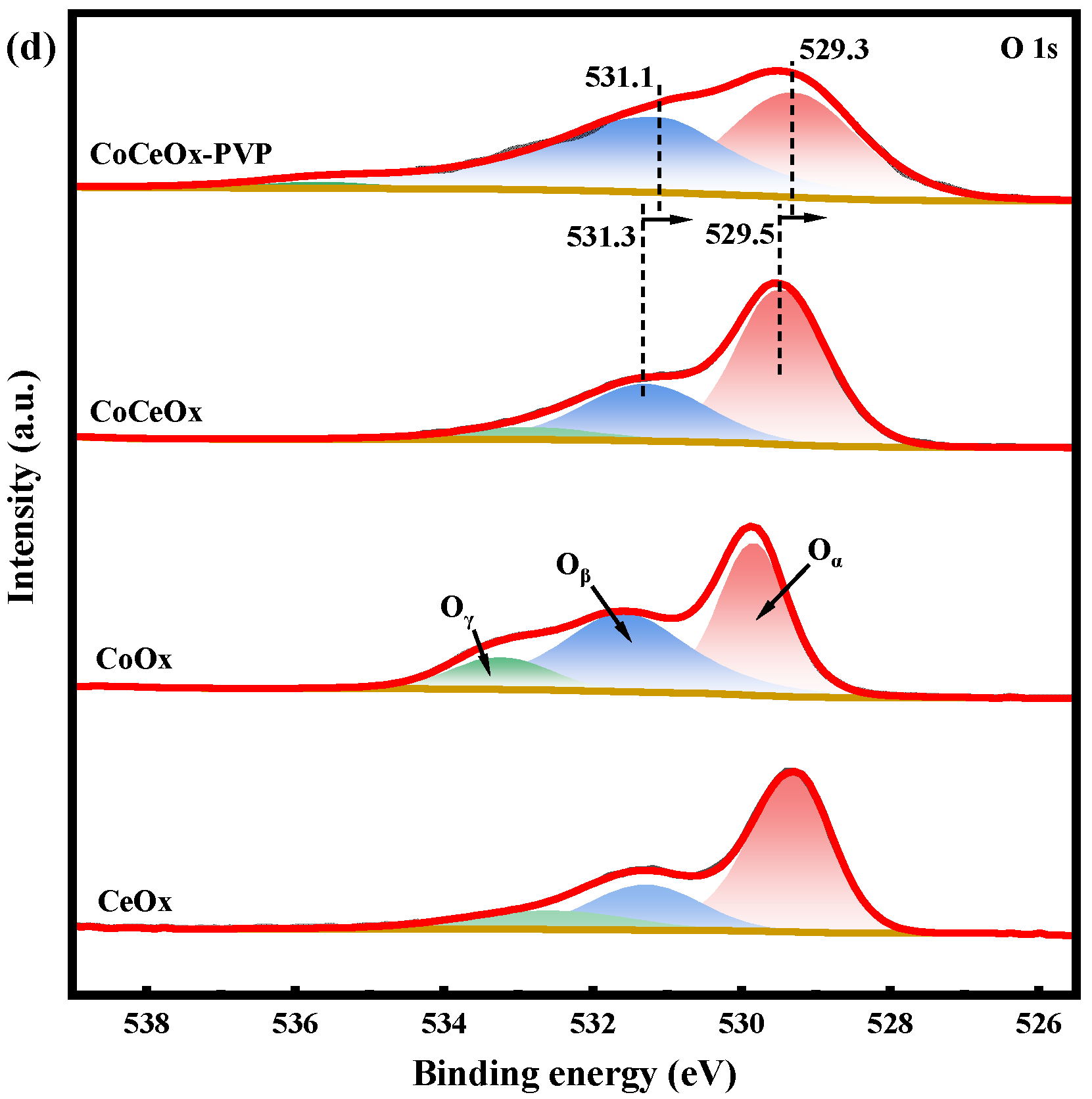
2.2.1. XPS
2.2.2. H2-TPR and O2-TPD
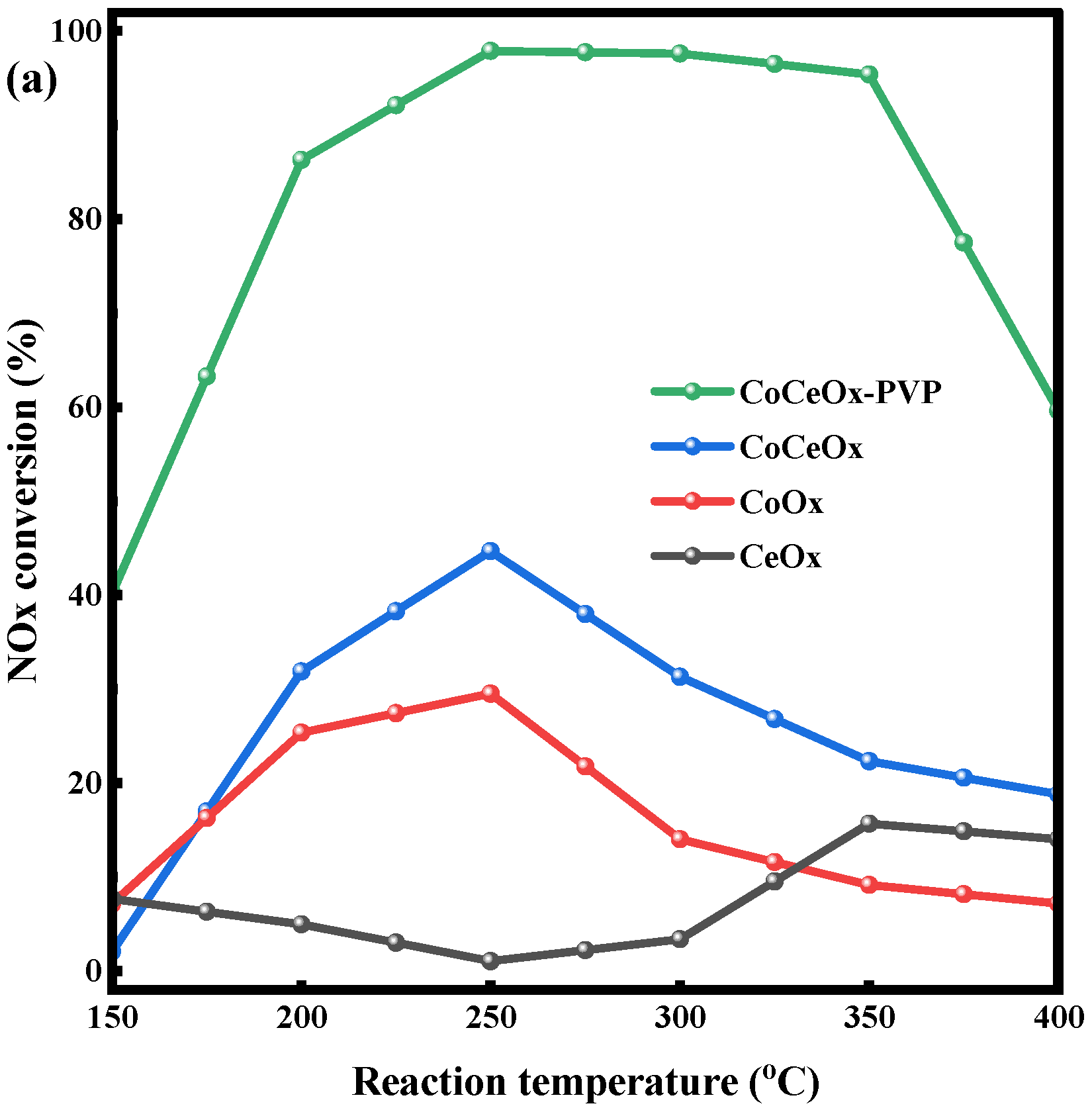
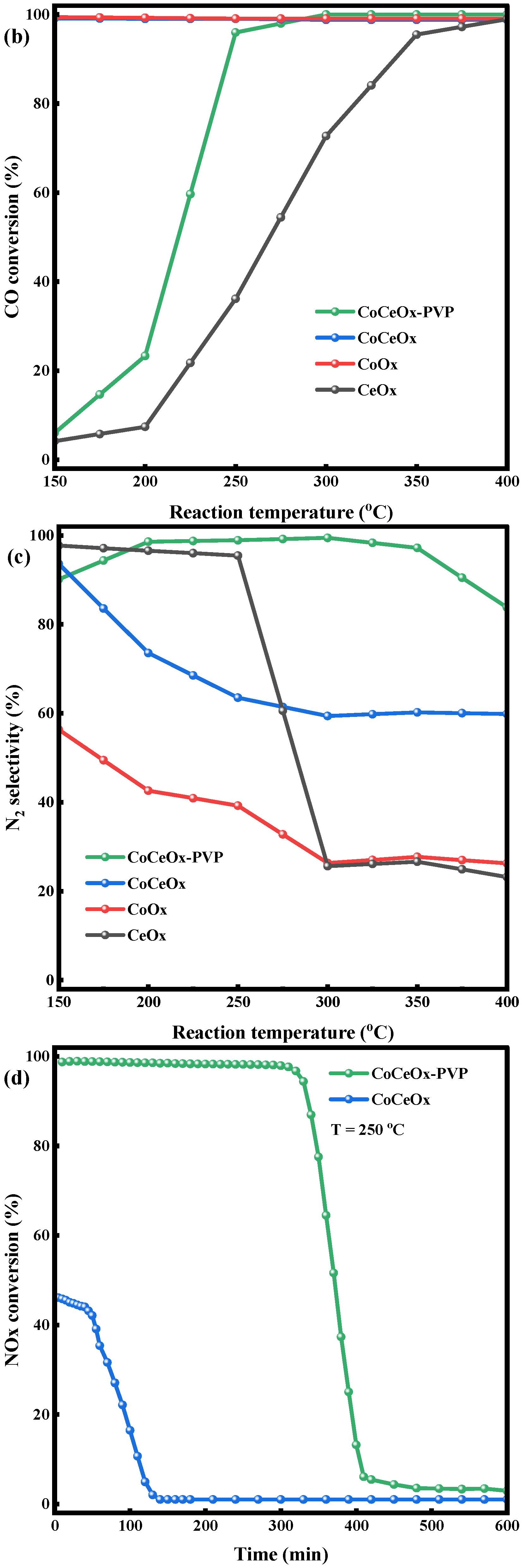
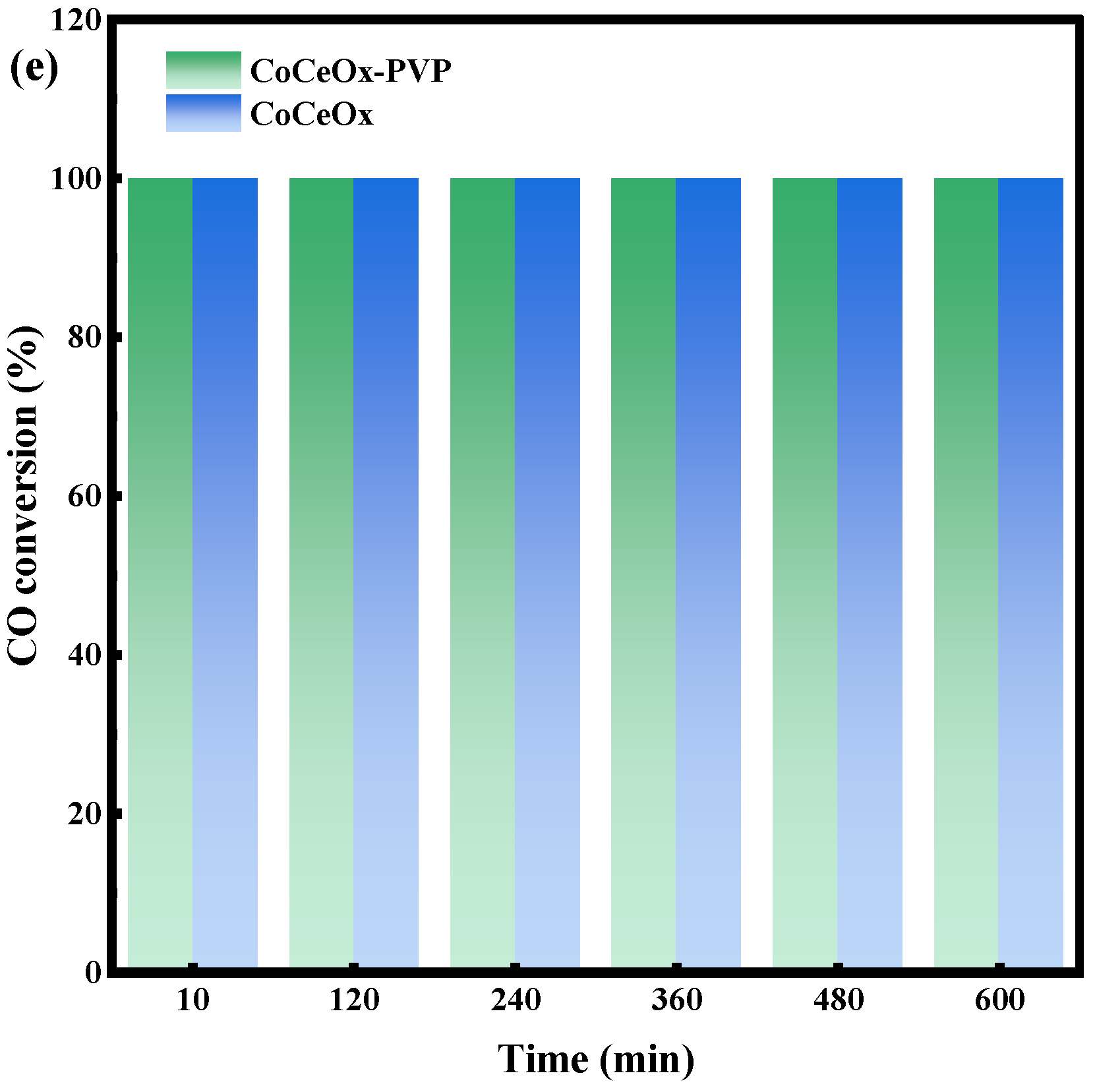
2.3. CO-SCR Performances of the Catalysts
2.4. In Situ DRIFTS Measurements
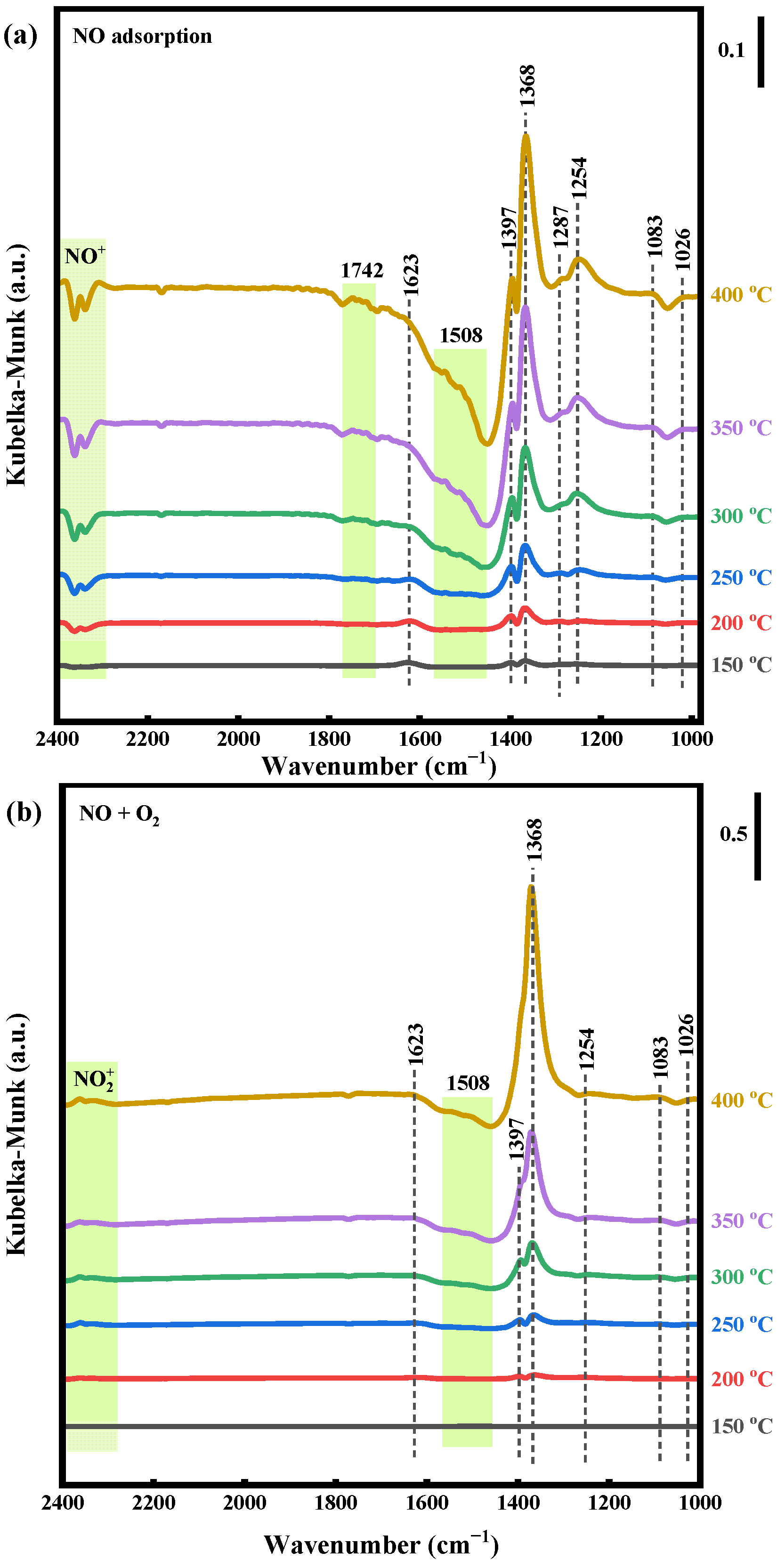
2.4.1. NO Adsorption and NO + O2 Reaction
2.4.2. CO Adsorption and CO + O2 Reaction
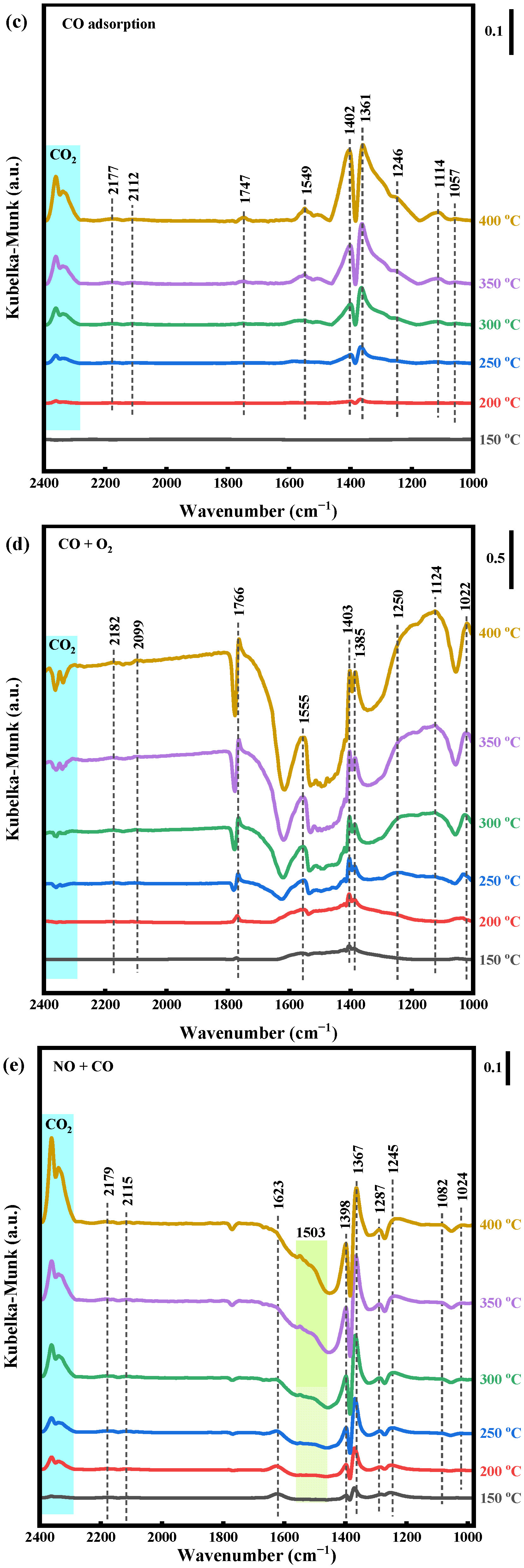
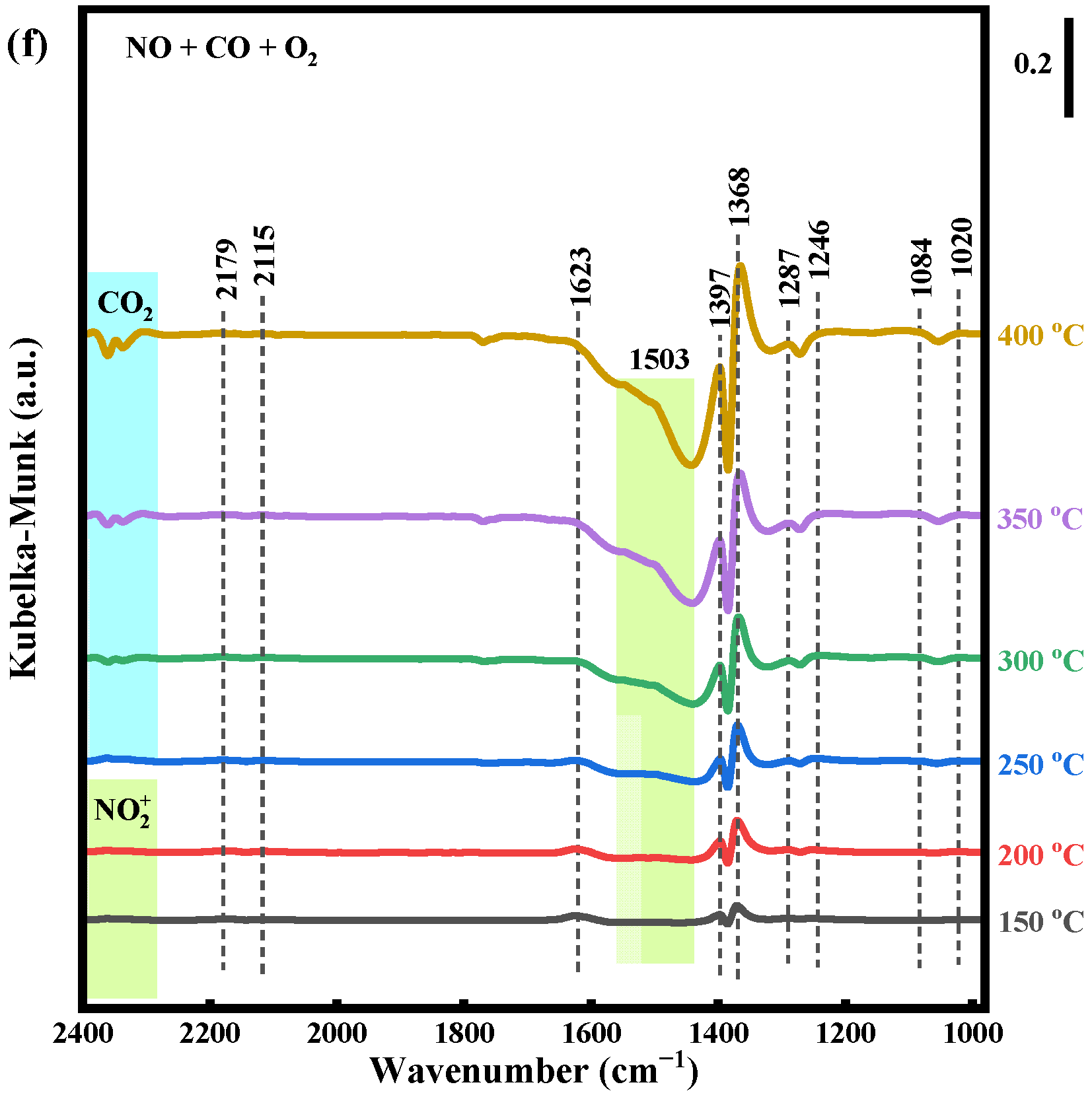
2.4.3. NO + CO and NO + CO + O2 Reaction
2.5. Proposed Reaction Mechanism
3. Materials and Methods
3.1. Materials
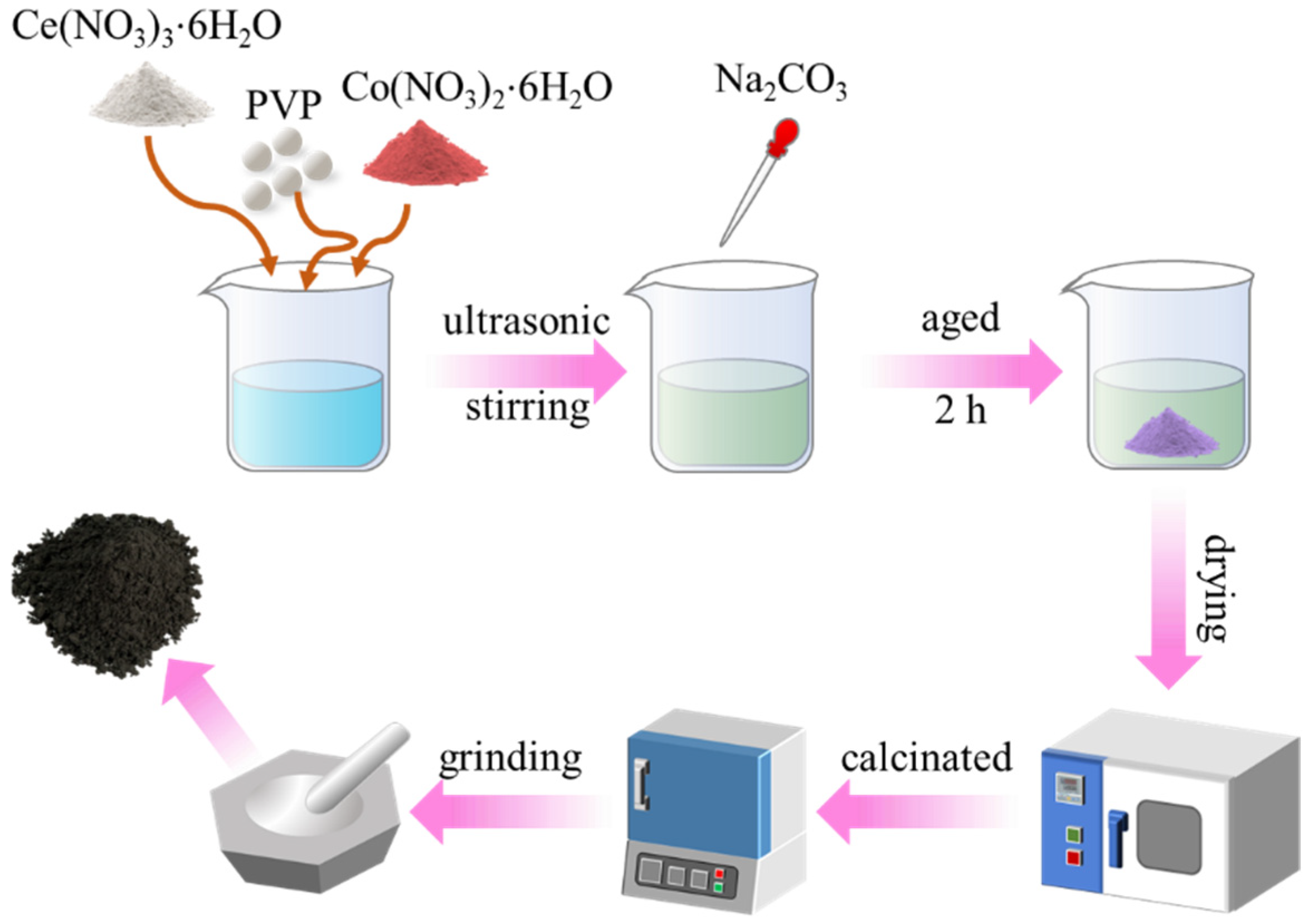
3.2. Catalyst Preparation
3.3. Catalyst Characterization
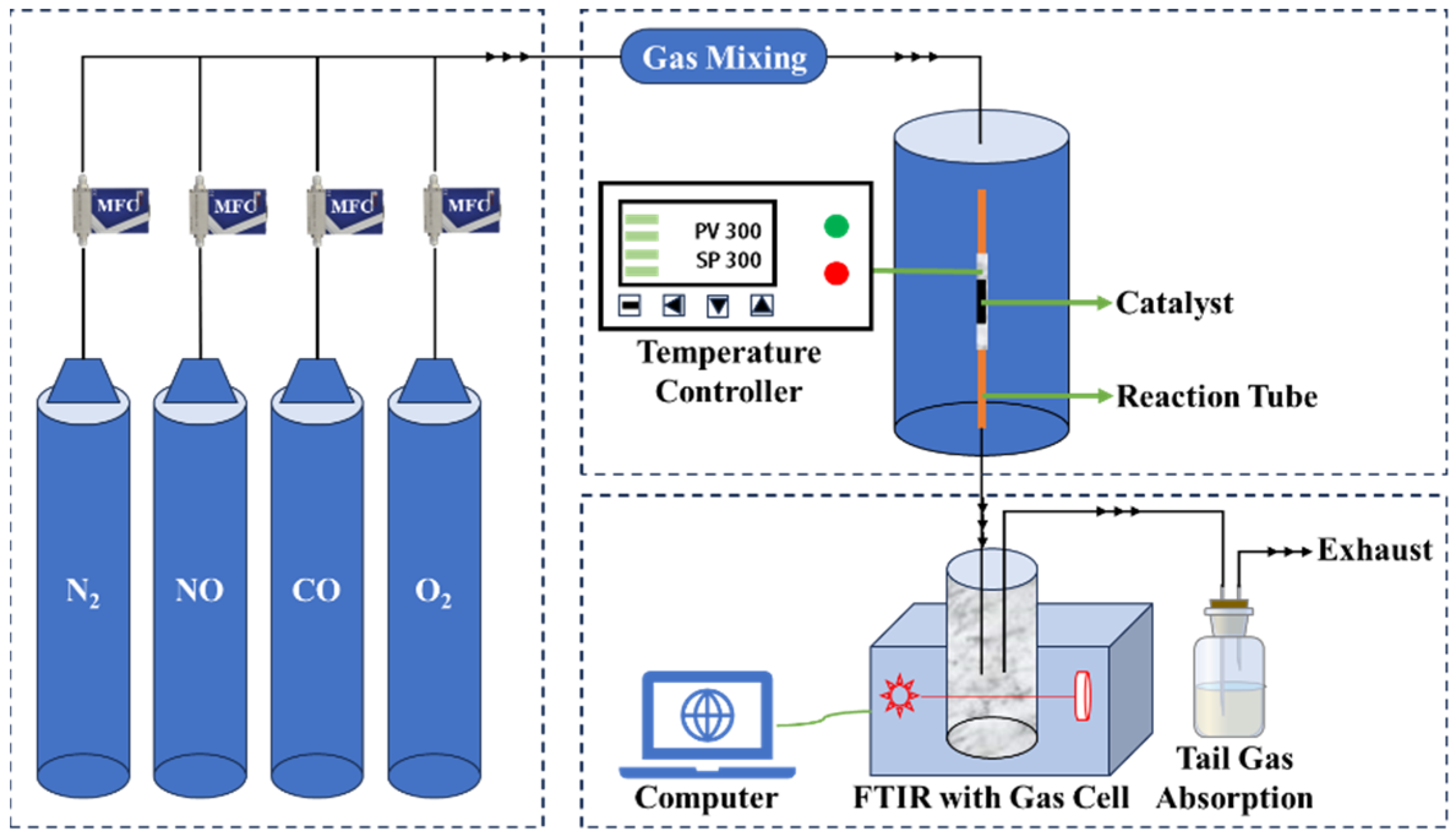
3.4. Catalytic Performance Evaluation
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Inomata, Y.; Kubota, H.; Hata, S.; Kiyonaga, E.; Morita, K.; Yoshida, K.; Sakaguchi, N.; Toyao, T.; Shimizu, K.I.; Ishikawa, S.; et al. Bulk tungsten-substituted vanadium oxide for low-temperature NOx removal in the presence of water. Nat. Commun. 2021, 12, 557. [Google Scholar] [CrossRef] [PubMed]
- Andana, T.; Rappé, K.G.; Gao, F.; Szanyi, J.; Pereira-Hernandez, X.; Wang, Y. Recent advances in hybrid metal oxide-zeolite catalysts for low-temperature selective catalytic reduction of NOx by ammonia. Appl. Catal. B-Environ. 2021, 291, 120054. [Google Scholar] [CrossRef]
- Boyle, E. Nitrogen pollution knows no bounds. Science 2017, 356, 700–701. [Google Scholar] [CrossRef]
- Anenberg, S.C.; Miller, J.; Injares, R.M.; Du, L.; Henze, D.K.; Lacey, F.; Malley, C.S.; Emberson, L.; Franco, V.; Klimont, Z.; et al. Impacts and mitigation of excess diesel-related NOx emissions in 11 major vehicle markets. Nature 2017, 545, 467–471. [Google Scholar] [CrossRef] [PubMed]
- Heo, I.; You, Y.W.; Lee, J.H.; Schmieg, S.J.; Yoon, D.; Kim, C.H. Urealess NOx Reduction by Carbon Monoxide in Simulated Lean-Burn Exhausts. Environ. Sci. Technol. 2020, 54, 8344–8351. [Google Scholar] [CrossRef]
- Leclercq, B.; Kluza, J.; Antherieu, S.; Sotty, J.; Alleman, L.Y.; Perdrix, E.; Loyens, A.; Coddeville, P.; Lo Guidice, J.M.; Marchetti, P.; et al. Air pollution-derived PM2.5 impairs mitochondrial function in healthy and chronic obstructive pulmonary diseased human bronchial epithelial cells. Environ. Pollut. 2018, 243, 1434–1449. [Google Scholar] [CrossRef]
- Lelieveld, J.; Evans, J.S.; Fnais, M.; Giannadaki, D.; Pozzer, A. The contribution of outdoor air pollution sources to premature mortality on a global scale. Nature 2015, 525, 367–371. [Google Scholar] [CrossRef]
- Song, J.H.; Park, D.C.; You, Y.-W.; Kim, Y.J.; Kim, S.M.; Heo, I.; Kim, D.H. Kinetic and DRIFTS studies of IrRu/Al2O3 catalysts for lean NOx reduction by CO at low temperature. Catal. Sci. Technol. 2020, 10, 8182–8195. [Google Scholar] [CrossRef]
- Gan, L.N.; Shi, W.B.; Li, K.Z.; Chen, J.J.; Peng, Y.; Li, J.H. Synergistic Promotion Effect between NOx and Chlorobenzene Removal on MnOx-CeO2 Catalyst. ACS Appl. Mater. Interfaces 2018, 10, 30426–30432. [Google Scholar] [CrossRef]
- Chen, R.Y.; Fang, X.Y.; Li, J.H.; Zhang, Y.; Liu, Z.M. Mechanistic investigation of the enhanced SO2 resistance of Co-modified MnOx catalyst for the selective catalytic reduction of NOx by NH3. Chem. Eng. J. 2023, 452, 139207. [Google Scholar] [CrossRef]
- Imanaka, N.; Masui, T. Advances in direct NOx decomposition catalysts. Appl. Catal. A Gen. 2012, 431, 1–8. [Google Scholar] [CrossRef]
- Boningari, T.; Pavani, S.M.; Ettireddy, P.R.; Chuang, S.S.C.; Smirniotis, P.G. Mechanistic investigations on NO reduction with CO over Mn/TiO2 catalyst at low temperatures. Mol. Catal. 2018, 451, 33–42. [Google Scholar] [CrossRef]
- Liu, Z.Y.; Zhou, Y.; Liu, J.; Chen, J.; Heck, A.J.R.; Wang, F.J. Reductive methylation labeling, from quantitative to structural proteomics. TrAC-Trends Anal. Chem. 2019, 118, 771–778. [Google Scholar] [CrossRef]
- Wang, Y.H.; Jiang, Q.K.; Xu, L.L.; Han, Z.K.; Guo, S.; Li, G.; Baiker, A. Effect of the Configuration of Copper Oxide-Ceria Catalysts in NO Reduction with CO: Superior Performance of a Copper-Ceria Solid Solution. ACS Appl. Mater. Interfaces 2021, 13, 61078–61087. [Google Scholar] [CrossRef]
- Yao, X.J.; Yu, Q.; Ji, Z.Y.; Lv, Y.Y.; Cao, Y.; Tang, C.J.; Gao, F.; Dong, L.; Chen, Y. A comparative study of different doped metal cations on the reduction, adsorption and activity of CuO/Ce0.67M0.33O2 (M = Zr4+, Sn4+, Ti4+) catalysts for NO plus CO reaction. Appl. Catal. B Environ. 2013, 130, 293–304. [Google Scholar] [CrossRef]
- Carlotto, S.; Natile, M.M.; Glisenti, A.; Vittadini, A. Catalytic Mechanisms of NO Reduction in a CO-NO Atmosphere at Co- and Cu-Doped SrTiO3(100) Surfaces. J. Phys. Chem. C 2018, 122, 449–454. [Google Scholar] [CrossRef]
- Xie, X.W.; Li, Y.; Liu, Z.Q.; Haruta, M.; Shen, W.J. Low-temperature oxidation of CO catalysed by Co3O4 nanorods. Nature 2009, 458, 746–749. [Google Scholar] [CrossRef]
- Ma, C.Y.; Mu, Z.; Li, J.J.; Jin, Y.G.; Cheng, J.; Lu, G.Q.; Hao, Z.P.; Qiao, S.Z. Mesoporous Co3O4 and Au/Co3O4 Catalysts for Low-Temperature Oxidation of Trace Ethylene. J. Am. Chem. Soc. 2010, 132, 2608–2613. [Google Scholar] [CrossRef]
- Wang, X.Y.; Li, X.Y.; Mu, J.C.; Fan, S.Y.; Chen, X.; Wang, L.; Yin, Z.F.; Tadé, M.; Liu, S.M. Oxygen Vacancy-rich Porous Co3O4 Nanosheets toward Boosted NO Reduction by CO and CO Oxidation: Insights into the Structure-Activity Relationship and Performance Enhancement Mechanism. ACS Appl. Mater. Interfaces 2019, 11, 41988–41999. [Google Scholar] [CrossRef]
- Wang, L.Y.; Cheng, X.X.; Wang, Z.Q.; Ma, C.Y.; Qin, Y.K. Investigation on Fe-Co binary metal oxides supported on activated semi-coke for NO reduction by CO. Appl. Catal. B Environ. 2017, 201, 636–651. [Google Scholar] [CrossRef]
- Zhang, Z.L.; Geng, H.R.; Zheng, L.S.; Du, B. Characterization and catalytic activity for the NO decomposition and reduction by CO of nanosized Co3O4. J. Alloys Compd. 2005, 392, 317–321. [Google Scholar] [CrossRef]
- Li, H.F.; Lu, G.Z.; Qiao, D.S.; Wang, Y.Q.; Guo, Y.; Guo, Y.L. Catalytic Methane Combustion over Co3O4/CeO2 Composite Oxides Prepared by Modified Citrate Sol-Gel Method. Catal. Lett. 2011, 141, 452–458. [Google Scholar] [CrossRef]
- Montini, T.; Melchionna, M.; Monai, M.; Fornasiero, P. Fundamentals and Catalytic Applications of CeO2-Based Materials. Chem. Rev. 2016, 116, 5987–6041. [Google Scholar] [CrossRef]
- Zhao, B.M.; Jian, Y.F.; Jiang, Z.Y.; Albilali, R.; He, C. Revealing the unexpected promotion effect of EuOx on Pt/CeO2 catalysts for catalytic combustion of toluene. Chin. J. Catal. 2019, 40, 543–552. [Google Scholar] [CrossRef]
- Wang, X.; Zhao, S.; Zhang, Y.; Wang, Z.; Feng, J.; Song, S.; Zhang, H. CeO2 nanowires self-inserted into porous Co3O4 frameworks as high-performance “noble metal free” hetero-catalysts. Chem. Sci. 2016, 7, 1109–1114. [Google Scholar] [CrossRef]
- Deng, C.S.; Huang, Q.Q.; Zhu, X.Y.; Hu, Q.; Su, W.L.; Qian, J.N.; Dong, L.H.; Li, B.; Fan, M.G.; Liang, C.Y. The influence of Mn-doped CeO2 on the activity of CuO/CeO2 in CO oxidation and NO plus CO model reaction. Appl. Surf. Sci. 2016, 389, 1033–1049. [Google Scholar] [CrossRef]
- Deng, Y.Q.; Shi, X.B.; Wei, L.Q.; Liu, H.; Li, J.; Ou, X.M.; Dong, L.H.; Li, B. Effect of intergrowth and coexistence CuO-CeO2 catalyst by grinding method application in the catalytic reduction of NOx by CO. J. Alloys Compd. 2021, 869, 159231. [Google Scholar] [CrossRef]
- Savereide, L.; Nauert, S.L.; Roberts, C.A.; Notestein, J.M. The effect of support morphology on CoOx/CeO2 catalysts for the reduction of NO by CO. J. Catal. 2018, 366, 150–158. [Google Scholar] [CrossRef]
- Liu, S.M.; Xue, W.J.; Ji, Y.J.; Xu, W.Q.; Chen, W.X.; Jia, L.H.; Zhu, T.Y.; Zhong, Z.Y.; Xu, G.W.; Mei, D.H.; et al. Interfacial oxygen vacancies at Co3O4-CeO2 heterointerfaces boost the catalytic reduction of NO by CO in the presence of O2. Appl. Catal. B Environ. 2023, 323, 122151. [Google Scholar] [CrossRef]
- Zeng, J.; Zhong, X.M.; Yu, J.; Zhang, T.; Wang, Y.Z.; Chang, H.Z. Promotional Effect of Preparation Methods on Catalytic Reduction of NO by CO over CoCeOx Catalysts. Ind. Eng. Chem. Res. 2020, 59, 34–41. [Google Scholar] [CrossRef]
- Guo, J.; Zhang, G.D.; Tang, Z.C.; Zhang, J.Y. Design of Prussian blue analogue-derived double-cone structure Ce-Fe catalysts and their enhanced performance for the selective catalytic reduction of NOx with NH3. New J. Chem. 2020, 44, 21244–21254. [Google Scholar] [CrossRef]
- Yan, L.J.; Liu, Y.Y.; Zha, K.W.; Li, H.R.; Shi, L.Y.; Zhang, D.S. Scale Activity Relationship of MnOx-FeOy Nanocage Catalysts Derived from Prussian Blue Analogues for Low-Temperature NO Reduction: Experimental and DFT Studies. ACS Appl. Mater. Interfaces 2017, 9, 2581–2593. [Google Scholar] [CrossRef]
- Passaro, J.; Imparato, C.; Parida, D.; Bifulco, A.; Branda, F.; Aronne, A. Electrospinning of PVP-based ternary composites containing SiO2 nanoparticles and hybrid TiO2 microparticles with adsorbed superoxide radicals. Compos. Part. B Eng. 2022, 238, 109874. [Google Scholar] [CrossRef]
- Xu, G.D.; Song, P.; Xia, L.X. Difunctional AuNPs@PVP with oxidase-like activity for SERRS detection of total antioxidant capacity. Talanta 2024, 270, 125554. [Google Scholar] [CrossRef]
- Xue, L.; Zhang, C.B.; He, H.; Teraoka, Y. Catalytic decomposition of N2O over CeO2 promoted CO3O4 spinel catalyst. Appl. Catal. B Environ. 2007, 75, 167–174. [Google Scholar] [CrossRef]
- Yokota, K.; Fukui, M.; Tanaka, T. Catalytic removal of nitric oxide with hydrogen and carbon monoxide in the presence of excess oxygen. Appl. Surf. Sci. 1997, 121, 273–277. [Google Scholar] [CrossRef]
- Takahashi, N.; Shinjoh, H.; Iijima, T.; Suzuki, T.; Yamazaki, K.; Yokota, K.; Suzuki, H.; Miyoshi, N.; Matsumoto, S.; Tanizawa, T.; et al. The new concept 3-way catalyst for automotive lean-burn engine: NOx storage and reduction catalyst. Catal. Today 1996, 27, 63–69. [Google Scholar] [CrossRef]
- Wang, X.; Li, X.; Mu, J.; Fan, S.; Wang, L.; Gan, G.; Qin, M.; Li, J.; Li, Z.; Zhang, D. Facile Design of Highly Effective CuCexCo1–xOy Catalysts with Diverse Surface/Interface Structures toward NO Reduction by CO at Low Temperatures. Ind. Eng. Chem. Res. 2019, 58, 15459–15469. [Google Scholar] [CrossRef]
- He, Y.J.; Liu, J.; Zhang, G.J.; Wang, Y.; Zhao, Y.Q.; Li, G.Q.; Zhang, Y.F.; Lv, D.K. Interfacial effects promote the catalytic performance of CuCoO2-CeO2 metal oxides for the selective reduction of NO by CO. Chem. Eng. J. 2023, 465, 142856. [Google Scholar] [CrossRef]
- Wang, J.; Xing, Y.; Jia, H.; Zhang, W.; Zhou, H.; Qian, D.; Su, W. TiO2-coated CeCoOx-PVP catalysts derived from Prussian blue analogue for synergistic elimination of NOx and o-DCB: The coupling of redox and acidity. Chem. Eng. J. 2023, 476, 146390. [Google Scholar] [CrossRef]
- Lopes, I.; El Hassan, N.; Guerba, H.; Wallez, G.; Davidson, A. Size-induced structural modifications affecting Co3O4 nanoparticles patterned in SBA-15 silicas. Chem. Mater. 2006, 18, 5826–5828. [Google Scholar] [CrossRef]
- Huang, J.; Sheng, H.; Ross, R.D.; Han, J.; Wang, X.; Song, B.; Jin, S. Modifying redox properties and local bonding of Co3O4 by CeO2 enhances oxygen evolution catalysis in acid. Nat. Commun. 2021, 12, 3036. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Shin, K.; Kawashima, K.; Youn, D.H.; Lin, J.; Hong, T.E.; Liu, Y.; Wygant, B.R.; Wang, J.; Henkelman, G.; et al. Enhanced Activity Promoted by CeOx on a CoOx Electrocatalyst for the Oxygen Evolution Reaction. ACS Catal. 2018, 8, 4257–4265. [Google Scholar] [CrossRef]
- Qiu, B.C.; Wang, C.; Zhang, N.; Cai, L.J.; Xiong, Y.J.; Chai, Y. CeO2-Induced Interfacial Co2+ Octahedral Sites and Oxygen Vacancies for Water Oxidation. ACS Catal. 2019, 9, 6484–6490. [Google Scholar] [CrossRef]
- Ling, T.; Yan, D.Y.; Jiao, Y.; Wang, H.; Zheng, Y.; Zheng, X.L.; Mao, J.; Du, X.W.; Hu, Z.P.; Jaroniec, M.; et al. Engineering surface atomic structure of single-crystal cobalt (II) oxide nanorods for superior electrocatalysis. Nat. Commun. 2016, 7, 12876. [Google Scholar] [CrossRef]
- Zhang, X.; Gao, C.; Wang, Z.; Wang, X.; Cheng, J.; Song, X.; Han, X.; Zhang, N.; Bao, J.; He, G. Co3O4 with ordered pore structure derived from wood vessels for efficient Hg0 oxidation. Chin. J. Chem. Eng. 2022, 50, 215–221. [Google Scholar] [CrossRef]
- Huang, H.; Dai, Q.G.; Wang, X.Y. Morphology effect of Ru/CeO2 catalysts for the catalytic combustion of chlorobenzene. Appl. Catal. B Environ. 2014, 158, 96–105. [Google Scholar] [CrossRef]
- Zhang, Q.L.; Liu, X.; Ning, P.; Song, Z.X.; Li, H.; Gu, J.J. Enhanced performance in NOx reduction by NH3 over a mesoporous Ce-Ti- MoOx catalyst stabilized by a carbon template. Catal. Sci. Technol. 2015, 5, 2260–2269. [Google Scholar] [CrossRef]
- Bao, T.; Zhao, Z.K.; Dai, Y.T.; Lin, X.L.; Jin, R.H.; Wang, G.R.; Muhammad, T. Supported Co3O4-CeO2 catalysts on modified activated carbon for CO preferential oxidation in H2-rich gases. Appl. Catal. B Environ. 2012, 119, 62–73. [Google Scholar] [CrossRef]
- Wang, J.Q.; Shen, M.Q.; Wang, J.; Gao, J.D.; Ma, J.; Liu, S.X. CeO2-CoOx mixed oxides: Structural characteristics and dynamic storage/release capacity. Catal. Today 2011, 175, 65–71. [Google Scholar] [CrossRef]
- Jampaiah, D.; Venkataswamy, P.; Coyle, V.E.; Reddy, B.M.; Bhargava, S.K. Low-temperature CO oxidation over manganese, cobalt, and nickel doped CeO2 nanorods. RSC Adv. 2016, 6, 80541–80548. [Google Scholar] [CrossRef]
- Luo, N.; Gao, F.; Liu, H.; Xiong, T.; Wen, J.; Duan, E.; Wang, C.; Zhao, S.; Yi, H.; Tang, X. Hierarchical structured Ti-doped CeO2 stabilized CoMn2O4 for enhancing the low-temperature NH3-SCR performance within highly H2O and SO2 resistance. Appl. Catal. B Environ. 2024, 343, 123442. [Google Scholar] [CrossRef]
- Liu, S.; Wu, X.; Tang, J.; Cui, P.; Jiang, X.; Chang, C.; Liu, W.; Gao, Y.; Li, M.; Weng, D. An exploration of soot oxidation over CeO2-ZrO2 nanocubes: Do more surface oxygen vacancies benefit the reaction? Catal. Today 2017, 281, 454–459. [Google Scholar] [CrossRef]
- Liu, Z.; Yu, F.; Pan, K.; Zhou, X.; Sun, R.; Tian, J.; Wan, Y.; Dan, J.; Dai, B. Two-dimensional vermiculite carried CuCoCe catalysts for CO-SCR in the presence of O2 and H2O: Experimental and DFT calculation. Chem. Eng. J. 2021, 422, 130099. [Google Scholar] [CrossRef]
- Deng, C.S.; Li, B.; Dong, L.H.; Zhang, F.Y.; Fan, M.G.; Jin, G.Z.; Gao, J.B.; Gao, L.W.; Zhang, F.; Zhou, X.P. NO reduction by CO over CuO supported on CeO2-doped TiO2: The effect of the amount of a few CeO2. Phys. Chem. Chem. Phys. 2015, 17, 16092–16109. [Google Scholar] [CrossRef]
- Venegas, F.; López, N.; Sánchez-Calderón, L.; Aguila, G.; Araya, P.; Guo, X.; Zhu, Y.; Guerrero, S. The transient reduction of NO with CO and naphthalene in the presence of oxygen using a core–shell SmCeO2@TiO2-supported copper catalyst. Catal. Sci. Technol. 2019, 9, 3408–3415. [Google Scholar] [CrossRef]
- Pan, K.L.; Young, C.W.; Pan, G.T.; Chang, M.B. Catalytic reduction of NO by CO with Cu-based and Mn-based catalysts. Catal. Today 2020, 348, 15–25. [Google Scholar] [CrossRef]
- Liu, S.M.; Ji, Y.J.; Liu, B.; Xu, W.Q.; Chen, W.X.; Yu, J.; Zhong, Z.Y.; Xu, G.W.; Zhu, T.Y.; Su, F.B. Co Single Atoms and CoOx Nanoclusters Anchored on Ce0.75Zr0.25O2 Synergistically Boosts the NO Reduction by CO. Adv. Funct. Mater. 2023, 33, 2303297. [Google Scholar] [CrossRef]
- Li, J.; Luo, G.H.; Chu, Y.; Wei, F. Experimental and modeling analysis of NO reduction by CO for a FCC regeneration process. Chem. Eng. J. 2012, 184, 168–175. [Google Scholar] [CrossRef]
- Sreekanth, P.M.; Smirniotis, P.G. Selective reduction of NO with CO over titania supported transition metal oxide catalysts. Catal. Lett. 2008, 122, 37–42. [Google Scholar] [CrossRef]
- Cheng, X.; Wang, L.; Wang, Z.; Zhang, M.; Ma, C. Catalytic Performance of NO Reduction by CO over Activated Semicoke Supported Fe/Co Catalysts. Ind. Eng. Chem. Res. 2016, 55, 12710–12722. [Google Scholar] [CrossRef]
- Li, S.H.; Chen, X.G.; Wang, F.; Xie, Z.L.; Hao, Z.R.; Liu, L.J.; Shen, B.X. Promotion effect of Ni doping on the oxygen resistance property of Fe/CeO catalyst for CO-SCR reaction: Activity test and mechanism investigation. J. Hazard. Mater. 2022, 431, 128622. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Wang, T.; Pan, X.H.; Ma, C.Y. Correlation of Physicochemical Characteristics of CoOx Supported by a CeO2 Nanorod with NO Removal by CO. J. Environ. Eng. 2020, 146, 04019100. [Google Scholar] [CrossRef]
- Li, S.H.; Wang, F.; Xie, Z.L.; Ng, D.; Shen, B.X. A novel core-shell structured Fe@CeO2-ZIF-8 catalyst for the reduction of NO by CO. J. Catal. 2023, 421, 240–251. [Google Scholar] [CrossRef]
- Liu, T.; Yao, Y.; Wei, L.; Shi, Z.; Han, L.; Yuan, H.; Li, B.; Dong, L.; Wang, F.; Sun, C. Preparation and Evaluation of Copper Manganese Oxide as a High-Efficiency Catalyst for CO Oxidation and NO Reduction by CO. J. Phys. Chem. C 2017, 121, 12757–12770. [Google Scholar] [CrossRef]
- Zhang, S.; Lee, J.; Kim, D.H.; Kim, T. NO reduction by CO over CoOx/CeO2 catalysts: Effect of support calcination temperature on activity. Mol. Catal. 2020, 482, 110703. [Google Scholar] [CrossRef]
- Wang, J.; You, R.; Qian, K.; Pan, Y.; Yang, J.; Huang, W. Effect of the modification of alumina supports with chloride on the structure and catalytic performance of Ag/Al2O3 catalysts for the selective catalytic reduction of NO with propene and H2/propene. Chin. J. Catal. 2021, 42, 2242–2253. [Google Scholar] [CrossRef]
- Liu, T.; Wei, L.; Yao, Y.; Dong, L.; Li, B. La promoted CuO-MnOx catalysts for optimizing SCR performance of NO with CO. Appl. Surf. Sci. 2021, 546, 148971. [Google Scholar] [CrossRef]
- Cheng, X.X.; Zhang, X.Y.; Su, D.X.; Wang, Z.Q.; Chang, J.C.; Ma, C.Y. NO reduction by CO over copper catalyst supported on mixed CeO2 and Fe2O3: Catalyst design and activity test. Appl. Catal. B Environ. 2018, 239, 485–501. [Google Scholar] [CrossRef]
- Hadjiivanov, K.I. Identification of Neutral and Charged NxOy Surface Species of IR Spectroscopy. Catal. Rev. 2007, 42, 71–144. [Google Scholar] [CrossRef]
- Liu, J.; Zang, P.C.; Liu, X.Q.; Mi, J.X.; Wang, Y.; Zhang, G.J.; Chen, J.J.; Zhang, Y.F.; Li, J.H. A novel highly active catalyst form CuFeMg layered double oxides for the selective catalytic reduction of NO by CO. Fuel 2022, 317, 123469. [Google Scholar] [CrossRef]
- Qi, L.; Yu, Q.; Dai, Y.; Tang, C.; Liu, L.; Zhang, H.; Gao, F.; Dong, L.; Chen, Y. Influence of cerium precursors on the structure and reducibility of mesoporous CuO-CeO2 catalysts for CO oxidation. Appl. Catal. B Environ. 2012, 119–120, 308–320. [Google Scholar] [CrossRef]
- Martínez-Arias, A.; Fernández-García, M.; Soria, J.; Conesa, J.C. Spectroscopic Study of a Cu/CeO2 Catalyst Subjected to Redox Treatments in Carbon Monoxide and Oxygen. J. Catal. 1999, 182, 367–377. [Google Scholar] [CrossRef]
- Dasireddy, V.; Likozar, B. Selective catalytic reduction of NOx by CO over bimetallic transition metals supported by multi-walled carbon nanotubes (MWCNT). Chem. Eng. J. 2017, 326, 886–900. [Google Scholar] [CrossRef]
- Zhao, R.Q.; Wei, X.L.; Chu, B.X.; Chen, K.A.; Qin, Q.J.; Liu, H.; Zhou, Y.M.; Li, B.; Dong, L.H. Multi-phase coexisting metal oxide derived by MOFs for the CO-SCR reaction at low temperature and in situ DRIFTS study on reaction mechanism. Appl. Surf. Sci. 2022, 580, 152277. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhao, L.; Kang, M.; Chen, Z.; Gao, S.; Hao, H. Insights into high CO-SCR performance of CuCoAlO catalysts derived from LDH/MOFs composites and study of H2O/SO2 and alkali metal resistance. Chem. Eng. J. 2021, 426, 131873. [Google Scholar] [CrossRef]
- Savva, P.; Efstathiou, A. The influence of reaction temperature on the chemical structure and surface concentration of active NOx in H2-SCR over Pt/MgOCeO2: SSITKA-DRIFTS and transient mass spectrometry studies. J. Catal. 2008, 257, 324–333. [Google Scholar] [CrossRef]
- Du, Y.; Gao, F.; Tang, X.; Yi, H.; Zhou, Y.; Zhao, S.; Duan, E.; Wang, J.; Qi, Z. Mechanistic insight into the enhanced NO reduction by CO over a pre-reduced CuxOy-CeO2 multiphase catalyst. J. Environ. Chem. Eng. 2023, 11, 110386. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalysts | BET Specific Surface Area a (m2/g) | Total Pore Volume b (cm3/g) | Average Pore Diameter b (nm) |
|---|---|---|---|
| CoCeOx-PVP | 24.6 | 0.13 | 20.89 |
| CoCeOx | 54.3 | 0.20 | 14.37 |
| CoOx | 25.6 | 0.11 | 17.65 |
| CeOx | 135.2 | 0.09 | 2.56 |
| Catalysts | Crystallite Size a (nm) | Lattice Parameter a (Å) | Estimated Crystallite Size b (nm) | Strain c (%) |
|---|---|---|---|---|
| CoCeOx-PVP | 9.4 | 5.4194 | 10.2 | 0.016 |
| CoCeOx | 5.2 | 5.4029 | 4.6 | −0.367 |
| CoOx | ---- | ---- | 26.8 | −0.022 |
| CeOx | 7 | 5.4141 | 9.8 | 0.506 |
| Catalyst | Co2+/Co (%) | Ce3+/Ce (%) | Oα/O (%) | Oβ/O (%) |
|---|---|---|---|---|
| CoCeOx-PVP | 49.58 | 43.37 | 44.57 | 53.14 |
| CoCeOx | 45.63 | 34.94 | 61.40 | 30.54 |
| CoOx | 36.44 | ---- | 43.13 | 44.61 |
| CeOx | ---- | 36.00 | 63.53 | 22.35 |
| Catalysts | H2-TPR | O2-TPD |
|---|---|---|
| Total H2 Consumption (mmol/g) | Total O2 Desorption (mmol/g) | |
| CoCeOx-PVP | 2.89 | 1.01 |
| CoCeOx | 3.10 | 0.66 |
| CoOx | 15.24 | 0.20 |
| CeOx | 0.47 | 0.93 |
| Catalyst | Reaction Conditions | Tmax (°C) | NO Conversion (%) | Ref. | |||
|---|---|---|---|---|---|---|---|
| NO (ppm) | CO (ppm) | O2 (%) | GHSV (h−1) | ||||
| CoCeOx-PVP | 1000 | 3000 | 5 | 20,000 | 250 | 100 | This work |
| K/Cu/SmCe@TiO2 | 500 | 10,000 | 10 | 10,000 | 330 | 97 | [56] |
| Cu-Ce-Fe-Co/TiO2 | 200 | 10,000 | 6 | 10,000 | 200 | 63 | [57] |
| CoSA+CoOxNC/CZO | 1000 | 2000 | 5 | 20,000 | 250 | 100 | [58] |
| Co3O4-CeO2-IOV | 1000 | 2000 | 5 | 20,000 | 250 | 100 | [29] |
| Co2.9Cu0.1O4 | 50,000 | 50,000 | 2.5 | 8000 | 700 | 88 | [59] |
| 10% MnO2/TiO2 | 400 | 400 | 2 | 50,000 | 200 | 95 | [60] |
| CuCoCe/2D-VMT | 500 | 1000 | 1 | 102,000 | 300 | 100 | [54] |
| Fe-Co/ASC | 1000 | 5000 | 1 | 20,000 | 300 | 45 | [61] |
| NiFe/CeO2 | 500 | 5000 | 0.5 | 22,800 | 225 | 100 | [62] |
| Co10-CeO2 | 1000 | 15,000 | 0.1 | 30,000 | 350 | 93 | [63] |
| H-CuCo-CeO2 | 500 | 1000 | 0 | 60,000 | 175 | 100 | [39] |
| CuCe0.2Co0.8Oy | 1000 | 2000 | 0 | 50,000 | 175 | 100 | [38] |
| Fe@CeO2-ZIF-8 | 500 | 1000 | 0 | 22,800 | 250 | 100 | [64] |
| CoCeOx-OA | 500 | 1000 | 2 | 80,000 mL/g∙h | 400 | 50 | [30] |
| Cu0.15Mn | 50,000 | 100,000 | 0 | 24,000 mL/g∙h | 275 | 100 | [65] |
| Co-AR CeO2 | 50,000 | 50,000 | 0 | 60,000 mL/g∙h | 300 | 100 | [66] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, Y.; Gao, F.; Yi, L.; Wang, J.; Yi, H.; Tang, X. CoCeOx-PVP Catalyst for Effective CO-SCR in the Presence of O2. Molecules 2025, 30, 1133. https://doi.org/10.3390/molecules30051133
Zhou Y, Gao F, Yi L, Wang J, Yi H, Tang X. CoCeOx-PVP Catalyst for Effective CO-SCR in the Presence of O2. Molecules. 2025; 30(5):1133. https://doi.org/10.3390/molecules30051133
Chicago/Turabian StyleZhou, Yuansong, Fengyu Gao, Lei Yi, Junyi Wang, Honghong Yi, and Xiaolong Tang. 2025. "CoCeOx-PVP Catalyst for Effective CO-SCR in the Presence of O2" Molecules 30, no. 5: 1133. https://doi.org/10.3390/molecules30051133
APA StyleZhou, Y., Gao, F., Yi, L., Wang, J., Yi, H., & Tang, X. (2025). CoCeOx-PVP Catalyst for Effective CO-SCR in the Presence of O2. Molecules, 30(5), 1133. https://doi.org/10.3390/molecules30051133