
Design and Binding Affinity of Antisense Peptides for Snake Venom Neutralization †


Abstract

1. Introduction

1.1. Composition of the Vipera ammodytes Venom
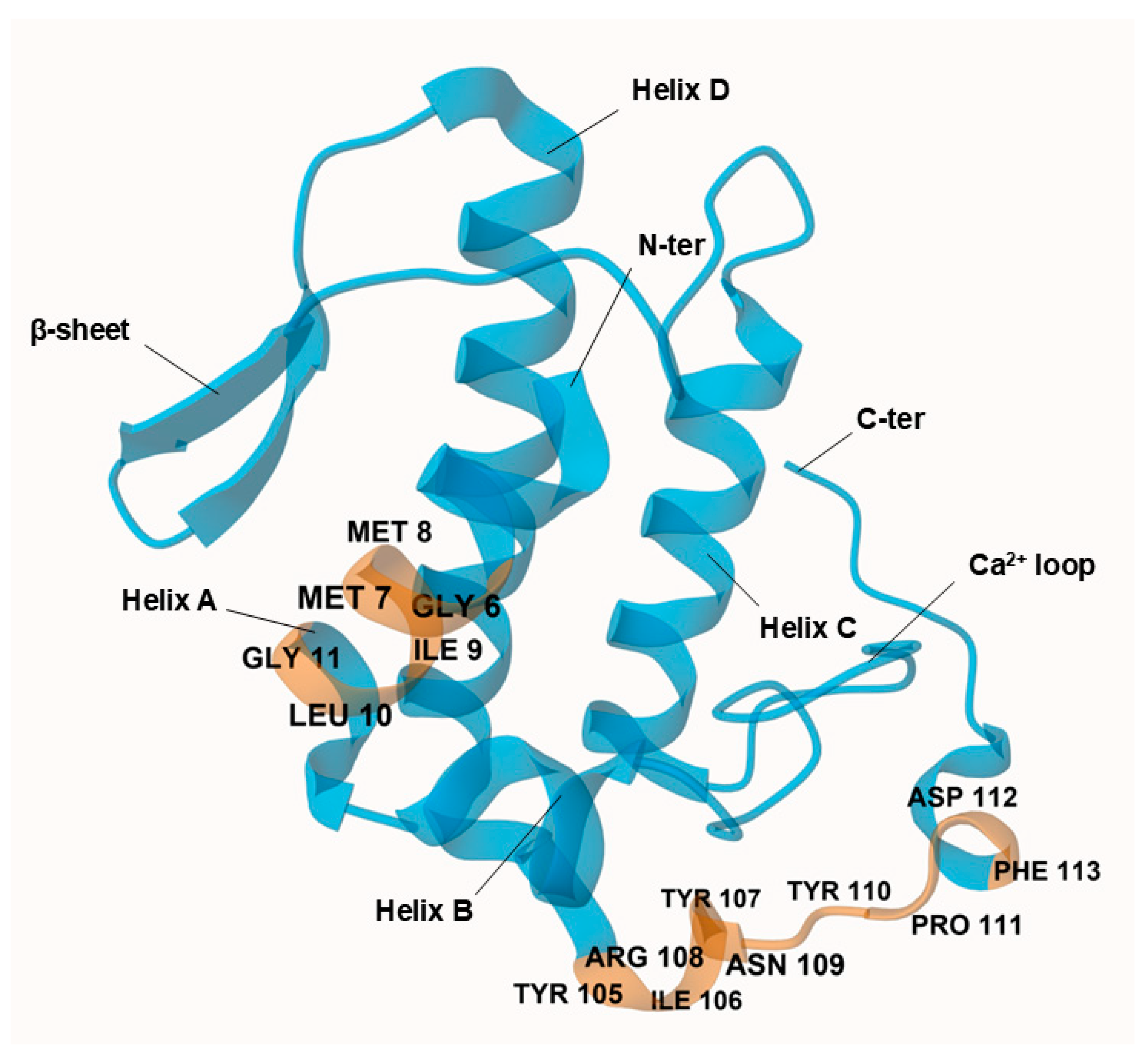
1.2. Molecular Recognition Theory
1.3. Scope and Justification
2. Results and Discussion
2.1. Physico-Chemical Properties of the Sense and Antisense Peptides
2.2. Fluorescence Assay of the Interaction Between Sense and Antisense Peptides
2.3. Binding Affinity of Sense and Antisense Peptides
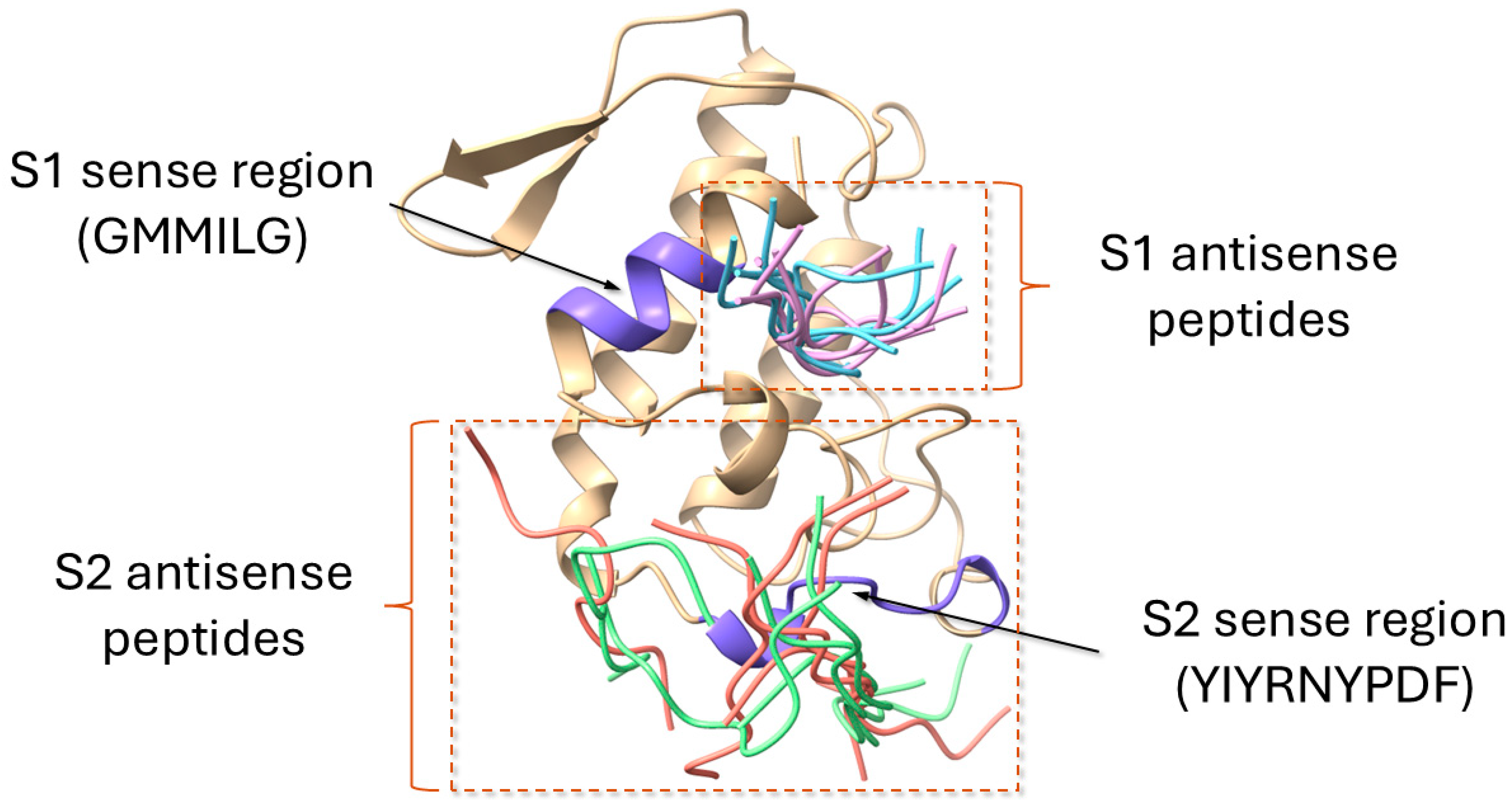
2.4. Molecular Docking of the Sense and Antisense Peptides
2.5. Molecular Docking of the Antisense Peptides and Ammodytoxin A
3. Materials and Methods
3.1. Materials
3.2. Antisense Peptide Design
3.3. Fluorescence Assay for the Binding of Sense and Antisense Peptides
3.4. Docking Simulations for the Binding of Sense and Antisense Peptides or Antisense Peptides and Ammodytoxin A
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
- Peptide Specifications: Detailed information on the synthesized peptides used in the study (GenScript Biotech B.V., Rijswijk, The Netherlands).
- Protparam Output: Computational analysis of peptide physico-chemical properties generated using the Expasy ProtParam tool.
- SPECFIT Data: Fluorescence titration results analyzed using SPECFIT software (version 2.12), including association constants, spectral fits, and residual plots.
- HPEPDOCK Outputs: Docking models and scores for the interaction of sense and antisense peptides or antisense peptides with AtxA (PDB: 3G8G). Initial 3D structures of the S1, S1A1 and S1A2 pentapeptides were generated using the PEP-FOLD3 server.
- ChimeraX Session Files: Molecular visualization sessions highlighting docking results, binding interfaces, and peptide conformations.
- Fluorescence Spectra: Experimental fluorescence spectra of stock solutions of sense and antisense peptides.
Acknowledgments
Conflicts of Interest
References
- Plavšić, F.; Žuntar, I. Uvod u Analitičku Toksikologiju; Školska Knjig: Zagreb, Croatia, 2006; pp. 206–222. [Google Scholar]
- Jelić, D.; Ajtić, R.; Bogoljub, S.; Crnobrnja-Isailović, J. Distribution of the genus Vipera in the western and Central Balkans (Squamata, Serpentes, Viperidae). Herpetozoa 2013, 25, 109–132. [Google Scholar]
- Radonić, V.; Budimir, D.; Bradarić, N.; Luksić, B.; Sapunar, D.; Vilović, K. Envenomation by the Horned Viper (Vipera ammodytes L.). Mil. Med. 1997, 162, 179–182. [Google Scholar] [CrossRef] [PubMed]
- Frangides, C.Y.; Koulouras, V.; Kouni, S.N.; Tzortzatos, G.V.; Nikolaou, A.; Pneumaticos, J.; Pierrakeas, C.; Niarchos, C.; Kounis, N.G.; Koutsojannis, C.M. Snake Venom Poisoning in Greece. Experiences with 147 Cases. Eur. J. Intern. Med. 2006, 17, 24–27. [Google Scholar] [CrossRef]
- Luksić, B.; Bradarić, N.; Prgomet, S. Venomous Snakebites in Southern Croatia. Coll. Antropol. 2006, 30, 191–197. [Google Scholar] [PubMed]
- Marinov, I.; Atanasov, V.N.; Stankova, E.; Duhalov, D.; Petrova, S.; Hubenova, A. Severe Coagulopathy after Vipera ammodytes ammodytes Snakebite in Bulgaria: A Case Report. Toxicon 2010, 56, 1066–1069. [Google Scholar] [CrossRef] [PubMed]
- Karabuva, S.; Vrkić, I.; Brizić, I.; Ivić, I.; Lukšić, B. Venomous Snakebites in Children in Southern Croatia. Toxicon 2016, 112, 8–15. [Google Scholar] [CrossRef]
- iNaturalist.org. © BY-NC 4.0 Alex Ville. Available online: https://www.inaturalist.org/photos/64687029 (accessed on 29 December 2024).
- Leonardi, A.; Sajevic, T.; Pungerčar, J.; Križaj, I. Comprehensive Study of the Proteome and Transcriptome of the Venom of the Most Venomous European Viper: Discovery of a New Subclass of Ancestral Snake Venom Metalloproteinase Precursor-Derived Proteins. J. Proteome Res. 2019, 18, 2287–2309. [Google Scholar] [CrossRef] [PubMed]
- Morita, T. Structures and Functions of Snake Venom CLPs (C-Type Lectin-like Proteins) with Anticoagulant-, Procoagulant-, and Platelet-Modulating Activities. Toxicon 2005, 45, 1099–1114. [Google Scholar] [CrossRef]
- Calvete, J.J.; Sanz, L.; Angulo, Y.; Lomonte, B.; Gutiérrez, J.M. Venoms, Venomics, Antivenomics. FEBS Lett. 2009, 583, 1736–1743. [Google Scholar] [CrossRef]
- Gopcevic, K.; Karadzic, I.; Izrael-Zivkovic, L.; Medic, A.; Isakovic, A.; Popović, M.; Kekic, D.; Stanojkovic, T.; Hozic, A.; Cindric, M. Study of the Venom Proteome of Vipera ammodytes ammodytes (Linnaeus, 1758): A Qualitative Overview, Biochemical and Biological Profiling. CBPD 2021, 37, 100776. [Google Scholar] [CrossRef] [PubMed]
- Six, D.A.; Dennis, E.A. The Expanding Superfamily of Phospholipase A2 Enzymes: Classification and Characterization. Biochim. Biophys. Acta 2000, 1488, 1488. [Google Scholar] [CrossRef]
- Ritonja, A.; Gubenek, F. Ammodytoxin A, a Highly Lethal Phospholipase A2 from Vipera ammodytes ammodytes Venom. Biochim. Biophys. Acta 1985, 828, 306–312. [Google Scholar] [CrossRef]
- Chang, C.C. Neurotoxins with Phospholipase A2 Activity in Snake Venoms. Proc. Natl. Sci. Counc. Repub. China B 1985, 9, 126–142. [Google Scholar]
- Pungerčar, J.; Križaj, I. Understanding the Molecular Mechanism Underlying the Presynaptic Toxicity of Secreted Phospholipases A2. Toxicon 2007, 50, 871–892. [Google Scholar] [CrossRef] [PubMed]
- Šribar, J.; Oberčkal, J.; Križaj, I. Understanding the Molecular Mechanism Underlying the Presynaptic Toxicity of Secreted Phospholipases A2: An Update. Toxicon 2014, 89, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Saul, F.A.; Prijatelj-Žnidarsić, P.; Vulliez-le Normand, B.; Villette, B.; Raynal, B.; Pungercar, J.; Križaj, I.; Faure, G. Comparative Structural Studies of Two Natural Isoforms of Ammodytoxin, Phospholipases A2 from Vipera ammodytes ammodytes Which Differ in Neurotoxicity and Anticoagulant Activity. J. Struct. Biol. 2010, 169, 360–369. [Google Scholar] [CrossRef]
- Prijatelj, P.; Jenko Pražnikar, Z.; Petan, T.; Križaj, I.; Pungerčar, J. Mapping the Structural Determinants of Presynaptic Neurotoxicity of Snake Venom Phospholipases A2. Toxicon 2008, 51, 1520–1529. [Google Scholar] [CrossRef] [PubMed]
- Petan, T.; Križaj, I.; Gelb, M.H.; Pungerčar, J. Ammodytoxins, Potent Presynaptic Neurotoxins, Are Also Highly Efficient Phospholipase A2 Enzymes. Biochemistry 2005, 44, 12535–12545. [Google Scholar] [CrossRef] [PubMed]
- Ivanovski, G.; Čopič, A.; Križaj, I.; Gubenšek, F.; Pungerčar, J. The Amino Acid Region 115–119 of Ammodytoxins Plays an Important Role in Neurotoxicity. Biochem. Biophys. Res. Commun. 2000, 276, 1229–1234. [Google Scholar] [CrossRef] [PubMed]
- Petan, T.; Križaj, I.; Gubenšek, F.; Pungerčar, J. Phenylalanine-24 in the N-Terminal Region of Ammodytoxins Is Important for Both Enzymic Activity and Presynaptic Toxicity. Biochem. J. 2002, 363, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Sehnal, D.; Bittrich, S.; Deshpande, M.; Svobodová, R.; Berka, K.; Bazgier, V.; Velankar, S.; Burley, S.K.; Koča, J.; Rose, A.S. Mol* Viewer: Modern Web App for 3D Visualization and Analysis of Large Biomolecular Structures. Nucleic Acids Res. 2021, 49, W431–W437. [Google Scholar] [CrossRef]
- Renetseder, R.; Brunie, S.; Dijkstra, B.W.; Drenth, J.; Sigler, P.B. A Comparison of the Crystal Structures of Phospholipase A2 from Bovine Pancreas and Crotalus Atrox Venom. J. Biol. Chem. 1985, 260, 11627–11634. [Google Scholar] [CrossRef] [PubMed]
- Štambuk, N.; Konjevoda, P.; Boban-Blagaić, A.; Pokrić, B. Molecular Recognition Theory of the Complementary (Antisense) Peptide Interactions. Theory Biosci. 2005, 123, 265–275. [Google Scholar] [CrossRef]
- Heal, J.R.; Roberts, G.W.; Raynes, J.G.; Bhakoo, A.; Miller, A.D. Specific Interactions Between Sense and Complementary Peptides: The Basis for the Proteomic Code. ChemBioChem 2002, 3, 136–151. [Google Scholar] [CrossRef] [PubMed]
- Kyte, J.; Doolittle, R.F. A Simple Method for Displaying the Hydropathic Character of a Protein. J. Mol. Biol. 1982, 157, 105–132. [Google Scholar] [CrossRef] [PubMed]
- Blalock, J.E. Genetic Origins of Protein Shape and Interaction Rules. Nat. Med. 1995, 1, 876–878. [Google Scholar] [CrossRef] [PubMed]
- Brentani, R.R. Biological Implications of Complementary Hydropathy of Amino Acids. J. Theor. Biol. 1988, 135, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Shai, Y.; Flashner, M.; Chaiken, I.M. Anti-Sense Peptide Recognition of Sense Peptides: Direct Quantitative Characterization with the Ribonuclease S-Peptide System Using Analytical High-Performance Affinity Chromatography. Biochemistry 1987, 26, 669–675. [Google Scholar] [CrossRef] [PubMed]
- Fassina, G.; Zamai, M.; Brigham-Burke, M.; Chaiken, I.M. Recognition Properties of Antisense Peptides to Arg8-Vasopressin/Bovine Neurophysin II Biosynthetic Precursor Sequences. Biochemistry 1989, 28, 8811–8818. [Google Scholar] [CrossRef] [PubMed]
- Root-Bernstein, R.S. Amino Acid Pairing. J. Theor. Biol. 1982, 94, 885–894. [Google Scholar] [CrossRef] [PubMed]
- WHO Expert Committee on Biological Standardization. Annex 5: Guidelines for the Production, Control and Regulation of Snake Antivenom Immunoglobulins; In 67th Report, WHO Technical Report Series No. 1004; WHO: Geneva, Switzerland, 2017; ISBN 978-92-4-121013-3. [Google Scholar]
- Štambuk, N.; Konjevoda, P.; Pavan, J. Antisense Peptide Technology for Diagnostic Tests and Bioengineering Research. Int. J. Mol. Sci. 2021, 22, 9106. [Google Scholar] [CrossRef] [PubMed]
- Gonda, D.K.; Bachmair, A.; Wünning, I.; Tobias, J.W.; Lane, W.S.; Varshavsky, A. Universality and Structure of the N-End Rule. J. Biol. Chem. 1989, 264, 16700–16712. [Google Scholar] [CrossRef] [PubMed]
- Guruprasad, K.; Reddy, B.V.B.; Pandit, M.W. Correlation between Stability of a Protein and Its Dipeptide Composition: A Novel Approach for Predicting in Vivo Stability of a Protein from Its Primary Sequence. Protein Eng. 1990, 4, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Ikai, A. Thermostability and Aliphatic Index of Globular Proteins. J. Biochem. 1980, 88, 1895–1898. [Google Scholar] [PubMed]
- Shi, S.M.; Di, L. Strategies to Optimize Peptide Stability and Prolong Half-Life. In Peptide Therapeutics; Jois, S.D., Ed.; AAPS Advances in the Pharmaceutical Sciences Series; Springer International Publishing: Cham, Switzerland, 2022; Volume 47, pp. 163–182. ISBN 978-3-031-04543-1. [Google Scholar]
- Thordarson, P. Determining Association Constants from Titration Experiments in Supramolecular Chemistry. Chem. Soc. Rev. 2011, 40, 1305–1323. [Google Scholar] [CrossRef] [PubMed]
- Weitner, T.; Friganović, T.; Šakić, D. Inner Filter Effect Correction for Fluorescence Measurements in Microplates Using Variable Vertical Axis Focus. Anal. Chem. 2022, 94, 7107–7114. [Google Scholar] [CrossRef] [PubMed]
- Alexander Ross, J.B.; Laws, W.R.; Rousslang, K.W.; Wyssbrod, H.R. Tyrosine Fluorescence and Phosphorescence from Proteins and Polypeptides. In Topics in Fluorescence Spectroscopy; Lakowicz, J.R., Ed.; Topics in Fluorescence Spectroscopy; Kluwer Academic Publishers: Boston, MA, USA, 2002; Volume 3, pp. 1–64. ISBN 978-0-306-43954-4. [Google Scholar]
- Lakowicz, J.R. Effects of Solvents on Fluorescence Emission Spectra. In Principles of Fluorescence Spectroscopy; Springer: Boston, MA, USA, 1983; pp. 187–215. ISBN 978-1-4615-7660-0. [Google Scholar]
- Liu, Z.; Li, Y.; Han, L.; Li, J.; Liu, J.; Zhao, Z.; Nie, W.; Liu, Y.; Wang, R. PDB-Wide Collection of Binding Data: Current Status of the PDBbind Database. Bioinformatics 2015, 31, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Landry, J.P.; Ke, Y.; Yu, G.-L.; Zhu, X.D. Measuring Affinity Constants of 1450 Monoclonal Antibodies to Peptide Targets with a Microarray-Based Label-Free Assay Platform. J. Immunol. Methods 2015, 417, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Gavade, A.; Nagraj, A.K.; Patel, R.; Pais, R.; Dhanure, P.; Scheele, J.; Seiz, W.; Patil, J. Understanding the Specific Implications of Amino Acids in the Antibody Development. Protein J. 2024, 43, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Marron, J.S.; Shen, H.; Zhu, Z. Singular Value Decomposition and Its Visualization. J. Comput. Graph. Stat. 2007, 16, 833–854. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure Visualization for Researchers, Educators, and Developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Jin, B.; Li, H.; Huang, S.-Y. HPEPDOCK: A Web Server for Blind Peptide–Protein Docking Based on a Hierarchical Algorithm. Nucleic Acids Res. 2018, 46, W443–W450. [Google Scholar] [CrossRef] [PubMed]
- Panuszko, A.; Pastwa, P.; Gajewski, J.; Bruździak, P. Characterizing Interactions Between Small Peptides and Dimethyl Sulfoxide Using Infrared Spectroscopy and Computational Methods. Molecules 2024, 29, 5869. [Google Scholar] [CrossRef]
- Tjernberg, A.; Markova, N.; Griffiths, W.J.; HalléN, D. DMSO-Related Effects in Protein Characterization. J. Biomol. Screen. 2006, 11, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Yadav, N.S.; Choudhury, D. Conformational Perturbation of Peptides in Presence of Polar Organic Solvents. J. Mol. Graph. Model. 2019, 89, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.; Wilkins, M.R.; Appel, R.D.; Bairoch, A. Protein Identification and Analysis Tools on the ExPASy Server. In The Proteomics Protocols Handbook; Walker, J.M., Ed.; Humana Press: Totowa, NJ, USA, 2005; pp. 571–607. ISBN 978-1-58829-343-5. [Google Scholar]
- Tetin, S.Y.; Hazlett, T.L. Optical Spectroscopy in Studies of Antibody–Hapten Interactions. Methods 2000, 20, 341–361. [Google Scholar] [CrossRef]
- Štambuk, N.; Manojlović, Z.; Turčić, P.; Martinić, R.; Konjevoda, P.; Weitner, T.; Wardega, P.; Gabričević, M. A Simple Three-Step Method for Design and Affinity Testing of New Antisense Peptides: An Example of Erythropoietin. Int. J. Mol. Sci. 2014, 15, 9209–9223. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.-Y.; Pownall, H.J.; Gotto, A.M. Identification of Peptides Containing Tryptophan, Tyrosine, and Phenylalanine Using Photodiode-Array Spectrophotometry. Anal. Biochem. 1985, 145, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Gampp, H.; Maeder, M.; Meyer, C.J.; Zuberbu¨hler, A.D. Calculation of Equilibrium Constants from Multiwavelength Spectroscopic Data—II. Talanta 1985, 32, 257–264. [Google Scholar] [CrossRef]
- Gampp, H.; Maeder, M.; Zuberbühler, A.D. Computerized Data Acquisition and Data Reduction in Spectrophotometric Analysis Part 2: Numerical Analysis with and without an Underlying Chemical Model. TrAC Trends Anal. Chem. 1988, 7, 147–150. [Google Scholar] [CrossRef]
- Huang, S.; Zou, X. An Iterative Knowledge-Based Scoring Function for Protein–Protein Recognition. Proteins 2008, 72, 557–579. [Google Scholar] [CrossRef] [PubMed]
- Weng, G.; Gao, J.; Wang, Z.; Wang, E.; Hu, X.; Yao, X.; Cao, D.; Hou, T. Comprehensive Evaluation of Fourteen Docking Programs on Protein–Peptide Complexes. J. Chem. Theory Comput. 2020, 16, 3959–3969. [Google Scholar] [CrossRef] [PubMed]
- Lamiable, A.; Thévenet, P.; Rey, J.; Vavrusa, M.; Derreumaux, P.; Tufféry, P. PEP-FOLD3: Faster de Novo Structure Prediction for Linear Peptides in Solution and in Complex. Nucleic Acids Res. 2016, 44, W449–W454. [Google Scholar] [CrossRef] [PubMed]
- Wohlwend, J.; Corso, G.; Passaro, S.; Reveiz, M.; Leidal, K.; Swiderski, W.; Portnoi, T.; Chinn, I.; Silterra, J.; Jaakkola, T.; et al. Boltz-1: Democratizing Biomolecular Interaction Modeling. bioRxiv 2024. [Google Scholar] [CrossRef]
- Žužek, M.C.; Ivanušec, A.; Herman, J.; Šribar, J.; Leonardi, A.; Frangež, R.; Križaj, I. Comparative Electrophysiological Characterization of Ammodytoxin A, a β-Neurotoxin from the Nose-Horned Viper Venom, and Its Enzymatically Inactive Mutant. Toxicon 2024, 247, 107833. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sense Amino Acid | Antisense Amino Acid (Direction 5′-3′) | Antisense Amino Acid (Direction 3′-5′) |
|---|---|---|
| Isoleucine (I) | Y, N, D | Y |
| Valine (V) | H, N, D, Y | H, Q |
| Leucine (L) | E, K, Q | N, E, D |
| Phenylalanine (F) | K, E | K |
| Cysteine (C) | T, A | T |
| Methionine (M) | H | Y |
| Alanine (A) | R, G, S, C | R |
| Glycine (G) | P, S, T, A | P |
| Threonine (T) | G, S, C, R | C, W |
| Tryptophan (W) | P | T |
| Serine (S) | R, G, T, A | S, R |
| Tyrosine (Y) | I, V | M, I |
| Proline (P) | G, W, R | M, R |
| Histidine (H) | V, M | V |
| Aspartate (D) | I, V | L |
| Glutamate (E) | L, F | L |
| Asparagine (N) | I, V | L |
| Glutamine (Q) | L | V |
| Lysine (K) | F, L | F |
| Arginine (R) | A, S, P, T | A, S |
| Sequence | Label | F a /f. u. | λmax/nm | pI | Half-Life b/h | II c | AI d | GRAVY e |
|---|---|---|---|---|---|---|---|---|
| GMMILG | S1 | 0.80 | 345 | 5.52 | 30 | 35.63 | 130 | 1.883 |
| YIYRNYPDF | S2 | 3.55 | 317 | 5.83 | 2.8 | 0.77 | 43.3 | −1.078 |
| SHHYEP | S1A1 | 2.37 | 320 | 5.93 | 1.9 | 101.1 | 0 | −2.267 |
| SHHYSP | S1A2 | 2.11 | 319 | 6.66 | 1.9 | 154.8 | 0 | −1.817 |
| INISIVGVK | S2A1 | 0.94 | 346 | 8.75 | 20 | 35.64 | 194.4 | 1.478 |
| VNISVIGIK | S2A2 | 0.75 | 343 | 8.72 | 100 | 38.84 | 194.4 | 1.478 |
| Sense peptide | Antisense Peptide | KD (σ)/μM |
|---|---|---|
| S1 | S1A1 | 4.9 (0.7) |
| S1A2 | 1.4 (0.5) | |
| S2 | S2A1 | 1.1 (0.1) |
| S2A2 | 8.9 (0.8) |
| Peptide Type | Sequence | Label * | M/g mol−1 | Purity/% |
|---|---|---|---|---|
| Sense | GMMILG | S1 | 621 | 95.9 |
| YIYRNYPDF | S2 | 1250 | 98.9 | |
| Antisense | SHHYEP | S1A1 | 769 | 98.5 |
| SHHYSP | S1A2 | 727 | 99.7 | |
| INISIVGVK | S2A1 | 942 | 95.3 | |
| VNISVIGIK | S2A2 | 942 | 95.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Biruš, I.; Šeba, T.; Marić, M.; Gabričević, M.; Weitner, T. Design and Binding Affinity of Antisense Peptides for Snake Venom Neutralization. Molecules 2025, 30, 903. https://doi.org/10.3390/molecules30040903
Biruš I, Šeba T, Marić M, Gabričević M, Weitner T. Design and Binding Affinity of Antisense Peptides for Snake Venom Neutralization. Molecules. 2025; 30(4):903. https://doi.org/10.3390/molecules30040903
Chicago/Turabian StyleBiruš, Ivan, Tino Šeba, Marin Marić, Mario Gabričević, and Tin Weitner. 2025. "Design and Binding Affinity of Antisense Peptides for Snake Venom Neutralization" Molecules 30, no. 4: 903. https://doi.org/10.3390/molecules30040903
APA StyleBiruš, I., Šeba, T., Marić, M., Gabričević, M., & Weitner, T. (2025). Design and Binding Affinity of Antisense Peptides for Snake Venom Neutralization. Molecules, 30(4), 903. https://doi.org/10.3390/molecules30040903