
Machine Learning Descriptors for CO2 Capture Materials

 , and
, and
Abstract
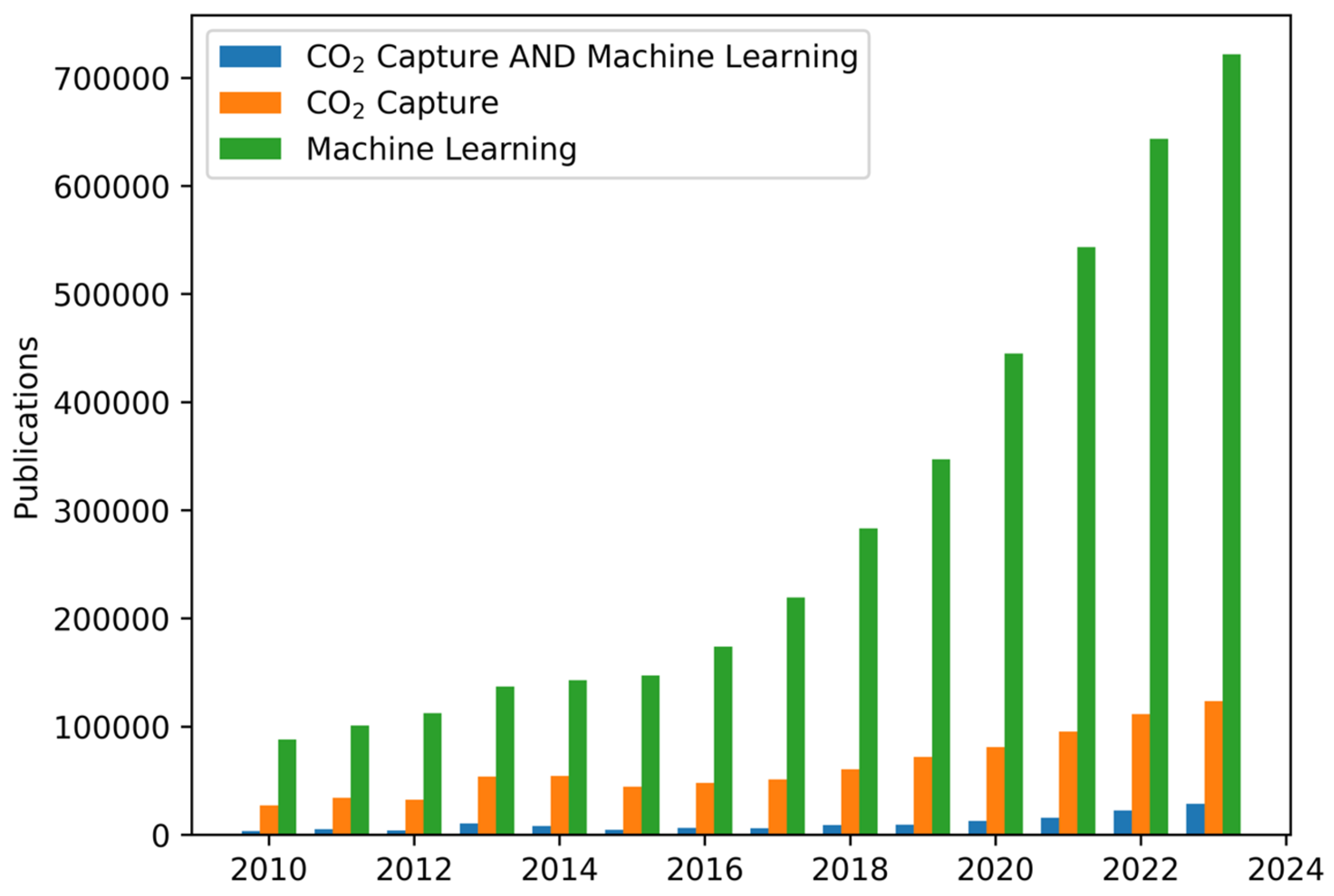
1. Introduction
2. Descriptors in ML for CO2 Capture
2.1. Operating Conditions
2.2. Charge- and Orbital-Based Descriptors
2.3. Thermodynamic Descriptors
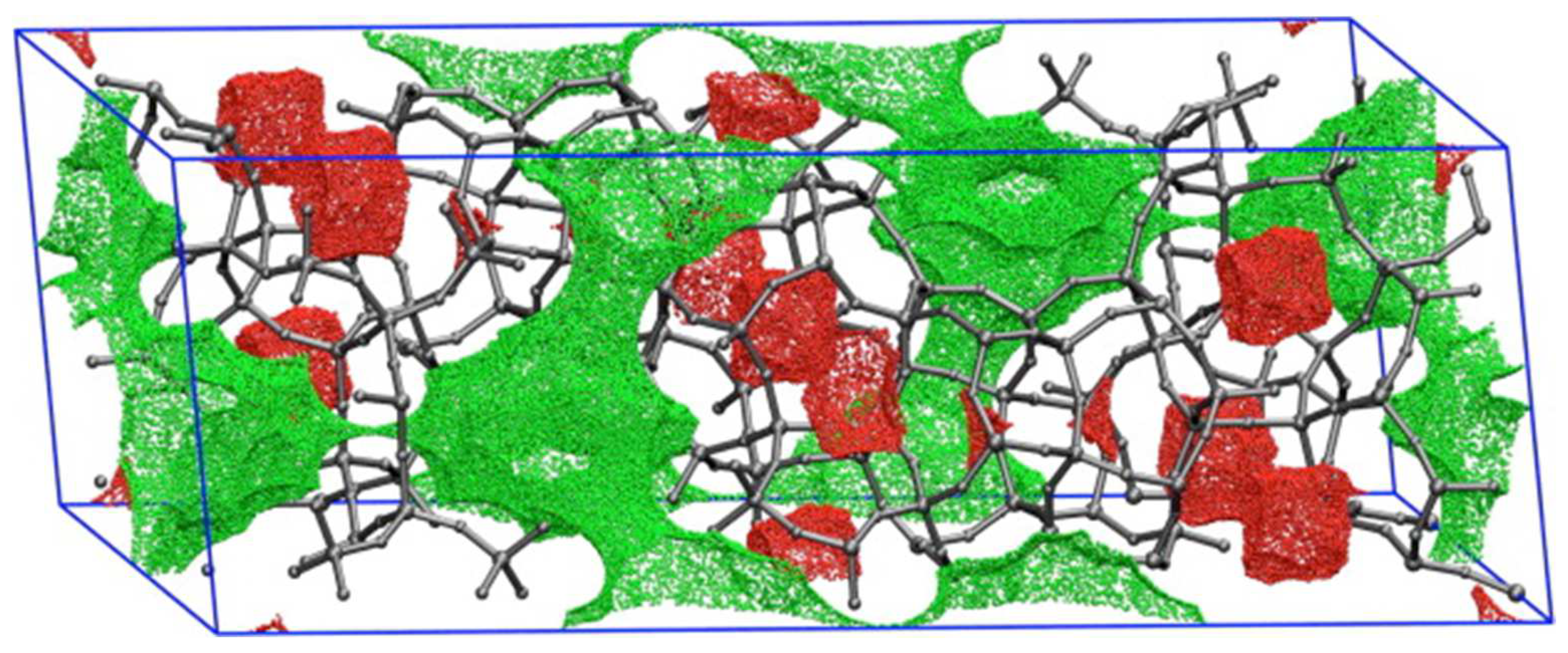
2.4. Geometric and Other Structural Descriptors
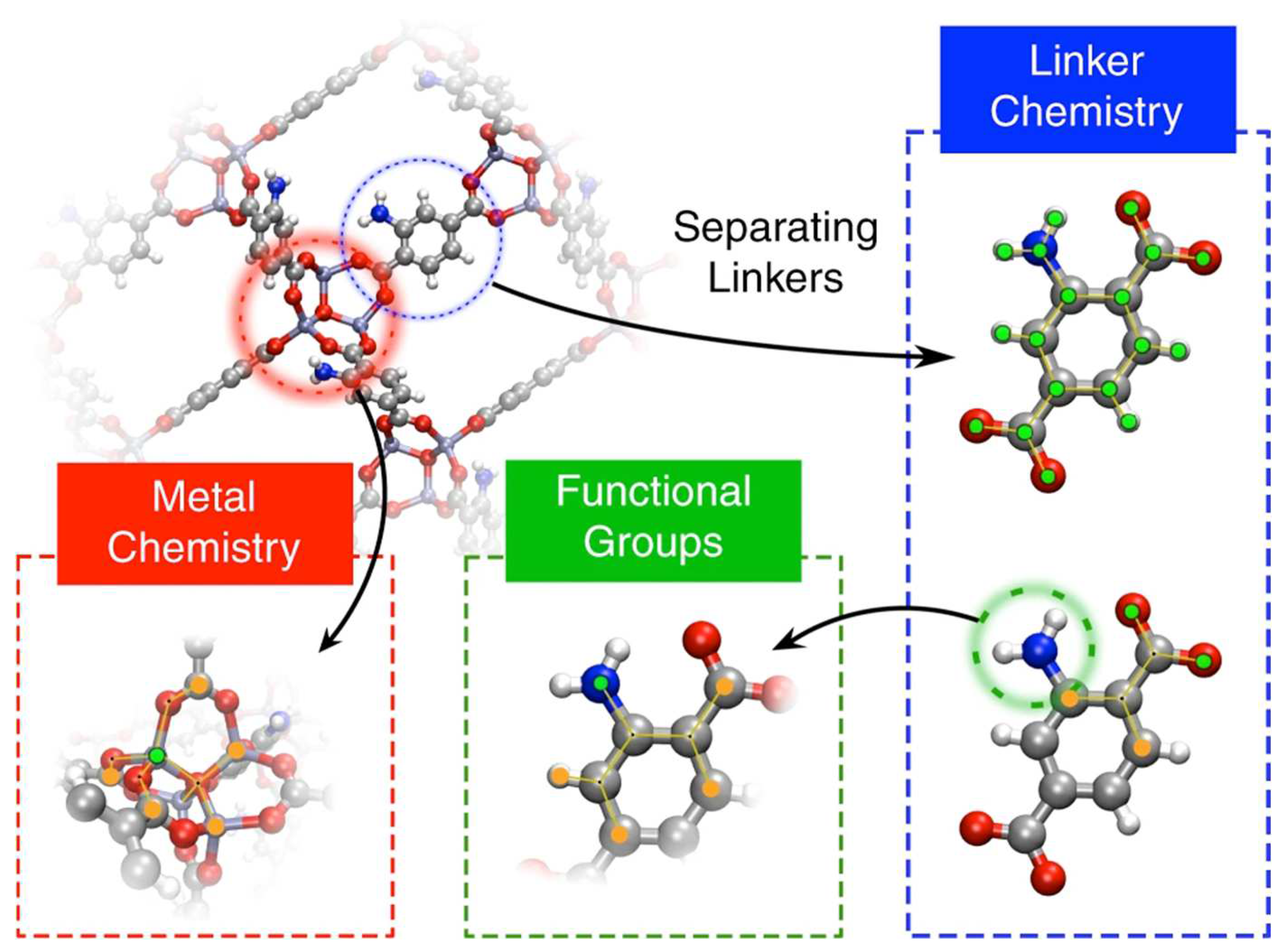
2.5. Chemical Composition-Based Descriptors
3. Descriptor Selection Strategies
4. Machine Learning Model Optimisation
4.1. Hyperparameter Tuning
4.2. Evaluation of the Performance of Models
4.2.1. Coefficient of Determination
4.2.2. Mean Absolute Error
4.2.3. Root Mean Square Error
4.2.4. Recall Rate
4.2.5. Spearman’s Rank Correlation Coefficient
4.2.6. Average Absolute Relative Deviation
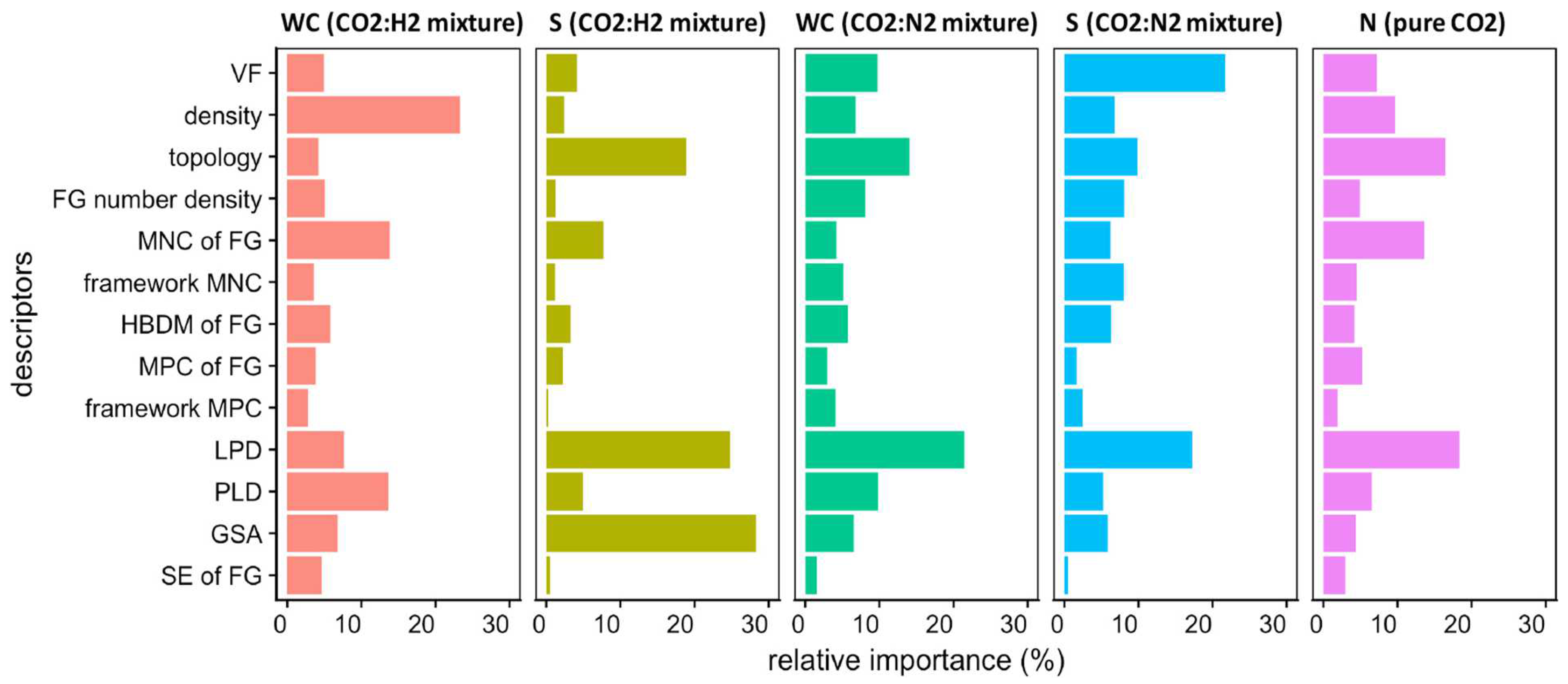
4.3. Descriptor Importance and Design Strategies
5. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Foote, E. On the Heat in the Sun’s Rays. Amer. J. Sci. Arts 1856, XXII, 377–382. [Google Scholar]
- Carbon Dioxide Now More Than 50% Higher Than Pre-Industrial Levels; National Oceanic and Atmospheric Administration—U.S. Department of Commerce; NOAA: Silver Spring, MD, USA.
- MacDowell, N.; Florin, N.; Buchard, A.; Hallett, J.; Galindo, A.; Jackson, G.; Adjiman, C.S.; Williams, C.K.; Shah, N.; Fennell, P. An overview of CO2 capture technologies. Energy Environ. Sci. 2010, 3, 1645–1669. [Google Scholar] [CrossRef]
- Abdulla, A.; Hanna, R.; Schell, K.R.; Babacan, O.; Victor, D.G. Explaining successful and failed investments in U.S. carbon capture and storage using empirical and expert assessments. Environ. Res. Lett. 2020, 15, 014036. [Google Scholar] [CrossRef]
- Dziejarski, B.; Serafin, J.; Andersson, K.; Krzyżyńska, R. CO2 capture materials: A review of current trends and future challenges. Mater. Today Sustain. 2023, 24, 100483. [Google Scholar] [CrossRef]
- Yang, R.T. Gas Separation by Adsorption Processes; Series on Chemical Engineering; World Scientific Publishing CO. 364; Imperial College Press: London, UK, 1997; Volume 1. [Google Scholar]
- Rouquerol, F.; Rouquerol, J.; Sing, K.S.W.; Llewellyn, P.; Maurin, G. Adsorption by Powders and Porous Solids, 2nd ed.; Oxford Academic Press: Oxford, UK, 2014. [Google Scholar]
- Figueroa, J.D.; Fout, T.; Plasynski, S.; McIlvried, H.; Srivastava, R.D. Advances in CO2 capture technology—The U.S. Department of Energy’s Carbon Sequestration Program. Int. J. Greenh. Gas Control 2008, 2, 9–20. [Google Scholar] [CrossRef]
- Lee, S.-Y.; Park, S.-J. A review on solid adsorbents for carbon dioxide capture. J. Ind. Eng. Chem. 2015, 23, 1–11. [Google Scholar] [CrossRef]
- Rombach, R.; Blattmann, A.; Lorenz, D.; Esser, P.; Ommer, B. High-Resolution Image Synthesis with Latent Diffusion Models. In Proceedings of the IEEE/CVF conference on computer vision and pattern recognition, New Orleans, LA, USA, 18–24 June 2022; pp. 10684–10695. [Google Scholar]
- Brown, T.; Mann, B.; Ryder, N.; Subbiah, M.; Kaplan, J.D.; Dhariwal, P.; Neelakantan, A.; Shyam, P.; Sastry, G.; Askell, A. Language models are few-shot learners. Adv. Neural Inf. Process. Syst. 2020, 33, 1877–1901. [Google Scholar]
- Falomir, Z.; Museros, L.; Sanz, I.; Gonzalez-Abril, L. Categorizing paintings in art styles based on qualitative color descriptors, quantitative global features and machine learning (QArt-Learn). Expert Syst. Appl. 2018, 97, 83–94. [Google Scholar] [CrossRef]
- Bahuleyan, H. Music Genre Classification using Machine Learning Techniques. arxiv 2018, arXiv:1804.01149. [Google Scholar]
- Sturm, B.L.; Ben-Tal, O.; Monaghan, Ú.; Collins, N.; Herremans, D.; Chew, E.; Hadjeres, G.; Deruty, E.; Pachet, F. Machine learning research that matters for music creation: A case study. J. New Music Res. 2019, 48, 36–55. [Google Scholar] [CrossRef]
- Karachun, I.; Vinnichek, L.; Tuskov, A. Machine learning methods in finance. SHS Web Conf. 2021, 110, 05012. [Google Scholar] [CrossRef]
- Warin, T.; Stojkov, A. Machine Learning in Finance: A Metadata-Based Systematic Review of the Literature. J. Risk Financ. Manag. 2021, 14, 302. [Google Scholar] [CrossRef]
- Sahu, S.K.; Mokhade, A.; Bokde, N.D. An Overview of Machine Learning, Deep Learning, and Reinforcement Learning-Based Techniques in Quantitative Finance: Recent Progress and Challenges. Appl. Sci. 2023, 13, 1956. [Google Scholar] [CrossRef]
- Shailaja, K.; Seetharamulu, B.; Jabbar, M.A. Machine Learning in Healthcare: A Review. In Proceedings of the 2018 Second International Conference on Electronics, Communication and Aerospace Technology (ICECA), Coimbatore, India, 29–31 March 2018; pp. 910–914. [Google Scholar]
- Tarca, A.L.; Carey, V.J.; Chen, X.-w.; Romero, R.; Drăghici, S. Machine learning and its applications to biology. PLoS Comput. Biol. 2007, 3, e116. [Google Scholar] [CrossRef]
- Gao, C.; Min, X.; Fang, M.; Tao, T.; Zheng, X.; Liu, Y.; Wu, X.; Huang, Z. Innovative Materials Science via Machine Learning. Adv. Funct. Mater. 2022, 32, 2108044. [Google Scholar] [CrossRef]
- Newen, C.; Müller, E. On the Independence of Adversarial Transferability to Topological Changes in the Dataset. In Proceedings of the 2023 IEEE 10th International Conference on Data Science and Advanced Analytics (DSAA), Thessaloniki, Greece, 9–13 October 2023; pp. 1–8. [Google Scholar]
- Thai, H.-T. Machine learning for structural engineering: A state-of-the-art review. Structures 2022, 38, 448–491. [Google Scholar] [CrossRef]
- Liu, Y.; Esan, O.C.; Pan, Z.; An, L. Machine learning for advanced energy materials. Energy AI 2021, 3, 100049. [Google Scholar] [CrossRef]
- Lu, W.; Xiao, R.; Yang, J.; Li, H.; Zhang, W. Data mining-aided materials discovery and optimization. J. Mater. 2017, 3, 191–201. [Google Scholar] [CrossRef]
- Ramprasad, R.; Batra, R.; Pilania, G.; Mannodi-Kanakkithodi, A.; Kim, C. Machine learning in materials informatics: Recent applications and prospects. NPJ Comput. Mater. 2017, 3, 54. [Google Scholar] [CrossRef]
- Khan, A.I.; Al-Badi, A. Open Source Machine Learning Frameworks for Industrial Internet of Things. Procedia Comput. Sci. 2020, 170, 571–577. [Google Scholar] [CrossRef]
- Sarker, I.H. Machine Learning: Algorithms, Real-World Applications and Research Directions. SN Comput. Sci. 2021, 2, 160. [Google Scholar] [CrossRef] [PubMed]
- Hussin, F.; Md Rahim, S.A.N.; Hatta, N.S.M.; Aroua, M.K.; Mazari, S.A. A systematic review of machine learning approaches in carbon capture applications. J. CO2 Util. 2023, 71, 102474. [Google Scholar] [CrossRef]
- Yan, Y.; Borhani, T.N.; Subraveti, S.G.; Pai, K.N.; Prasad, V.; Rajendran, A.; Nkulikiyinka, P.; Asibor, J.O.; Zhang, Z.; Shao, D.; et al. Harnessing the power of machine learning for carbon capture, utilisation, and storage (CCUS)—A state-of-the-art review. Energy Environ. Sci. 2021, 14, 6122–6157. [Google Scholar] [CrossRef]
- Yang, Z.; Chen, B.; Chen, H.; Li, H. A critical review on machine-learning-assisted screening and design of effective sorbents for carbon dioxide (CO2) capture. Front. Energy Res. 2023, 10, 1043064. [Google Scholar] [CrossRef]
- Naseem, U.; Dunn, A.G.; Khushi, M.; Kim, J. Benchmarking for biomedical natural language processing tasks with a domain specific ALBERT. BMC Bioinform. 2022, 23, 144. [Google Scholar] [CrossRef]
- Boiko, D.A.; MacKnight, R.; Kline, B.; Gomes, G. Autonomous chemical research with large language models. Nature 2023, 624, 570–578. [Google Scholar] [CrossRef]
- Shalaby, A.; Elkamel, A.; Douglas, P.L.; Zhu, Q.; Zheng, Q.P. A machine learning approach for modeling and optimization of a CO2 post-combustion capture unit. Energy 2021, 215, 119113. [Google Scholar] [CrossRef]
- Venkatraman, V.; Alsberg, B.K. Predicting CO2 capture of ionic liquids using machine learning. J. CO2 Util. 2017, 21, 162–168. [Google Scholar] [CrossRef]
- Zhang, Z.; Cao, X.; Geng, C.; Sun, Y.; He, Y.; Qiao, Z.; Zhong, C. Machine learning aided high-throughput prediction of ionic liquid@MOF composites for membrane-based CO2 capture. J. Membr. Sci. 2022, 650, 120399. [Google Scholar] [CrossRef]
- Meng, M.; Qiu, Z.; Zhong, R.; Liu, Z.; Liu, Y.; Chen, P. Adsorption characteristics of supercritical CO2/CH4 on different types of coal and a machine learning approach. Chem. Eng. J. 2019, 368, 847–864. [Google Scholar] [CrossRef]
- Sipöcz, N.; Tobiesen, F.A.; Assadi, M. The use of Artificial Neural Network models for CO2 capture plants. Appl. Energy 2011, 88, 2368. [Google Scholar] [CrossRef]
- Fernandez, M.; Boyd, P.G.; Daff, T.D.; Aghaji, M.Z.; Woo, T.K. Rapid and Accurate Machine Learning Recognition of High Performing Metal–Organic Frameworks for CO2 Capture. J. Phys. Chem. Lett. 2014, 5, 3056. [Google Scholar] [CrossRef] [PubMed]
- Baghban, A.; Bahadori, A.; Mohammadi, A.H.; Behbahaninia, A. Prediction of CO2 loading capacities of aqueous solutions of absorbents using different computational schemes. Int. J. Greenh. Gas Control 2017, 57, 143–161. [Google Scholar] [CrossRef]
- Baghban, A.; Mohammadi, A.H.; Taleghani, M.S. Rigorous modeling of CO2 equilibrium absorption in ionic liquids. Int. J. Greenh. Gas Control 2017, 58, 19–41. [Google Scholar] [CrossRef]
- Kim, Y.; Jang, H.; Kim, J.; Lee, J. Prediction of storage efficiency on CO2 sequestration in deep saline aquifers using artificial neural network. Appl. Energy 2017, 185, 916–928. [Google Scholar] [CrossRef]
- Anderson, R.; Rodgers, J.; Argueta, E.; Biong, A.; Gómez-Gualdrón, D.A. Role of Pore Chemistry and Topology in the CO2 Capture Capabilities of MOFs: From Molecular Simulation to Machine Learning. Chem. Mater. 2018, 30, 6325–6337. [Google Scholar] [CrossRef]
- Burns, T.D.; Pai, K.N.; Subraveti, S.G.; Collins, S.P.; Krykunov, M.; Rajendran, A.; Woo, T.K. Prediction of MOF Performance in Vacuum Swing Adsorption Systems for Postcombustion CO2 Capture Based on Integrated Molecular Simulations, Process Optimizations, and Machine Learning Models. Environ. Sci. Technol. 2020, 54, 4536–4544. [Google Scholar] [CrossRef]
- Song, Z.; Shi, H.; Zhang, X.; Zhou, T. Prediction of CO2 solubility in ionic liquids using machine learning methods. Chem. Eng. Sci. 2020, 223, 115752. [Google Scholar] [CrossRef]
- Zhu, X.; Tsang, D.C.W.; Wang, L.; Su, Z.; Hou, D.; Li, L.; Shang, J. Machine learning exploration of the critical factors for CO2 adsorption capacity on porous carbon materials at different pressures. J. Clean. Prod. 2020, 273, 122915. [Google Scholar] [CrossRef]
- Daryayehsalameh, B.; Nabavi, M.; Vaferi, B. Modeling of CO2 capture ability of [Bmim][BF4] ionic liquid using connectionist smart paradigms. Environ. Technol. Innov. 2021, 22, 101484. [Google Scholar] [CrossRef]
- Demir, H.; Aksu, G.O.; Gulbalkan, H.C.; Keskin, S. MOF Membranes for CO2 Capture: Past, Present and Future. Carbon Capture Sci. Technol. 2022, 2, 100026. [Google Scholar] [CrossRef]
- Dureckova, H.; Krykunov, M.; Aghaji, M.Z.; Woo, T.K. Robust Machine Learning Models for Predicting High CO2 Working Capacity and CO2/H2 Selectivity of Gas Adsorption in Metal Organic Frameworks for Precombustion Carbon Capture. J. Phys. Chem. C 2019, 123, 4133. [Google Scholar] [CrossRef]
- Rahimi, M.; Abbaspour-Fard, M.H.; Rohani, A.; Yuksel Orhan, O.; Li, X. Modeling and Optimizing N/O-Enriched Bio-Derived Adsorbents for CO2 Capture: Machine Learning and DFT Calculation Approaches. Ind. Eng. Chem. Res. 2022, 61, 10670–10688. [Google Scholar] [CrossRef]
- Mazari, S.A.; Siyal, A.R.; Solangi, N.H.; Ahmed, S.; Griffin, G.; Abro, R.; Mubarak, N.M.; Ahmed, M.; Sabzoi, N. Prediction of thermo-physical properties of 1-Butyl-3-methylimidazolium hexafluorophosphate for CO2 capture using machine learning models. J. Mol. Liq. 2021, 327, 114785. [Google Scholar] [CrossRef]
- Zhou, Z.; Davoudi, E.; Vaferi, B. Monitoring the effect of surface functionalization on the CO2 capture by graphene oxide/methyl diethanolamine nanofluids. J. Environ. Chem. Eng. 2021, 9, 106202. [Google Scholar] [CrossRef]
- Fathalian, F.; Aarabi, S.; Ghaemi, A.; Hemmati, A. Intelligent prediction models based on machine learning for CO2 capture performance by graphene oxide-based adsorbents. Sci. Rep. 2022, 12, 21507. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, K.; Lee, Y. Machine Learning Enabled Tailor-Made Design of Application-Specific Metal–Organic Frameworks. ACS Appl. Mater. Interfaces 2020, 12, 734–743. [Google Scholar] [CrossRef]
- Majumdar, S.; Moosavi, S.M.; Jablonka, K.M.; Ongari, D.; Smit, B. Diversifying Databases of Metal Organic Frameworks for High-Throughput Computational Screening. ACS Appl. Mater. Interfaces 2021, 13, 61004–61014. [Google Scholar] [CrossRef]
- Leperi, K.T.; Yancy-Caballero, D.; Snurr, R.Q.; You, F. 110th Anniversary: Surrogate Models Based on Artificial Neural Networks To Simulate and Optimize Pressure Swing Adsorption Cycles for CO2 Capture. Ind. Eng. Chem. Res. 2019, 58, 18241–18252. [Google Scholar] [CrossRef]
- Lei, Z.; Dai, C.; Chen, B. Gas solubility in ionic liquids. Chem. Rev. 2014, 114, 1289–1326. [Google Scholar] [CrossRef]
- Venkatraman, V.; Alsberg, B.K. KRAKENX: Software for the generation of alignment-independent 3D descriptors. J. Mol. Model. 2016, 22, 93. [Google Scholar] [CrossRef] [PubMed]
- Fukui, K. Theory of Orientation and Stereoselection. In Orientation and Stereoselection; Springer: Berlin/Heidelberg, Germany, 1970; pp. 1–85. [Google Scholar]
- Carvalho, P.J.; Kurnia, K.A.; Coutinho, J.A.P. Dispelling some myths about the CO2 solubility in ionic liquids. Phys. Chem. Chem. Phys. 2016, 18, 14757–14771. [Google Scholar] [CrossRef] [PubMed]
- Klähn, M.; Seduraman, A. What Determines CO2 Solubility in Ionic Liquids? A Molecular Simulation Study. J. Phys. Chem. B 2015, 119, 10066–10078. [Google Scholar] [CrossRef] [PubMed]
- Orhan, I.B.; Le, T.C.; Babarao, R.; Thornton, A.W. Accelerating the prediction of CO2 capture at low partial pressures in metal-organic frameworks using new machine learning descriptors. Commun. Chem. 2023, 6, 214. [Google Scholar] [CrossRef]
- Chung, Y.G.; Haldoupis, E.; Bucior, B.J.; Haranczyk, M.; Lee, S.; Zhang, H.; Vogiatzis, K.D.; Milisavljevic, M.; Ling, S.; Camp, J.S.; et al. Advances, Updates, and Analytics for the Computation-Ready, Experimental Metal–Organic Framework Database: CoRE MOF 2019. J. Chem. Eng. Data 2019, 64, 5985. [Google Scholar] [CrossRef]
- Blatov, V.A.; Shevchenko, A.P.; Proserpio, D.M. Applied Topological Analysis of Crystal Structures with the Program Package ToposPro. Cryst. Growth Des. 2014, 14, 3576. [Google Scholar] [CrossRef]
- Willems, T.F.; Rycroft, C.H.; Kazi, M.; Meza, J.C.; Haranczyk, M. Algorithms and tools for high-throughput geometry-based analysis of crystalline porous materials. Microporous Mesoporous Mater. 2012, 149, 134–141. [Google Scholar] [CrossRef]
- Colón, Y.J.; Gómez-Gualdrón, D.A.; Snurr, R.Q. Topologically Guided, Automated Construction of Metal–Organic Frameworks and Their Evaluation for Energy-Related Applications. Cryst. Growth Des. 2017, 17, 5801. [Google Scholar] [CrossRef]
- Dubbeldam, D.; Calero, S.; Ellis, D.E.; Snurr, R.Q. RASPA: Molecular Simulation Software for Adsorption and Diffusion in Flexible Nanoporous Materials. Mol. Simul. 2016, 42, 81. [Google Scholar] [CrossRef]
- Ma, X.; Xu, W.; Su, R.; Shao, L.; Zeng, Z.; Li, L.; Wang, H. Insights into CO2 capture in porous carbons from machine learning, experiments and molecular simulation. Sep. Purif. Technol. 2023, 306, 122521. [Google Scholar] [CrossRef]
- Zhu, X.; Wang, X.; Ok, Y.S. The application of machine learning methods for prediction of metal sorption onto biochars. J. Hazard. Mater. 2019, 378, 120727. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Chen, R.; Zhou, K.; Wu, Q.; Li, H.; Zeng, Z.; Li, L. Activated Porous Carbon with an Ultrahigh Surface Area Derived from Waste Biomass for Acetone Adsorption, CO2 Capture, and Light Hydrocarbon Separation. ACS Sustain. Chem. Eng. 2020, 8, 11721–11728. [Google Scholar] [CrossRef]
- Li, J.; Cheng, K.; Wang, S.; Morstatter, F.; Trevino, R.P.; Tang, J.; Liu, H. Feature Selection: A Data Perspective. ACM Comput. Surv. 2017, 50, 94. [Google Scholar] [CrossRef]
- Jović, A.; Brkić, K.; Bogunović, N. A review of feature selection methods with applications. In Proceedings of the 2015 38th International Convention on Information and Communication Technology, Electronics and Microelectronics (MIPRO), Opatija, Croatia, 25–29 May 2015; pp. 1200–1205. [Google Scholar]
- Orhan, I.B.; Daglar, H.; Keskin, S.; Le, T.C.; Babarao, R. Prediction of O2/N2 Selectivity in Metal–Organic Frameworks via High-Throughput Computational Screening and Machine Learning. ACS Appl. Mater. Interfaces 2022, 14, 736–749. [Google Scholar] [CrossRef]
- Pardakhti, M.; Moharreri, E.; Wanik, D.; Suib, S.L.; Srivastava, R. Machine Learning Using Combined Structural and Chemical Descriptors for Prediction of Methane Adsorption Performance of Metal Organic Frameworks (MOFs). ACS Comb. Sci. 2017, 19, 640–645. [Google Scholar] [CrossRef]
- Moosavi, S.M.; Nandy, A.; Jablonka, K.M.; Ongari, D.; Janet, J.P.; Boyd, P.G.; Lee, Y.; Smit, B.; Kulik, H.J. Understanding the diversity of the metal-organic framework ecosystem. Nat. Commun. 2020, 11, 4068. [Google Scholar] [CrossRef]
- Wilmer, C.E.; Leaf, M.; Lee, C.Y.; Farha, O.K.; Hauser, B.G.; Hupp, J.T.; Snurr, R.Q. Large-Scale Screening of Hypothetical Metal-Organic Frameworks. Nat. Chem. 2012, 4, 83. [Google Scholar] [CrossRef]
- Moosavi, S.M.; Novotny, B.Á.; Ongari, D.; Moubarak, E.; Asgari, M.; Kadioglu, Ö.; Charalambous, C.; Ortega-Guerrero, A.; Farmahini, A.H.; Sarkisov, L.; et al. A data-science approach to predict the heat capacity of nanoporous materials. Nat. Mater. 2022, 21, 1419–1425. [Google Scholar] [CrossRef]
- Kira, K.; Rendell, L.A. A. A Practical Approach to Feature Selection. In Machine Learning Proceedings 1992; Sleeman, D., Edwards, P., Eds.; Morgan Kaufmann: San Francisco, CA, USA, 1992; pp. 249–256. [Google Scholar]
- Venkatesh, B.; Anuradha, J. A Review of Feature Selection and Its Methods. Cybern. Inf. Technol. 2019, 19, 3. [Google Scholar] [CrossRef]
- Colaco, S.; Kumar, S.; Tamang, A.; Biju, V.G. A Review on Feature Selection Algorithms. In Proceedings of the Emerging Research in Computing, Information, Communication and Applications, Singapore, 11 September 2019; pp. 133–153. [Google Scholar]
- Oh, D.-H.; Dat Vo, N.; Lee, J.-C.; You, J.K.; Lee, D.; Lee, C.-H. Prediction of CO2 capture capability of 0.5 MW MEA demo plant using three different deep learning pipelines. Fuel 2022, 315, 123229. [Google Scholar] [CrossRef]
- Khaire, U.M.; Dhanalakshmi, R. Stability of feature selection algorithm: A review. J. King Saud Univ. Comput. Inf. Sci. 2022, 34, 1060–1073. [Google Scholar] [CrossRef]
- Yang, L.; Shami, A. On hyperparameter optimization of machine learning algorithms: Theory and practice. Neurocomputing 2020, 415, 295–316. [Google Scholar] [CrossRef]
- Deng, X.; Yang, W.; Li, S.; Liang, H.; Shi, Z.; Qiao, Z. Large-Scale Screening and Machine Learning to Predict the Computation-Ready, Experimental Metal-Organic Frameworks for CO2 Capture from Air. Appl. Sci. 2020, 10, 569. [Google Scholar] [CrossRef]
- Jian, Y.; Wang, Y.; Barati Farimani, A. Predicting CO2 Absorption in Ionic Liquids with Molecular Descriptors and Explainable Graph Neural Networks. ACS Sustain. Chem. Eng. 2022, 10, 16681–16691. [Google Scholar] [CrossRef]
- Liashchynskyi, P.; Liashchynskyi, P. Grid Search, Random Search, Genetic Algorithm: A Big Comparison for NAS. arXiv 2019, arXiv:1912.06059. [Google Scholar]
- Burner, J.; Schwiedrzik, L.; Krykunov, M.; Luo, J.; Boyd, P.G.; Woo, T.K. High-Performing Deep Learning Regression Models for Predicting Low-Pressure CO2 Adsorption Properties of Metal–Organic Frameworks. J. Phys. Chem. C 2020, 124, 27996–28005. [Google Scholar] [CrossRef]
- Bergstra, J.; Bengio, Y. Random Search for Hyper-Parameter Optimization. J. Mach. Learn. Res. 2012, 13, 281–305. [Google Scholar]
- Handelman, G.S.; Kok, H.K.; Chandra, R.V.; Razavi, A.H.; Huang, S.; Brooks, M.; Lee, M.J.; Asadi, H. Peering Into the Black Box of Artificial Intelligence: Evaluation Metrics of Machine Learning Methods. Am. J. Roentgenol. 2019, 212, 38–43. [Google Scholar] [CrossRef]
- Shcherbakov, M.; Brebels, A.; Shcherbakova, N.L.; Tyukov, A.; Janovsky, T.A.; Kamaev, V.A. A survey of forecast error measures. World Appl. Sci. J. 2013, 24, 171–176. [Google Scholar] [CrossRef]
- Alexander, D.L.J.; Tropsha, A.; Winkler, D.A. Beware of R2: Simple, Unambiguous Assessment of the Prediction Accuracy of QSAR and QSPR Models. J. Chem. Inf. Model. 2015, 55, 1316–1322. [Google Scholar] [CrossRef]
- Hodson, T.O. Root-mean-square error (RMSE) or mean absolute error (MAE): When to use them or not. Geosci. Model Dev. 2022, 15, 5481–5487. [Google Scholar] [CrossRef]
- Cort, J.W.; Kenji, M. Advantages of the mean absolute error (MAE) over the root mean square error (RMSE) in assessing average model performance. Clim. Res. 2005, 30, 79–82. [Google Scholar]
- Hyndman, R.J.; Koehler, A.B. Another look at measures of forecast accuracy. Int. J. Forecast. 2006, 22, 679–688. [Google Scholar] [CrossRef]
- Powers, D. Evaluation: From Precision, Recall and F-Factor to ROC, Informedness, Markedness & Correlation. Mach. Learn. Technol. 2008, 2. [Google Scholar] [CrossRef]
- Spearman, C. The Proof and Measurement of Association between Two Things. Am. J. Psychol. 1904, 15, 72–101. [Google Scholar] [CrossRef]
- Kader, G.D. Means and MADs. Math. Teach. Middle Sch. 1999, 4, 398–403. [Google Scholar] [CrossRef]
- Rahimi, M.; Moosavi, S.M.; Smit, B.; Hatton, T.A. Toward smart carbon capture with machine learning. Cell Rep. Phys. Sci. 2021, 2, 100396. [Google Scholar] [CrossRef]
- Boyd, P.G.; Chidambaram, A.; García-Díez, E.; Ireland, C.P.; Daff, T.D.; Bounds, R.; Gładysiak, A.; Schouwink, P.; Moosavi, S.M.; Maroto-Valer, M.M.; et al. Data-driven design of metal–organic frameworks for wet flue gas CO2 capture. Nature 2019, 576, 253–256. [Google Scholar] [CrossRef]
- Collins, S.P.; Daff, T.D.; Piotrkowski, S.S.; Woo, T.K. Materials Design by Evolutionary Optimization of Functional Groups in Metal-Organic Frameworks. Sci. Adv. 2016, 2, e1600954. [Google Scholar] [CrossRef]
- Guan, J.; Huang, T.; Liu, W.; Feng, F.; Japip, S.; Li, J.; Wu, J.; Wang, X.; Zhang, S. Design and prediction of metal organic framework-based mixed matrix membranes for CO2 capture via machine learning. Cell Rep. Phys. Sci. 2022, 3, 100864. [Google Scholar] [CrossRef]
- Lu, C.; Wan, X.; Ma, X.; Guan, X.; Zhu, A. Deep-Learning-Based End-to-End Predictions of CO2 Capture in Metal–Organic Frameworks. J. Chem. Inf. Model. 2022, 62, 3281–3290. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Wu, J.; Yoo, H.; Lee, Y. Machine Learning-based approach for Tailor-Made design of ionic Liquids: Application to CO2 capture. Sep. Purif. Technol. 2021, 275, 119117. [Google Scholar] [CrossRef]
- Makarov, D.M.; Krestyaninov, M.A.; Dyshin, A.A.; Golubev, V.A.; Kolker, A.M. CO2 capture using choline chloride-based eutectic solvents. An experimental and theoretical investigation. J. Mol. Liq. 2024, 413, 125910. [Google Scholar] [CrossRef]
- Choudhary, K.; Yildirim, T.; Siderius, D.W.; Kusne, A.G.; McDannald, A.; Ortiz-Montalvo, D.L. Graph neural network predictions of metal organic framework CO2 adsorption properties. Comput. Mater. Sci. 2022, 210, 111388. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Title | Year | Type of Machine Learning | Descriptors |
|---|---|---|---|
| The use of Artificial Neural Network models for CO2 capture plants | 2011 | ANN | Temperature, Mass Flow, Mass Fraction, Solvent Lean Load, Solvent Circulation Rate, Removal Efficiency |
| Rapid and Accurate Machine Learning Recognition of High Performing Metal Organic Frameworks for CO2 Capture | 2014 | Support Vector Machine (SVM) | Chemical Descriptors via Atomic Property-Weighted Radial Distribution Function (AP-RDF) |
| Rigorous modelling of CO2 equilibrium absorption in ionic liquids | 2017 | Least Square Support Vector Machine, Adaptive Neuro-Fuzzy Inference System, Multi-Layer Perceptron Artificial Neural Network, and Radial Basis Function Artificial Neural Network | Operating Temperature, Operating Pressure, Critical Temperature, Critical Pressure, Acentric Factor |
| Prediction of storage efficiency on CO2 sequestration in deep saline aquifers using artificial neural network | 2017 | ANN | Porosity, Thickness, Permeability, Depth, Time, Residual Gas Saturation |
| Prediction of CO2 loading capacities of aqueous solutions of absorbents using different computational schemes | 2017 | MLP-ANN, Radial Basis Function ANN, LSSVM, and ANFIS | Temperature, Concentration, Molecular Weight, Pressure |
| Role of Pore Chemistry and Topology in the CO2 Capture Capabilities of MOFs: From Molecular Simulation to Machine Learning | 2018 | Multiple Linear Regression, SVM, Decision Trees, Random Forests, Neural Networks, and Gradient Boosting Machines. | Functional Group (FG) Number Density, Void Fraction, Highest Dipole Moment of FG, Most Positive Charge, Most Negative Charge, Largest Pore Diameter, Limiting Pore Diameter, Sum of Epsilons, Gravimetric Surface Area |
| Prediction of CO2 solubility in ionic liquids using machine learning methods | 2020 | ANN and SVM | Temperature, Pressure, Building Groups (similar to SBUs) |
| Prediction of MOF Performance in Vacuum Swing Adsorption Systems for Post-combustion CO2 Capture Based on Integrated Molecular Simulations, Process Optimizations, and Machine Learning Models | 2020 | ANN and Gradient-Boosted Decision Tree Model | Adsorption Metrics (e.g., Henry’s Selectivity, Heat of Adsorption, Working Capacity), Geometric Descriptors |
| Machine learning exploration of the critical factors for CO2 adsorption capacity on porous carbon materials at different pressures | 2020 | Random Forest | Textural Properties, Chemical Composition, Pressure |
| Modeling of CO2 capture ability of [Bmim][BF4] ionic liquid using connectionist smart paradigms | 2021 | ANN, Cascade Feed-Forward Neural Network, SVM, ANFIS | Temperature, Pressure |
| Material | Title | Descriptors | Algorithm | Performance | Target |
|---|---|---|---|---|---|
| MOF and Zeolite | Rapid and Accurate Machine Learning Recognition of High Performing Metal Organic Frameworks for CO2 Capture | AP-RDF | SVM | Up to 99.9% recall rate of the top 1000 MOFs at 0.15 bar and 96.8% at 1 bar | Classification of MOF CO2 adsorption capacity (>1 mmol/g at 0.15 bar and >4 mmol/g at 1 bar) |
| Role of Pore Chemistry and Topology in the CO2 Capture Capabilities of MOFs: From Molecular Simulation to Machine Learning | Topological, geometric, charge-based | DT, RF, MLR, GBM, SVM, NN | [CO2/N2 Selectivity] R2 = 0.905, SRCC = 0.921 [CO2 Loading] R2 = 0.905, SRCC = 0.950, [CO2/H2 selectivity] R2 = 0.855, SRCC = 0.938 | CO2 loading, CO2/N2 selectivity, CO2/H2 selectivity | |
| Prediction of MOF Performance in Vacuum Swing Adsorption Systems for Postcombustion CO2 Capture Based on Integrated Molecular Simulations, Process Optimizations, and Machine Learning Models | Geometric, adsorption Metrics, figures of merit (Yang’s FOM, Wiersum’s FOM, etc.) | Random Forest | [Productivity] Correlation R2 = 0.41 [PE] Correlation R2 = 0.18 | Productivity of a material (i.e., how much CO2 the sorbent can extract per unit volume of the material per unit time), parasitic energy | |
| Robust Machine Learning Models for Predicting High CO2 Working Capacity and CO2/H2 Selectivity of Gas Adsorption in Metal Organic Frameworks for Precombustion Carbon Capture | Geometric, AP-RDF | Gradient boosted trees | [CO2 working capacities] R2 = 0.944 [CO2/H2 selectivities] R2 = 0.872 | CO2 working capacities, CO2/H2 selectivity | |
| A data-science approach to predict the heat capacity of nanoporous materials | Geometric, atomic, chemical | XGB | MAE = 0.02 RMAE = 2.89% SRCC = 0.98 | Heat capacity (J g−1 K−1) | |
| Large-Scale Screening and Machine Learning to Predict the Computation-Ready, Experimental Metal-Organic Frameworks for CO2 Capture from Air | Five structural parameters: volumetric surface area (VSA), largest cavity diameter (LCD), pore-limiting diameter (PLD), porosity φ, density ρ, and an energy parameter: heat of adsorption | BPNN, RF, DT, SVM | Train R = 0.994, Test R = 0.981 (RF Model) | Adsorption selectivity (CO2/N2+O2) | |
| Design and prediction of metal organic framework-based mixed matrix membranes for CO2 capture via machine learning | Operating conditions, polymer type, geometric, gas adsorption metrics (selectivity, permeability, etc.) | RF | [Permeability] R2 = 0.77, RMSE = 1.45 [Selectivity] R2 = 0.7, RMSE = 0.31 | CO2 permeability, CO2/CH4 selectivity | |
| High-Performing Deep Learning Regression Models for Predicting Low-Pressure CO2 Adsorption Properties of Metal Organic Frameworks | AP-RDF, chemical motif, and geometric descriptors | ANN (MLP) | [CO2 working capacity] Pearson r2 = 0.958, SRCC = 0.965, RMSE = 0.13 [CO2/N2 selectivity] r2 = 0.948, SRCC = 0.975, RMSE = 10 | CO2/N2 selectivity | |
| Ionic Liquids | Modeling of CO2 capture ability of [Bmim][BF4] ionic liquid using connectionist smart paradigms | Temperature, pressure | ANN, LS-SVM, ANFIS | AARD (%), MSE, RMSE, R2 = 7.01, 0.00115, 0.03396, 0.98408 | Solubility of CO2 in the 1-n-butyl-3- methylimidazolium tetrafluoroborate ([Bmim][BF4]) |
| Predicting CO2 capture of ionic liquids using machine learning | Semi-empirical (PM6) electronic, thermodynamic, and geometrical descriptors | SVM, RF, XGB, MLP, graph-based networks | [Dataset-1] R2, RMSE (MAE) = 0.96 0.05 (0.03) [Dataset-2] R2, RMSE (MAE) = 0.85 0.10 (0.06) | CO2 solubility in 1-Butyl-3-methylimidazolium hexafluorophosphate ([Bmim][PF6]) | |
| Prediction of thermo-physical properties of 1-Butyl-3-methylimidazolium hexafluorophosphate for CO2 capture using machine learning models | Temperature, CO2 partial pressure and water wt% | Gaussian process regression | R2 = 0.992; AARD% = 0.137976 | CO2 solubility in [Bmim][PF6] | |
| Machine learning aided high-throughput prediction of ionic liquid@MOF composites for membrane-based CO2 capture | Structural and chemical | RF | R2 = 0.728, RMSE = 0.365, MAE = 0.277 | CO2/N2 Selectivity | |
| Predicting CO2 Absorption in Ionic Liquids with Molecular Descriptors and Explainable Graph Neural Networks | Morgan fingerprints, temperature, pressure | PLSR, CTREE, RF | MAE of 0.0137, R2 of 0.9884 | CO2 absorption/solubility in ILs | |
| Others (Graphene, Graphite, and Activated Carbon) | Monitoring the effect of surface functionalization on the CO2 capture by graphene oxide/methyl diethanolamine nanofluids | Temperature, pressure, functionalized group, graphene oxide dosage | CFF-NN | AARD = 1.78%, MSE = 0.007, RMSE = 0.08, and R2 = 0.9906 | CO2 solubility in graphene oxide/methyl diethanolamine |
| Intelligent prediction models based on machine learning for CO2 capture performance by graphene oxide-based adsorbents | Geometric (surface area, pore volume), temperature, pressure | SVM, GBR, RF, extra trees, XGB, ANN | R2 > 0.99 | CO2 uptake capacity | |
| Modeling and Optimizing N/O-Enriched Bio-Derived Adsorbents for CO2 Capture: Machine Learning and DFT Calculation Approaches | Physicochemical and structural features of biomass-based activated carbon | RBF-NN | R2 = 0.99, 0.974, 0.995, 0.9658, 0.9476, 0.9891 for test set predictions at (298 K and 273 K) 0.15 bar, (298 K and 273 K) 0.6 bar, and (298 K and 273 K) 1 bar, respectively | CO2 adsorption |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Orhan, I.B.; Zhao, Y.; Babarao, R.; Thornton, A.W.; Le, T.C. Machine Learning Descriptors for CO2 Capture Materials. Molecules 2025, 30, 650. https://doi.org/10.3390/molecules30030650
Orhan IB, Zhao Y, Babarao R, Thornton AW, Le TC. Machine Learning Descriptors for CO2 Capture Materials. Molecules. 2025; 30(3):650. https://doi.org/10.3390/molecules30030650
Chicago/Turabian StyleOrhan, Ibrahim B., Yuankai Zhao, Ravichandar Babarao, Aaron W. Thornton, and Tu C. Le. 2025. "Machine Learning Descriptors for CO2 Capture Materials" Molecules 30, no. 3: 650. https://doi.org/10.3390/molecules30030650
APA StyleOrhan, I. B., Zhao, Y., Babarao, R., Thornton, A. W., & Le, T. C. (2025). Machine Learning Descriptors for CO2 Capture Materials. Molecules, 30(3), 650. https://doi.org/10.3390/molecules30030650