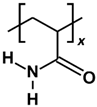
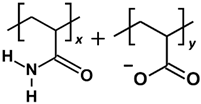
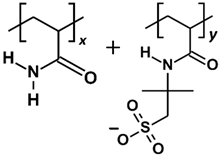
2.1. Effect of Polymer Molecular Weight
The polymer adsorption, measured through both simulations and experiments at different molecular weights (MW), is depicted in
Table 1, along with the simulated
and
. In simulations, polymer MW was represented in terms of repeat units (213 Da per repeat unit) as a surrogate for experimental candidates with varying MW. Despite experimental polymers having significantly higher MW, potentially reaching millions of Daltons, there was good agreement between simulations and experiments, with increasing polymer adsorption observed as molecular weight increased. This trend was expected because longer molecular chains experience stronger interactions with the calcite surface, increasing the contact area between the polymer and the surface and enhancing adsorption strength. In addition, since HPAM is a hydrophilic polymer, the shortest HPAM chains prefer to remain in the water and require a longer time to stabilise their adsorption onto the calcite surface.
While polymer MW positively impacts adsorption, both the experimental and simulation literature indicate that adsorption will reach an equilibrium value. Once the polymer adsorption reaches saturation, further increases in polymer MW or concentration do not enhance adsorption [
7,
8,
39] and may even reduce stability [
13]. This is attributed to changes in the adsorption mechanism with increasing polymer MW. Experimentally, it has been observed that polymer adsorption via polymer bridging is the strongest mechanism, followed by electrostatic bare patching [
40]. At low MW, polymers primarily adopt a flat, extended conformation on the calcite surface, with electrostatic bare patching being the dominant adsorption mechanism. As MW or concentration increases, the polymer chains become compressed with a reduced effective contact area for each chain. This leads to the formation of more loops, with polymer bridging becoming the predominant adsorption mechanism [
7,
8,
39]. Further increases in MW do not enhance adsorption due to the limited contact area and may inhibit existing polymer bridging between particles, potentially affecting adsorption stability.
In our simulation, given that only a single polymer chain was modelled and the calcite surface area was not saturated with polymer adsorption, the equilibrium adsorbed amount was not observed but was expected to be reached with higher polymer repeat units. Nevertheless, the polymer conformation with different MWs can be characterised by
, as shown in
Table 1. The increasing
with higher MW was expected due to the larger polymer size. In contrast, the decrease in
with increasing MW indicates that the polymer transitions from an extended conformation to a coiled conformation, showing excellent agreement with theoretical expectations.
Since both the polymer adsorbed amount and
increase with increasing MW, it is interesting to determine if a proportional relationship between them can be established in this study. We propose a simple relation to relate polymer
with the polymer repeat unit. Polymer
can be considered as the radius of the polymer globule, where the globular volume,
, can be approximated as the volume of a sphere:
The total polymer mass,
, is directly proportional to the number of repeat units,
, and the mass of a single repeat unit,
:
Assuming the polymer density,
, is constant, the polymer mass can be expressed in terms of
and
, approximated as shown in Equation (3). The number of repeat units thus has a proportional relationship with the
as indicated in Equation (4).
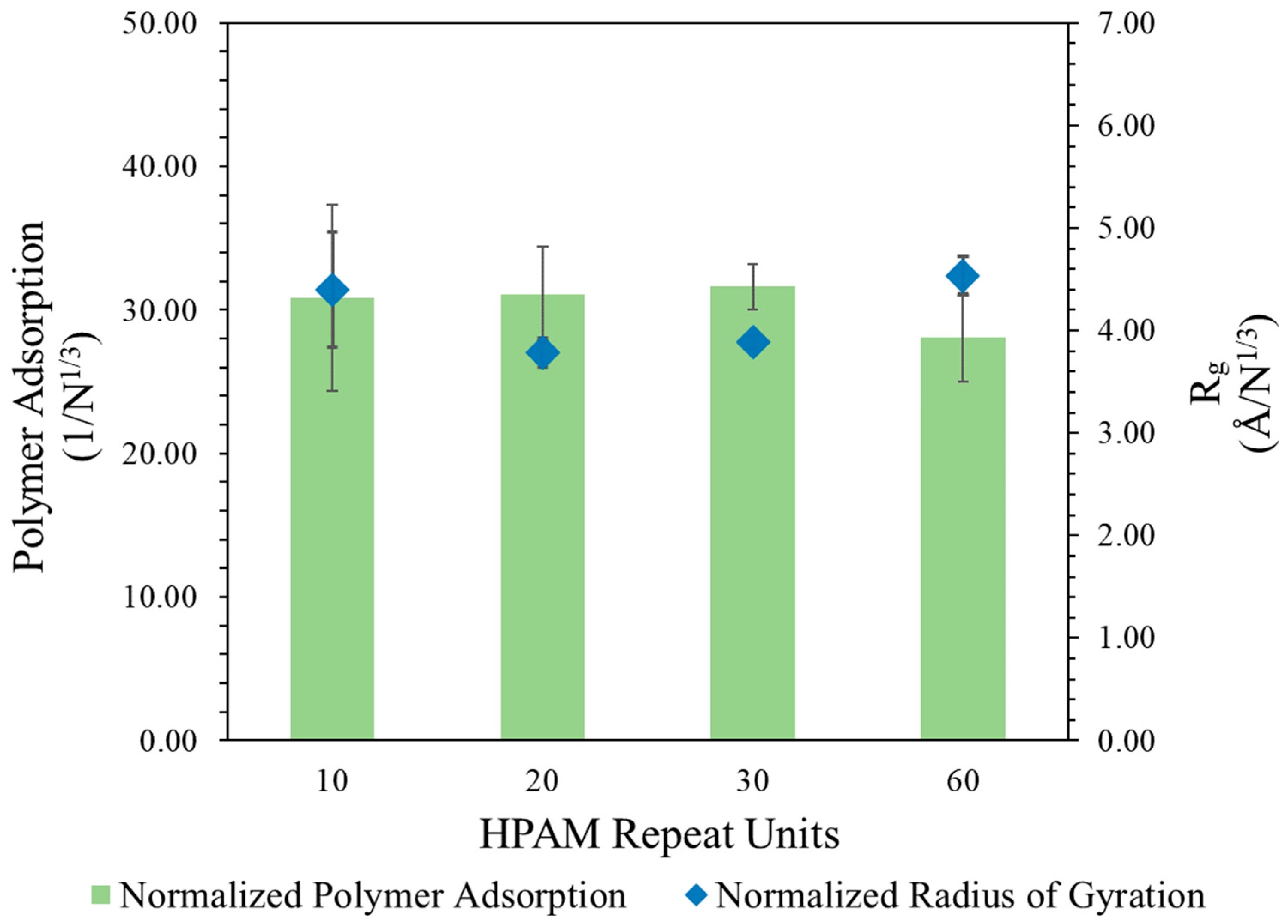
This proportionality was investigated by dividing the polymer
and number of adsorbed atoms using the normalization factor
, with the results presented in
Figure 1. To further validate this proportionality, another case of HPAM with 60 repeat units was also modelled in a larger water–calcite system to avoid system size effects. After normalization, it could be observed that the polymer
and adsorbed amount reached a constant range with varying polymer repeat units, suggesting that their values increased proportionally with the cubic root of polymer repeat units during adsorption.
This implies that the
of the polymer scales with MW at an exponent of 0.33 during adsorption to calcite surfaces, which diverges from the expected scaling relationship of greater than 0.5 typically seen in free solution under good solvent conditions [
41]. This discrepancy can be attributed to the conformational changes that occur when HPAM interacts with the surface. Upon adsorption, the HPAM exhibits reduced flexibility, transitioning to a more coiled or globular conformation that is influenced by the increasing MW (as observed in
< 0.5, indicating nearly spherical shape). The observed lower exponent is consistent with findings in the literature, which suggest that globular polymers demonstrate a scaling behaviour characterized by lower exponents [
41]. This indicates that the adsorption environment significantly alters the polymer conformation, resulting in a unique scaling relationship that highlights the complexities of polymer behaviour at interfaces.
2.3. Effect of Temperature
The results of polymer adsorption under the influence of temperature are interesting. As shown in
Table 3, experimental data indicate that polymer adsorption deteriorates with increasing temperature, whereas simulations show an opposite trend. Theoretically, polymer adsorption is generally exothermic, and increasing temperature shifts the equilibrium state towards desorption [
25]. This effect is further exacerbated by polymer degradation at high temperatures, causing the polymer to lose its effectiveness in bridging particles. The experimental results align with this theory, indicating that the adsorption effectiveness of HPAM could be inhibited by increasing temperature.
Meanwhile, the discrepancy in simulations with increasing polymer adsorption under higher temperature, though interesting, is not uncommon and has been reported in other studies. For example, a literature study on molecular adsorption on liposome surfaces observed rapid molecular transport kinetics at high temperatures with more negative adsorption free energy [
43]. At higher temperatures, liposomes experience increased kinetic energy, which can influence adsorption behaviour depending on the enthalpic and entropic contributions to Gibbs free energy. Although adsorption typically results in decreased entropy due to reduced disorder, the author noticed a positive change in entropy. This is attributed to the replacement of water and counterions from the surface by adsorbate molecules, leading to an overall increase in entropy upon adsorption. To investigate the transport properties of HPAM, polymer mean squared displacement, MSD, and self-diffusion coefficients,
, are computed below [
44]:
where
is the coordinate at the initial time and
is the coordinate at different times.
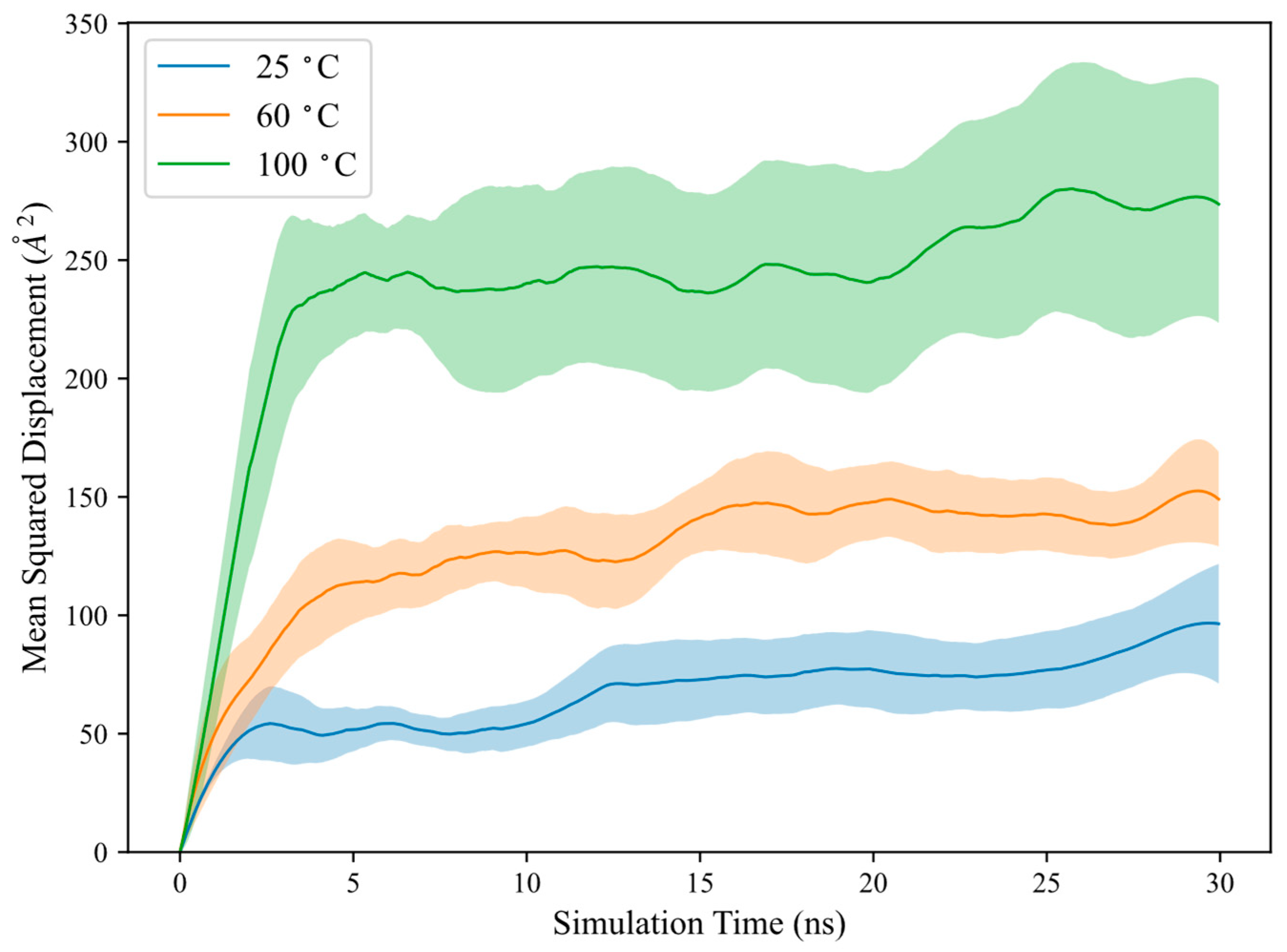
can be measured from the slope fitted over the linear region of the MSD versus the time plot, as depicted in
Figure 2 for polymer MSD at different temperatures. It is important to note that
is typically valid only for free polymers in solution. In this study,
is computed to characterise the polymer transport behaviour during adsorption and is measured over the linear slope at the initial timesteps. The computed
, along with polymer
and
, are presented in
Table 4.
The results indicate that polymer MSD and are significantly higher at elevated temperatures, suggesting that the polymer could diffuse more rapidly towards the calcite surface with increasing temperatures. The polymer could more vigorously explore its surrounding configurational space, leading to an increased contact area with the calcite surface and enhanced adsorption. This is further supported by the increase in and with temperature, signifying that the polymer could adopt a more extended conformation to maximize adsorption contact with the calcite surface. Additionally, solvent quality may change with temperature, which could also influence the polymer–calcite interaction, although this is not considered in the current model.
In the literature, the influence of temperature on polymer adsorption has shown different trends in both experiments and simulations. Some studies report that polymers experienced enhanced adsorption with increasing temperatures [
27,
28,
43,
45], while others indicate increased desorption with higher temperatures [
13,
17,
25]. The polymer–surface interactions can further complicate this inconsistency and require careful examination. In addition, a limitation of the simulation approach is its inability to account for polymer degradation at high temperatures. Our simulations do not consider structural changes in the polymer, which could be modelled only with a reactive forcefield. Consequently, the observed discrepancy between the simulation and experimental results may be attributed to the simulation focusing solely on thermodynamic factors without incorporating polymer degradation effects.
2.5. Effect of Salinity
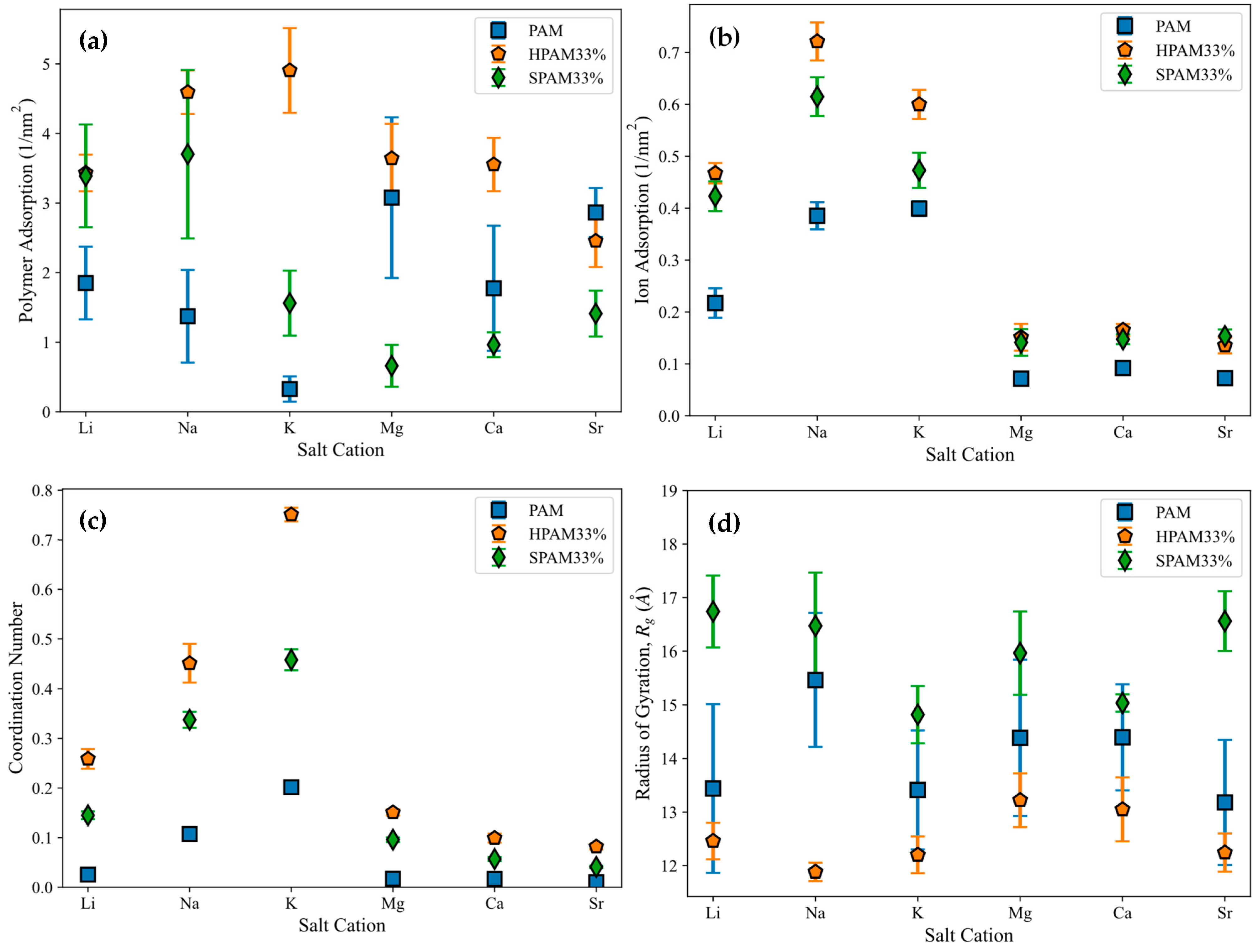
In this section, the adsorption of various PAM-based polymers, including HPAM 33%, SPAM 33%, and neutral PAM, are investigated under varying cation valencies and ion sizes to observe the effect of their functional groups in saline environments, as presented in
Figure 3a. The cation adsorption density on the surface is analysed similarly to polymer adsorption, with results displayed in
Figure 3b. Additionally, radial distribution analysis is conducted to examine the adsorption of cations onto the polymer, as shown in
Figure 3c. This is computed via the coordination number of the cations with respect to the carbonyl oxygen atom of the polymer amide group (C(=O)NH
2). Lastly, the polymer conformational size is analysed using
in
Figure 3d.
The polymer adsorption results show that HPAM generally performs better than SPAM 33% and neutral PAM, with relatively high adsorption amounts measured across all cation cases. As explained in the previous adsorption free energy analysis, this can be attributed to the presence of carboxyl functional groups in HPAM, which experience stronger electrostatic attraction with the calcite surface compared to the bulkier functional groups in SPAM and the neutral ionicity of PAM. When comparing monovalent cations (Li+, Na+, K+) and divalent cations (Mg2+, Ca2+, Sr2+), interesting trends emerge among different polymers. HPAM adsorption is higher in monovalent cation cases than in divalent cation cases, with increasing monovalent cation sizes leading to increased adsorption, while increasing divalent cation sizes result in decreased adsorption. SPAM also shows reduced adsorption in divalent cation cases compared to monovalent cations. However, unlike HPAM, increasing monovalent cation sizes leads to decreased adsorption, while increasing divalent cation sizes results in increased adsorption. On the other hand, PAM adsorption is less influenced by cations. PAM shows better adsorption in divalent cation cases than in monovalent cation cases, with increasing monovalent cation sizes resulting in decreased adsorption and no significant trend observed for increasing divalent cation sizes.
The differing adsorption behaviours among various polymers can be attributed to their distinct adsorption mechanisms influenced by cations. In the literature, the roles of cations in enhancing or inhibiting polymer adsorption have yielded contradictory results, with different mechanisms proposed to explain these behaviours. For polymer adsorption enhancement, some authors suggest that salt ions can form an Electric Double Layer (EDL) on the surface, facilitating salt bridging that attracts the polymer to the surface regardless of the polymer’s ionicities [
17,
23]. Conversely, polymer inhibition is attributed to the charge-screening effect of ions, which suppresses the polymer charge or causes surface charge reversal, thereby reducing the polymer’s adsorption strength to the surface [
11,
35]. In a saline environment, the adsorption of PAM-based polymers onto the calcite surface, focusing on HPAM, can be illustrated with the schematic diagram in
Figure 4, with an electrostatic surface potential diagram provided as
Figure S2 in the
Supplementary Information.
Effective polymer adsorption to the calcite surface results from the competing effects of salt bridging and charge screening. In experiments, the calcite surface is positively charged under normal reservoir pH conditions [
47], where cations may weaken the charge of an anionic polymer and screen its attraction to the surface. However, the calcite surface in the simulation is neutral, consisting of alternating CO
32− and Ca
2+ ions. HPAM can form hydrogen bonds with calcite CO
32− via hydrogen atoms from the amide groups or significantly stronger charge interactions between calcite Ca
2+ and deprotonated oxygen atoms from the carboxyl groups. While cations may neutralise the charge attraction between Ca
2+ and deprotonated carboxyl groups, they can also function as salt bridges between CO
32− and carboxyl groups, enhancing adsorption.
Simulation results suggest that salt bridging is more dominant in HPAM adsorption. HPAM’s higher charge density makes it more susceptible to cation influence than SPAM and PAM [
31]. This claim agrees with the density profile analysis of the relevant oxygen atoms of the polymers and the cation (using Na
+ as an example) during adsorption, as shown in
Figure S3 in the
Supplementary Information. This enhances the role of salt bridging, contributing to its higher adsorption across various cation cases. The differing adsorption trends between monovalent and divalent cations can be attributed to the hydration energy of the cations. Cations with stronger charge density form a more robust hydrogen-bonding network with water, exhibiting stronger hydration energy and preferring to remain in the bulk water phase. Such cations are known as water structure makers, while those with weaker hydration energy are termed water structure breakers [
23].
Divalent cations exhibit significantly higher charge density than monovalent cations, while increasing ion sizes within the same valency can reduce charge density. Consequently, the hydration energy follows the following trend: Mg
2+ > Ca
2+ > Sr
2+ > Li
+ > Na
+ > K
+ [
48]. This trend is reflected in the cation adsorption density shown in
Figure 3b, where divalent cations, with stronger hydration energy, exhibit lower surface adsorption and prefer to remain in the aqueous phase compared to monovalent cations. The trend observed for monovalent cations also aligns with the hydration energy trend. In HPAM and SPAM cases, the lower adsorption of K
+ compared to Na
+ may be attributed to its larger ionic radius, which positions the adsorption peak further from the surface and may extend beyond the defined 4 Å adsorption layer [
49].
The variation in cation adsorption density on the surface across different polymers indicates the influence of the polymer on cation adsorption. On the other hand, cation adsorption to the surface also affects the density of cations adsorbed onto the polymer. Higher cation adsorption to the surface creates a more concentrated salt layer, which attracts the polymer more effectively and increases the cation adsorption onto the polymer. As shown in
Figure 3c, the cation adsorption onto the polymer exhibits a trend similar to that in
Figure 3b, highlighting the interplay between cations and polymers. Consequently, these two factors contribute to the adsorption trends observed for different polymers under varying cation conditions depicted in
Figure 3a.
Since the adsorption of HPAM to the surface is primarily driven by salt bridging, the observed increase in adsorption with larger monovalent cations is due to stronger cation adsorption onto the polymer (K
+ > Na
+ > Li
+). Conversely, the decrease in adsorption with larger divalent cations is attributed to the reduced cation adsorption density on the polymer (Mg
2+ > Ca
2+ > Sr
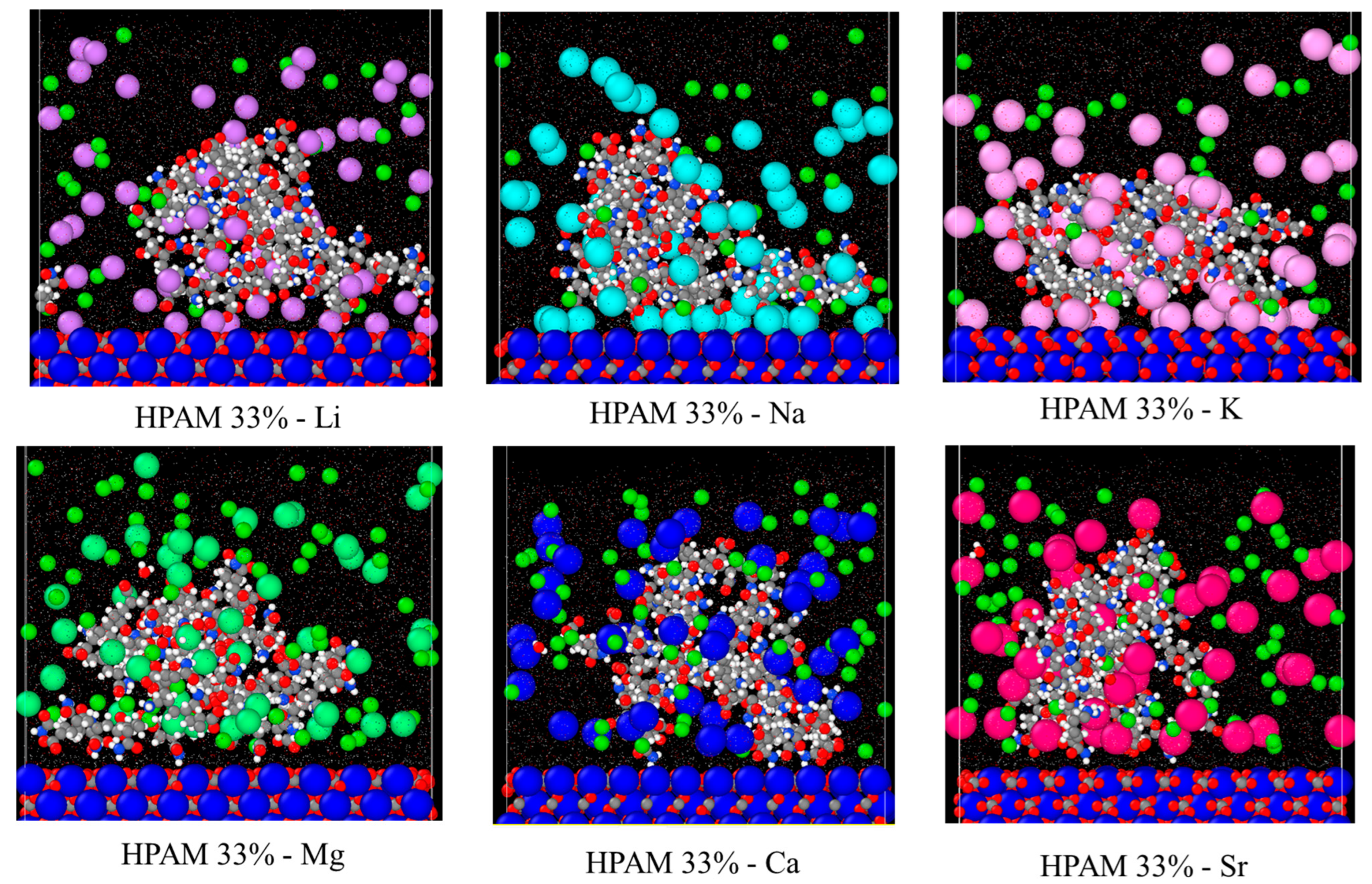
2+). The simulation snapshots in
Figure 5 further illustrate this. Closer inspection reveals that a stable EDL is formed for monovalent cations, with cations creating the Stern layer (the first EDL layer) and Cl
− anions constituting the diffuse layer (the second EDL layer), stabilising the adsorption. In contrast, no stable EDL exists for divalent cations, resulting in lower adsorption. This observation aligns with previously reported cation interfacial interaction behaviours in calcite–water systems [
49].
On the other hand, the differing trends observed for SPAM may be attributed to its lower charge strength, where cations have a reduced influence on polymer adsorption [
31] or might even inhibit it through charge-screening effects. Compared to monovalent ions, SPAM exhibits lower adsorption to a calcite surface in divalent ions due to the stronger charge-screening effects. The decreasing adsorption trend with increasing monovalent cation size is attributed to stronger cation adsorption and greater charge screening. Conversely, the increasing adsorption trend with larger divalent cations results from a relatively lower charge-screening effect combined with reduced cation adsorption density.
Meanwhile, PAM, which has a neutral charge, primarily adsorbs onto the calcite surface through hydrogen bonding from amide groups, with adsorption governed mainly by surface oxygen densities [
29]. Cations can inhibit this adsorption by screening the interaction between hydrogen atoms and calcite CO
32−. Consequently, the lower cation adsorption density on both the polymer and surface for divalent cations leads to higher PAM adsorption than monovalent cations. The decreasing trend in adsorption with increasing monovalent cation size is due to increasing cation adsorption density, resulting in more pronounced charge-screening effects. While cations inhibit adsorption in the case of PAM, they function as salt bridges between the anionic polymer and calcite CO
32−. For neutral PAM, anions may thus play a more dominant role in bridging interactions between the hydrogen atoms of the amide group and Ca
2+. Although not presented here, a published study investigated the role of anions, showing that nitrate ions (NO
3−) with a smaller hydrated radius and hydration energy than Cl
− can enhance the adsorption of neutral PAM on the calcite surface [
50].
As illustrated in
Figure 3d, polymer
shows no significant trend across different cation cases. Among the polymers, SPAM has the largest size, followed by PAM and HPAM. Since all polymers have the same degree of polymerization, SPAM naturally exhibits the largest size due to its bulkier functional groups. Conversely, HPAM, with the smallest
, suggests a more compact structure. This is expected, as the cations strongly adsorb onto HPAM, neutralising its charge and causing the polymer to adopt a more compact conformation.
We conclude this section by comparing the experimental and simulation results for HPAM adsorption in salt-free conditions (control), NaCl, and CaCl
2 environments, as presented in
Table 6. Both experimental and simulation results show a similar trend for salt-free and Na
+ cases, indicating that salt cations can enhance polymer adsorption through salt bridging. However, for divalent Ca
2+, the simulation shows lower adsorption compared to Na
+, while experiments reveal significantly higher adsorption. This discrepancy may be attributed to different interpretations of polymer adsorption. In experiments, Ca
2+ provided more charges to neutralise the polymer, causing it to adopt a more coiled conformation and enabling more polymer chains to adsorb onto calcite particles, thereby increasing overall adsorption. In contrast, since only a single polymer chain is modelled in the simulation, the effects of multiple polymer chains adsorbing to the surface cannot be captured, leading to different observations.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}