Abstract
In organic synthesis, the solvent is the chemical compound that represents the largest proportion of the process. However, conventional solvents are often toxic and dangerous for the environment, and an interesting alternative is to replace them by water. In this context, catalyst surfactants allow both organic reagents in water to be solubilized and organic reactions to be catalyzed. This article describes the synthesis of new biobased organocatalytic surfactants soluble in water, composed of a hydrocarbon chain grafted onto an imidazolidinone moiety. The imidazolidinone moiety acts as catalyst, but also as the polar head of the surfactant, while the fatty chain constitutes the hydrophobic tail. The five steps of the synthesis were optimized, respecting the principles of green chemistry, and two organocatalytic surfactants were obtained with a good selectivity. Surface properties in an aqueous medium were then evaluated with the use of tensiometric analysis. Their molecular organization in vesicles was characterized by Dynamic Light Scattering. The presence of vesicles allows reactions to be carried out in an organized aqueous medium. Model catalytic reactions performed in aqueous medium validated the feasibility of replacing conventional hazardous organic solvents. The newly synthesized biobased surfactants showed satisfactory catalytic activity and allowed the expected products to be obtained with good enantioselectivity.
1. Introduction
One of the chemical industry’s most frequently cited problems is the production of large quantities of waste, due, in particular, to the use of organic solvents, most of which are not recycled. In the pharmaceutical industry, for example, solvents account for more than 80% by mass of the compounds involved [1]. In addition, most of the organic solvents commonly used in laboratories and industry are toxic to humans and/or the environment. A major challenge is, therefore, to find alternatives solvents, in line with the principles of green chemistry [2].
However, the insolubility of many organic compounds limits the application of water as a solvent. In particular, low yields or selectivities have been obtained when water is used as a reaction medium in catalyzed reactions. To overcome these drawbacks, strategies based on the use of surfactants can be envisaged. The organization of surfactants in water allows the solubilization of organic compounds in the surrounding aqueous phase [3], eliminating the need for organic solvents, as the reactions take place in the hydrophobic core of the formed aggregates (micelles, vesicles, etc.).
Moreover, organocatalysis is an extremely powerful tool that has been developing steadily since the early 2000s, due to the lower toxicity of organocatalysts compared to metal catalysts, their lower cost, and better compatibility with water [4].
In recent years, a great deal of research has been carried out into the design of molecules that can play both the role of a surfactant and an organocatalyst [5,6,7,8,9]. The advantage of these molecules is that they reduce the amount of chemical compounds involved (no excess reagent), while allowing organocatalyzed reactions to take place in water.
The vast majority of organocatalytic surfactants are proline or threonine derivatives. They have the advantage that they can be used directly in water. While these surface-active organocatalysts allowed enantio-enriched molecules to be obtained, demonstrating the effectiveness of the concept, the results obtained in terms of yield and enantiomeric excess seem to be substrate-dependent. However, the development of new efficient asymmetric synthesis methods is crucial, since a very large number of molecules, whether biologically active compounds for the pharmaceutical and agrochemical industries, food additives, or flavoring agents, require pure enantiomers [10]. This opens the way for the development of new organocatalytic surfactants (other than proline or threonine derivatives) that would allow good yields and enantiomeric excesses for a wide range of reactions.
Imidazolidinones represent a specific type of organocatalyst developed by D. MacMillan. In recognition of their pioneering work in this field, he and B. List were awarded the Nobel Prize in Chemistry in 2021. The imidazolidinone moiety behaves like a Lewis acid via the formation of an iminium [11] and can be obtained from a phenylalanine derivative (amino acid). The geometry of imidazolidinones also allows asymmetric catalysis to be performed on a wide range of chemical reactions.
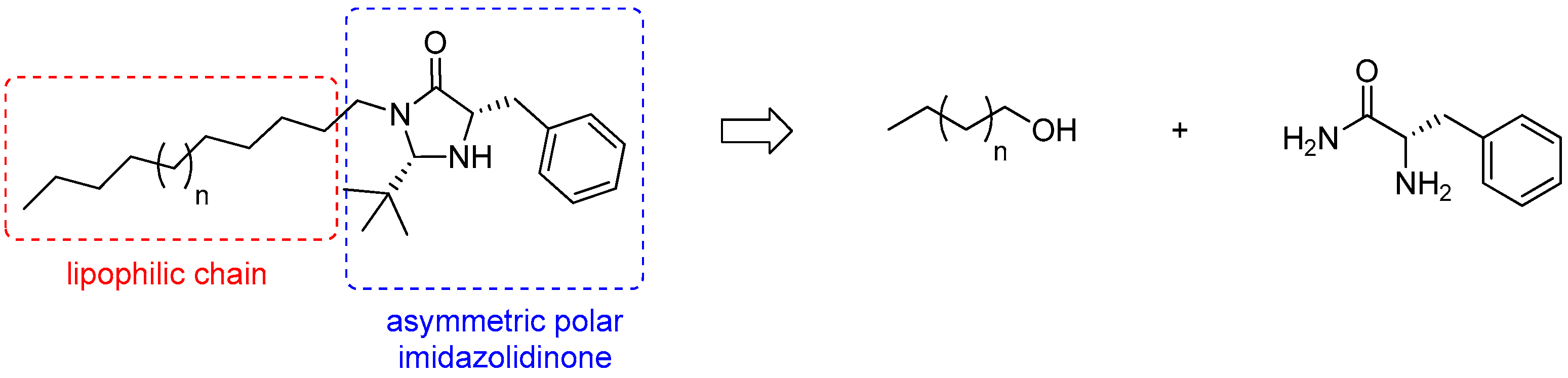
In this context, the aim of this work is to synthesize chiral organocatalysts with surfactant properties for asymmetric reactions in water, thus, meeting green chemistry criteria. We, thus, propose herein the synthesis of a new asymmetric organocatalytic surfactant, consisting of the imidazolidinone organocatalyst moiety and a fatty chain (Figure 1). This lipophilic moiety is readily available in the lipids of brown algae and was chosen as a follow-up to previous work on the valorization of these algae [12].

Figure 1.
Retrosynthetic pathway of the targeted surfactant.
Syntheses work carried out according to the principles of green chemistry led to the production of two molecules whose surface-active and catalytic properties for enantioselective reactions were evaluated.
2. Results and Discussion
2.1. Synthesis
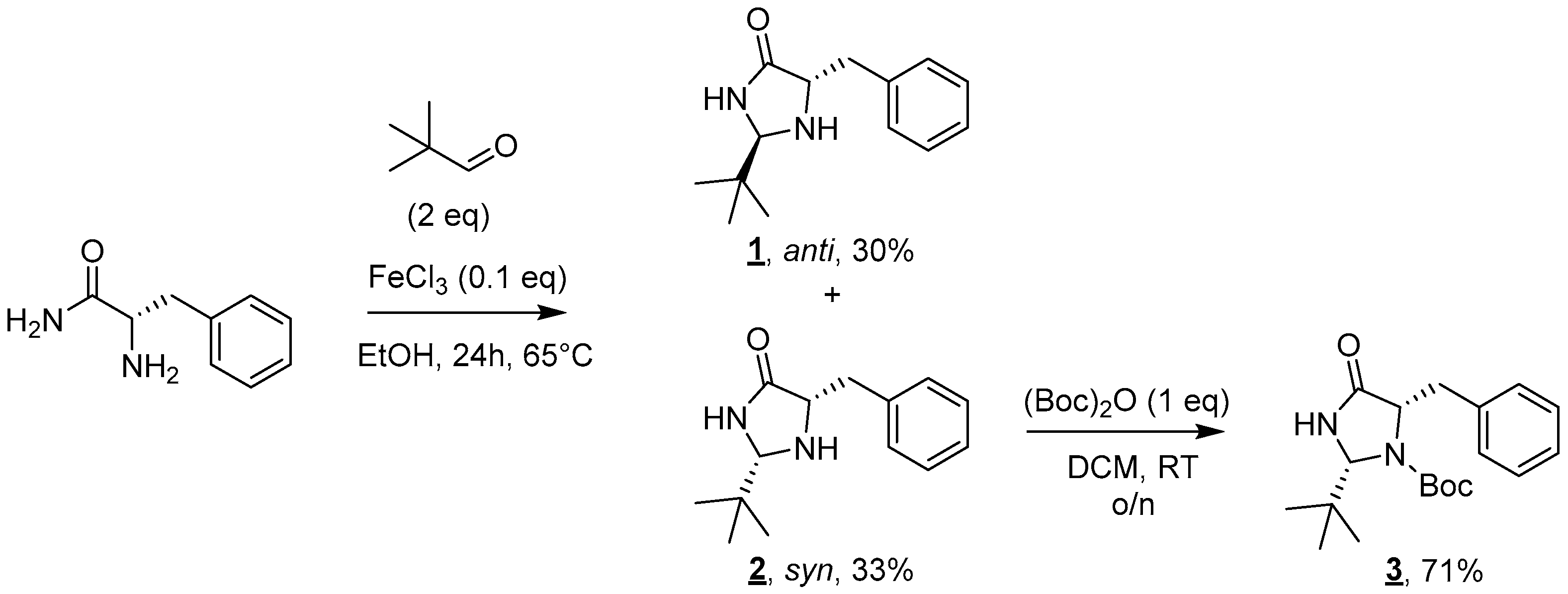
The proposed synthesis relies on the grafting of a lipophilic chain on a polar catalytic moiety, here a 4-imidazolidinone [13,14]. The first step of this convergent synthesis involves the formation of the imidazolidinone moiety (Figure 2). The reaction of L-phenylalaninamide with two equivalents of pivaldehyde in the presence of a catalytic amount of iron(III) chloride led to the formation of a 1:1 syn/anti mixture of diastereoisomers 1 and 2 after 24 h at 65 °C [15]. Diastereoisomers 1 and 2 were efficiently separated on a silica gel column and, respectively, obtained in 30% and 33% yields.
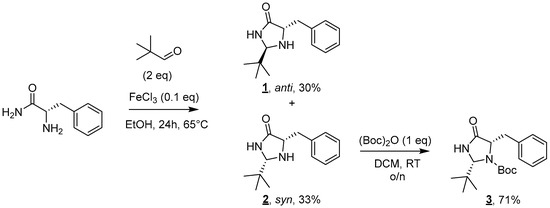
Figure 2.
Synthesis of imidazolidinones 1, 2, and 3.
As only the syn diastereoisomer was described in the literature to present an interesting catalytic activity [11], molecule 1 was discarded for the rest of the synthesis. The amine moiety of imidazolidinone 2 was then Boc-protected to deactivate this group prior to any further functionalization. This protection was performed using one equivalent of Boc2O in DCM at room temperature overnight. Molecule 3 was obtained after purification in 71% yield [16].
In order to graft a lipophilic chain on the obtained imidazolidione 3, two fatty alcohols, decan-1-ol and hexadecan-1-ol, first needed to be converted into reactive mesylated alcohols (Table 1). The corresponding reactions were run in the presence of a 1.5 equivalent of MsCl and a 2 equivalent of a base in DMC for 7 h [17]. After an acidic treatment, mesylated alcohols 4 and 5 were, respectively, obtained in 95% and 70% yields without further purification.

Table 1.
Synthesis of mesylated alcohol 4 and 5.
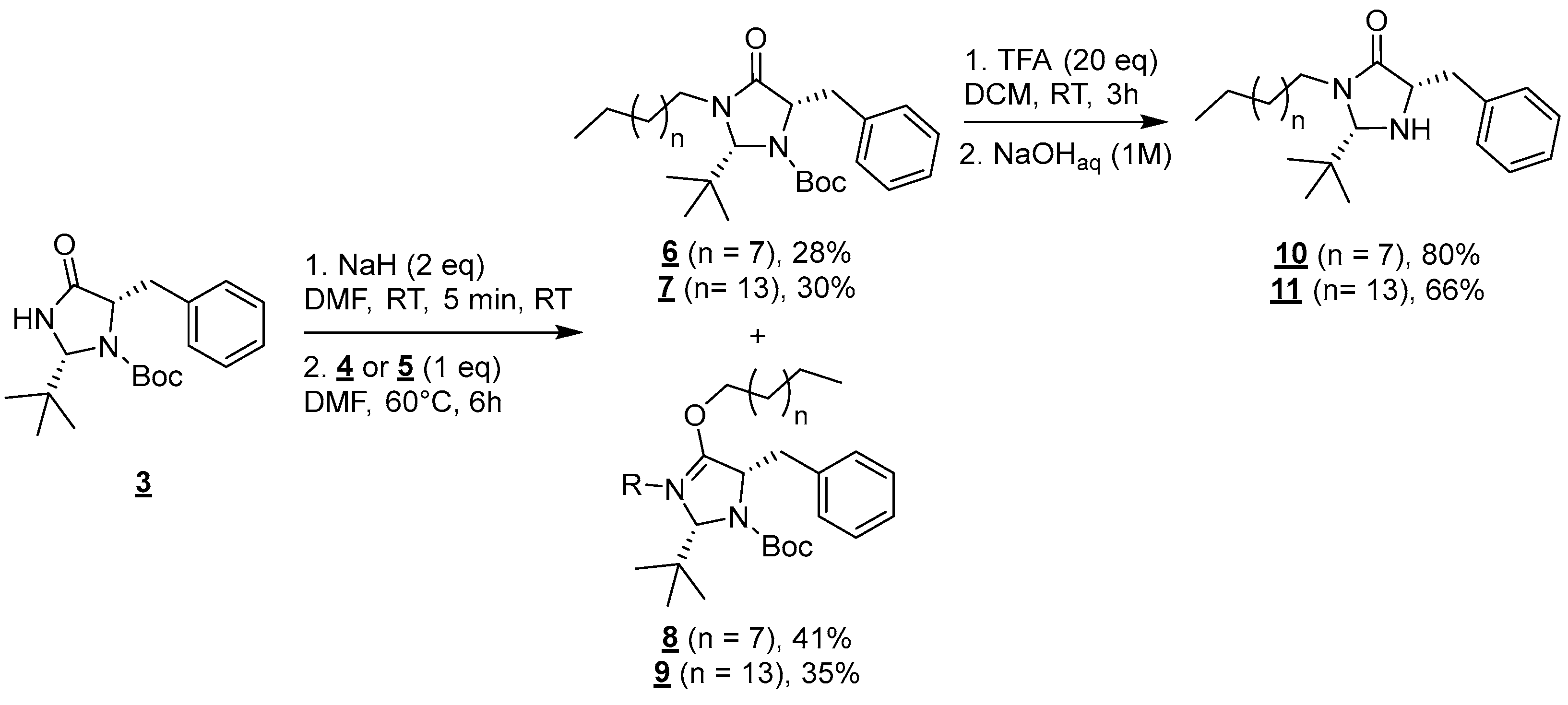
Molecules 4 and 5 were then engaged in the grafting step: the nucleophilic substitution between amide 3 and mesylated alcohols 4 or 5 was carried out in the presence of NaH (60% dispersed in mineral oil) in anhydrous DMF at 60 °C for 6 h (Figure 3) [18].
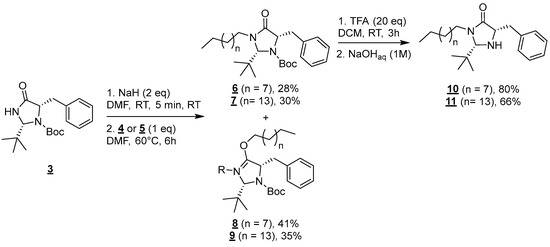
Figure 3.
Formation of compounds 6 to 11.
In both cases, this reaction led to the formation of a mixture of regioisomers resulting from competitive N (6 and 7) and O-alkylations (8 and 9) [19]. Those regioisomers were separable on silica gel columns and compounds 6 and 8 (n = 7) and 7, and 9 (n = 13) were, respectively, obtained in 28, 41, 30, and 35% yields. In this paper, we chose to focus on the synthesis of surfactants derived from the N-alkylation products 6 and 7. The functionalization of the O-alkylation derivatives 8 and 9 will be published in due time.
Finaly, the deprotection of amine groups of compounds 6 and 7 was carried out using trifluoroacetic acid (TFA) in DCM for 1 h at room temperature [20]. Targeted molecules 10 and 11 were, respectively, obtained after a base workup in 80 and 66% yields.
We then decided to evaluate the surfactant properties of the synthesized molecules.
2.2. Solubility in Water
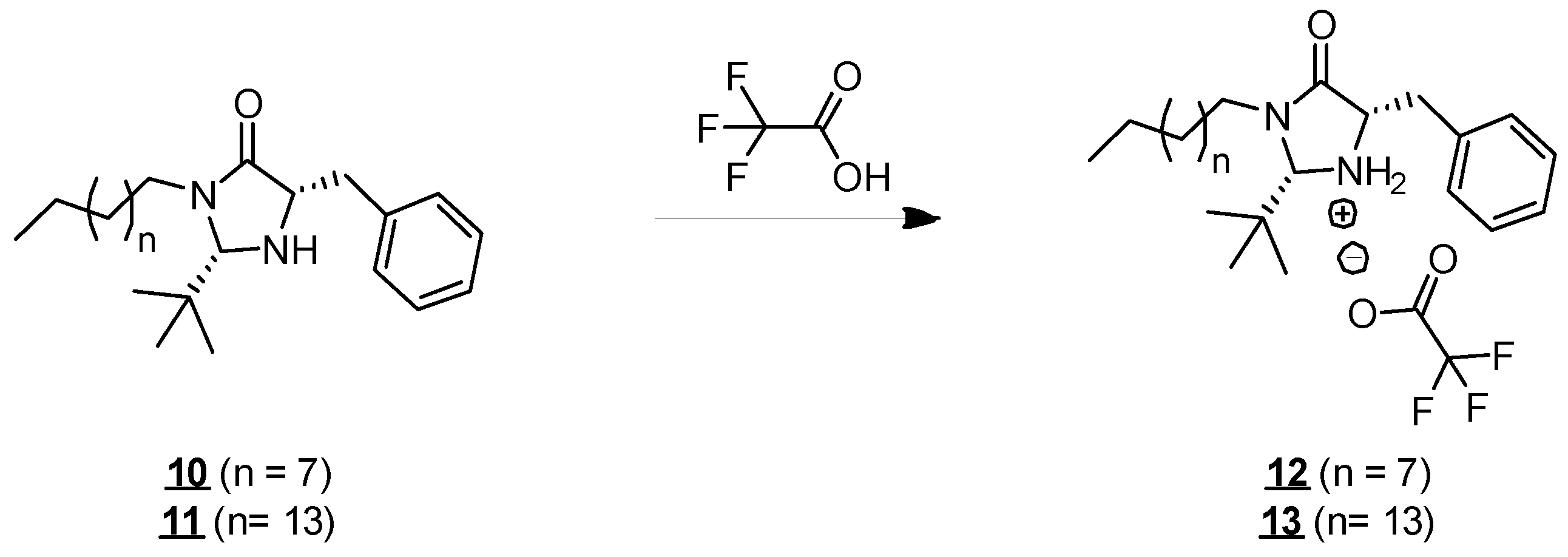
Molecules 10 and 11 have an atypical structure for surfactants. They have two main hydrophobic groups (C10 and C16 hydrocarbon chains and an aromatic group) and a hydrophilic center (imidazolidinone unit). They are, therefore, molecules with a more lipophilic character. Their solubility in organic solvents, such as cyclohexane, ethyl acetate, and dichloromethane was observed during the various reactions in which they were involved and the corresponding purification stages. However, the molecules remained insoluble in water from 25 to 45 °C, even after vigorous stirring, forming a fatty organic immiscible phase above the water. In order to make the molecules soluble in water, we protonated them by an acid treatment (Figure 4). The effect of protonation on the solubility of molecules in water has already been described in the literature, particularly for long-chain amines, such as hexadecanamide, which is poorly soluble in water [21] but could be solubilized at higher concentrations in its protonated form [22,23]. In our case, the addition of trifluoroacetic acid allowed us to convert the amine function into its ammonium form, thus, giving a positive charge to the imidazolidinone unit. The resulting molecules (12 and 13, respectively) presented solubility in water at 25 °C up to 3 g·L−1 and 1 g·L−1 for molecules 12 and 13, respectively, allowing the study of their surface activity.
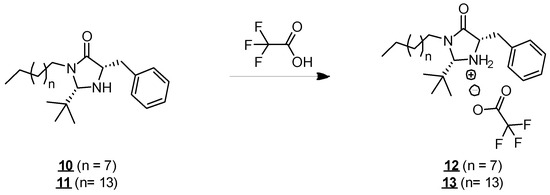
Figure 4.
Formation of compounds 12 and 13.
2.3. Surface Tension
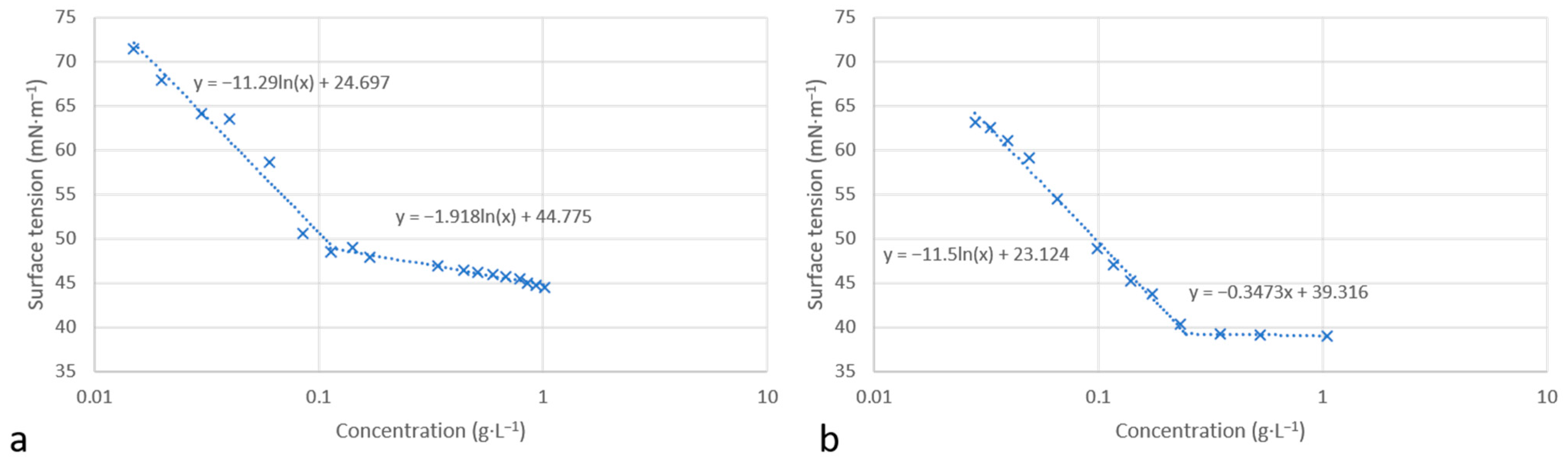
The surface tension in water was determined using the Wilhelmy plate method at 25 °C. Figure 5 presents the variation in the surface tension versus concentration for molecules 12 and 13. Similar curves were obtained in both cases, showing the decrease in water/air surface tension with increasing surfactant concentration. An inflection point was observed for each molecule corresponding to a critical aggregation concentration, CACw, in water. Above this concentration, the surface tension remains constant.
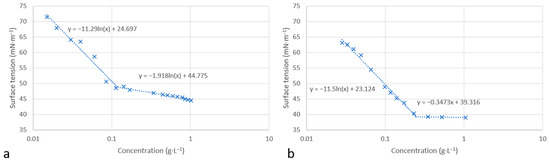
Figure 5.
Surface tension versus concentration for compounds 12 (a) and 13 (b).
In order to investigate the adsorption of molecules at the water/air interface, we used the Gibbs equation to calculate the maximum saturated adsorption capacity Γmax and the minimum molecular occupied area Amin (Equations (1) and (2)).
where C is the surfactant concentration (M), γ is the surface tension (mN·m−1), and n takes the value of 2 for an ionic surfactant with a monovalent cation; T represents the temperature (K), and R is the ideal gas constant (8.314 J·mol−1·K−1). Na is the Avogadro constant (6.022 × 1023 mol−1).
As shown in Table 2, the CACw values for molecules 12 and 13 are quite close to those of conventional surfactants, in particular Tween 20 and Triton X100. The measured values of the surface tension, γw, at the critical association concentration of both molecules also show a surface activity similar to the three conventional surfactants (Tween 20, Triton X 100, and CTAB).
Table 2.
Critical association concentration (CACw), surface tension at the CAC (γw), and surface adsorption parameters (Γmax, Amin) for compounds 12 and 13 and three other conventional surfactants.
It can be observed that molecule 13, with a longer alkyl chain, has a higher occupied molecular area Amin compared to molecule 12. A similar effect of the hydrocarbon chain length on the Amin value has already been described by Man [30]. The long chains are more flexible and can induce a larger occupied area at the water/air interface. Furthermore, the calculated values of Amin and Γmax are in the same order of magnitude as those of the three conventional surfactants chosen as references (Tween 20, Triton X 100 and CTAB).
2.4. Aggregation Behavior in Water
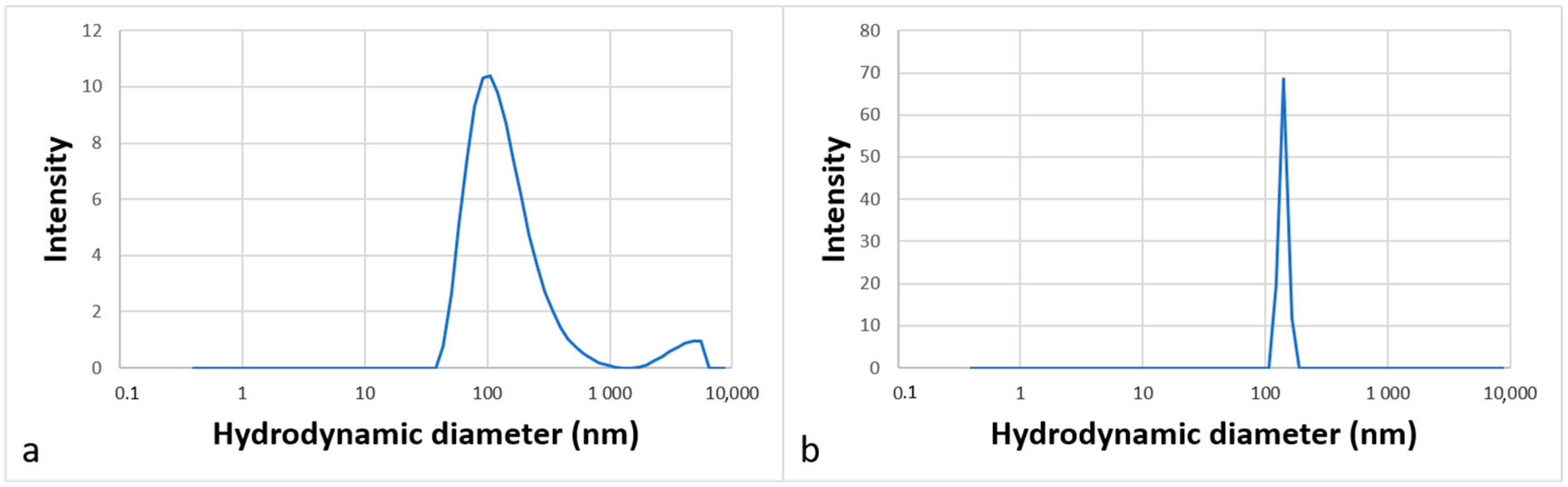
The aggregation behavior was studied by DLS analysis in water, which corresponds to the medium of the future catalytic reactions. Figure 6 shows the intensity-weighted size distribution (a) for molecule 12 at 3 g·L−1 and (b) for molecule 13 at 1 g·L−1. The curves show unimodal size distributions.
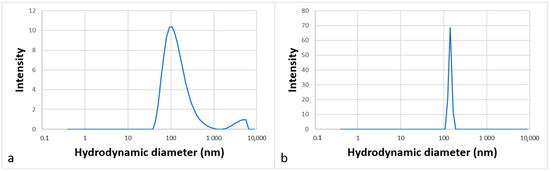
Figure 6.
Partial size distribution of aggregates formed in an aqueous solution of surfactants 12 (a) and 13 (b) at 3 g·L−1 and 1 g·L−1, respectively.
The diameters of the particles measured were around 120 ± 2 nm for molecule 12 and around 140 ± 21 nm for molecule 13, indicating that molecules can self-assemble in water to form large aggregates that can be assimilated to vesicles. Such organized structures dispersed in water are suitable for carrying out organic reactions at their core [4].
2.5. Asymmetric Catalytic Activity of Compounds 12 and 13 in Water
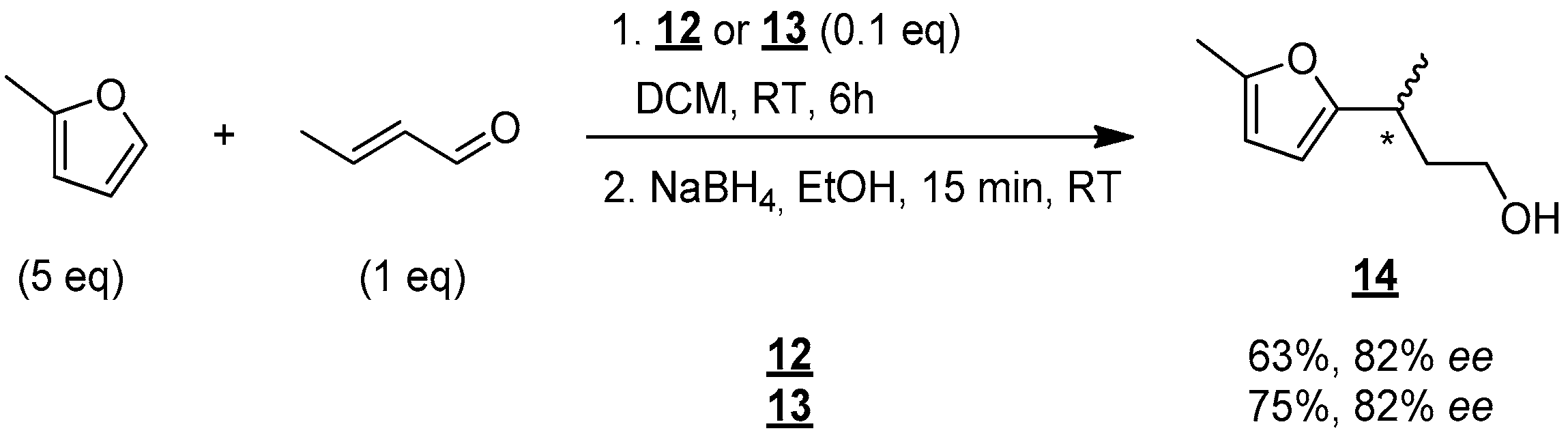
The performance of catalysts 12 and 13 were tested in enantioselective Friedel–Crafts reaction (Figure 7) [15]: crotonaldehyde (1 equiv), 2-methoxythiophene (5 equiv) were stirred in the presence of a catalytic amount of 12 or 13 (0.1 equiv) in DCM at room temperature for 6 h. The resulting aldehyde was then reduced to give alcohol 14, which was finally analyzed to determine the enantioselectivity of the reaction.
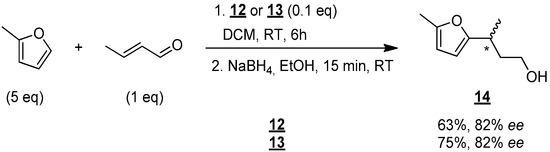
Figure 7.
Enantioselective synthesis of compound 14 in DCM. * indicates the asymmetric carbon.
Alcohol 14 was then obtained in good yield of 63% and an encouraging enantiomeric excess of 82% with catalyst 12. The use of catalyst 13 led to the isolation of 14 in 75% yield and an identical enantiomeric excess of 82%. These results, therefore, demonstrate that molecules 12 and 13 do indeed catalyze the Friedel–Crafts reaction, and they do so stereoselectively.
Given these promising results, the use of these new organocatalytic surfactants, 12 and 13, was tested in water (Figure 8).
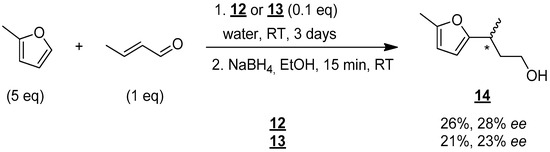
Figure 8.
Enantioselective synthesis of compound 14 in water. * indicates the asymmetric carbon.
Under the same conditions described above (number of equivalents, temperature), vigorous stirring was required when water was used as the solvent in order to obtain a good emulsion. 1H NMR monitoring of the reaction also showed that a reaction time of 3 days was required to obtain reasonable yields. Alcohol 14 was then isolated in yields of 26% and 21% using molecules 12 and 13, respectively. These results show that 12 and 13 really act as organocatalytic surfactants. However, a decrease in chiral activity was observed as enantiomeric excesses of 28% and 23% were obtained with 12 and 13, respectively. Despite this reduction in enantiomeric excess, the results are still very encouraging, and a more detailed study is underway to modify the reaction conditions to obtain enantiomerically pure compounds. The results will be published in due time.
3. Materials and Methods
3.1. Experimental Apparatus
1H NMR spectra were acquired with a FOURIER300 (300 MHz) or an Avance III HD 500 (500 MHz) spectrometer from Brüker Coporation (Karlsruhe, Germany). The chemical shifts, δ, are expressed in parts per million (ppm) and referenced to tetramethylsilane at 0 ppm. Coupling constants, J, are expressed in Hertz (Hz). Data are reported as follows: δ, multiplicity (br: broad, s: singlet, d: doublet, dd: doublet of doublet, t: triplet, q, quadruplet, and m: multiplet), J, integration, and assignment. All samples were diluted in deuterochloroform.
13C NMR spectra were recorded on the same instrument at 75 or 126 MHz. Chemical shifts are referenced to the central peak of residual CDCl3 (77.2 ppm).
Infrared (IR) spectra were recorded on a Spectrum 65 FT-IT spectrometer from Perkin SPE 263 IR, PerkinElmer (Villepinte, France) in ATR mode. Wavenumbers are expressed in cm−1.
High Resolution Mass Spectra (HRMS) were obtained on a GCT Premier (Waters, Milford, MA, USA) mass spectrometer via direct introduction. Analyses were performed at the Institut Chimique de Toulouse, Université Paul Sabatier, Toulouse, France.
The tensiometer was an automatic 3S GBX scientific instrument (Loire, France). Particle size analyses were performed on a Zetasizer Nano-ZS, Malvern Panalytical Instrument, Ltd. (Westborough, MA, USA).
3.2. Synthesis Experimental Procedure
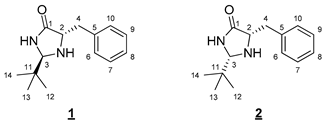
- (2R,5S)-5-Benzyl-2-(tert-butyl)imidazolidin-4-one (1) and (2S,5S)-5-benzyl-2-(tert-butyl)imidazolidin-4-one (2)
To a solution of 0.998 g (6.08 mmol, 1 equiv) of L-phenylalaninamide in 30 mL of ethanol, the following were added: 0.105 g (0.61 mmol, 0.1 equiv) of FeCl3 and 1.45 mL (13.40 mmol, 2.2 equiv) of pivaldehyde. The resulting mixture was stirred at 65 °C for 24 h, and then the volatile compounds were evaporated in a rotary evaporator. Ethyl acetate was added to the medium, which was then filtered through Celite. The volatile compounds were then evaporated in a rotary evaporator. The crude product was finally purified by flash chromatography on silica gel (cyclohexane/EtOAc 5:5 (v/v)) to give 0.423 mg of anti 1 diastereoisomer (30%, white solid) and 0.471 mg of syn 2 diastereoisomer (33%, white solid).
- Anti 1 diastereoisomer (Rf 0.40 (cyclohexane/EtOAc 5:5 (v/v)): 1H NMR (300 MHz, CDCl3) δ 7.40 (br s, 1H, NH), 7.35–7.21 (m, 5H, H6–10), 4.03 (d, J = 1.9 Hz, 1H, H3), 3.80 (ddd, J = 6.6, 4.4, 1.9 Hz, 1H, H2), 3.10 (dd, J = 14.1, 4.4 Hz, 1H, H4), 2.94 (dd, J = 14.1, 6.6 Hz, 1H, H4), 0.86 (s, 9H, H12–14). 13C NMR (75 MHz, CDCl3) δ 177.8 (C1), 137.2 (C5), 129.5 (C6, C 10), 128.7 (C7, C9), 126.8 (C8), 77.8 (C3), 60.0 (C2), 38.0 (C4), 36.1 (C11), 24.2 (s, C12–14). IR (KBr): 3357, 3196, 2967, 2866, 1688, 1454, 1376, 756, 700 cm−1. HRMS (DCI-CH4) Calculated for C14H21N2O 233.1654 [M]+, found 233.1642.
- Syn 2 diastereoisomer (Rf 0.15 (cyclohexane/EtOAc 5:5 (v/v)): 1H NMR (300 MHz, CDCl3) δ 7.81 (br s, 1H, NH), 7.36–7.17 (m, 5H, H6–10) 4.25 (d, J = 1.5 Hz, 1H, H3), 3.81 (ddd, J = 7.5, 4.0, 1.5 Hz, 1H, H2), 3.12 (dd, J = 13.7, 4.0 Hz, 1H, H4), 2.92 (dd, J = 13.7, 7.5 Hz, 1H, H4), 0.80 (s, 9H, H12–14). 13C NMR (75 MHz, CDCl3) δ 177.6 (C1), 137.9 (C5), 129.6 (C6, C10), 128.6 (C7, C9), 126.7 (C8), 77.3 (C3), 60.3 (C2), 37.7 (C4), 34.0 (C11), 24.3 (C12–14). IR (KBr): 3342, 3229, 2955, 2865, 1705, 1454, 1341, 730, 699 cm−1. HRMS (DCI-CH4): Calculated for C14H21N2O 233.1654 [M]+, found 233.1650.
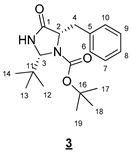
- tert-Butyl (2R,5S)-5-benzyl-2-(tert-butyl)-4-oxoimidazolidine-1-carboxylate (3)
To a solution of 0.471 g (2.03 mmol, 1 equiv) of 2 in 10 mL of DCM, the following were added: 0.443 g (2.03 mmol, 1 equiv) of di-tert-butyl dicarbonate and 0.82 mL (6.090 mmol, 3 equiv) of triethylamine. The resulting mixture was stirred at room temperature overnight, and then the volatile compounds were evaporated in a rotary evaporator. The crude product was purified by flash chromatography on silica gel (cyclohexane/EtOAc 6:4 (v/v)) to give 0.479 g of 3 as a white solid (71%).
- Rf 0.40 (cyclohexane/EtOAc 6:4 (v/v)). 1H NMR (300 MHz, CDCl3) δ 7.35–7.20 (m, 5H, H6–10), 6.68 (br s, 1H, NH), 5.02 (s, 1H, H3), 4.34 (t, J = 6.7 Hz, 1H, H2), 3.17 (dd, J = 13.8, 6.7 Hz, 1H, H4), 3.03 (dd, J = 13.8, 6.7 Hz, 1H, H4), 1.30 (s, 9H, H17–19), 0.98 (s, 9H, H12–14). 13C NMR (75 MHz, CDCl3) δ 173.7 (C1), 156.1 (C15), 138.1 (C5), 129.7 (C6, C10), 128.3 (C7, C9), 126.5 (C8), 81.3 (C16), 77.5 (C3), 61.6 (C2), 39.6 (C4), 36.7 (C3), 28.0 (C17–19), 25.8 (C12–14). IR (KBr): 3294, 2975, 1721, 1709, 1371, 1357, 1304, 1167, 1076, 953, 805, 780, 749, 737, 702 cm−1. HRMS (DCI-CH4) Calculated for C19H29N2O3 333.2178 [M+H]+, found 333.2174.
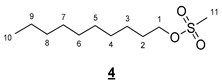
- Decyl methanesulfonate (4)
To a solution of 5.000 g (31.59 mmol, 1 equiv) of decanol in 50 mL of DCM, the following were added: 3.67 mL (47.38 mmol, 1.5 equiv) of mesyl chloride and 8.53 mL (63.18 mmol, 2 equiv) of trimethylamine. The resulting mixture was stirred for 7 h at room temperature, and then 30 mL of HCl (2N) was added. The medium was vigorously stirred overnight. The organic phase was then washed 3 times with 30 mL of water. The organic phases were combined, dried (MgSO4) and concentrated in vacuo to give 7.119 g of 4 as yellow oil (95%). The structure of 4 was confirmed by comparison with literature reported data [31].
- 1H NMR (300 MHz, CDCl3) δ 4.22 (t, J = 6.6 Hz, 2H, H1), 3.00 (s, 3H, H11), 1.86–1.65 (m, 2H, H2), 1.46–1.16 (m, 14H, H3–9), 0.88 (t, J = 6.6 Hz, 3H, H10). IR (KBr): 2929, 2856, 1467, 1354, 1177, 975, 836, 746, 722 cm−1.
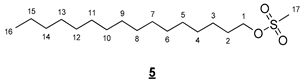
- Hexadecyl methanesulfonate (5)
To a solution of 4.740 g (19.550 mmol, 1 equiv) of hexadecanol in 30 mL of DCM, the following were added: 2.27 mL (29.330 mmol, 1.5 equiv) of mesyl chloride and 3.16 mL (39.100 mmol, 2 equiv) of pyridine. The resulting mixture was stirred for 7 h at room temperature, and then 20 mL of HCl (2N) was added. The medium was vigorously stirred overnight. The organic phase was then washed with 20 mL of an aqueous solution of NaHCO3 (5%) and 20 mL of water. The organic phases were combined, dried (MgSO4), and concentrated in vacuo to give 4.380 g of 5 as a white solid (70%). The structure of 5 was confirmed by comparison with literature reported data [31].
- 1H NMR (300 MHz, CDCl3) δ 4.22 (t, J = 6.6 Hz, 2H, H1), 3.00 (s, 3H, H17), 1.86–1.64 (m, 2H, H2), 1.46–1.16 (m, 26H, H3–15), 0.88 (t, J = 6.7 Hz, 3H, H16). IR (KBr): 2918, 2851, 1474, 1344, 1328, 1168, 1160, 984, 942, 851, 751, 718 cm−1.
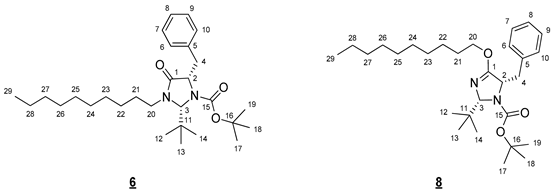
- tert-Butyl (2R,5S)-5-benzyl-2-(tert-butyl)-4-(decyloxy)-2,5-dihydro-1H-imidazole-1-carboxylate (6) et tert-butyl (2R,5S)-5-benzyl-2-(tert-butyl)-3-decyl-4-oxoimidazolidine-1-carboxylate (8)
To a solution of 1.000 g (3.01 mmol, 1 equiv) of 3 in 20 mL of DMF, 0.241 g (6.02 mmol, 2 equiv) of sodium hydride (60% in mineral oil) was added. The resulting mixture was stirred for 5 min. A solution of 0.712 g (3.01 mmol, 1 equiv) of 4 in 5 mL of DMF was then added and the medium was stirred at 60 °C for 6 h. It was cooled to room temperature and hydrolyzed with 20 mL of water. The resulting aqueous phase was extracted with 50 mL of ethyl acetate. The combined organic phases were washed five times with 20 mL of water, dried (MgSO4), and concentrated in vacuo. The crude product was purified by flash chromatography on silica gel (cyclohexane/AcOEt 95:5 (v/v)) to give 0.404 g (28%) of 6 and 0.581 g (41%) of 8 as colorless oils.
- Compound 6 (Rf 0.19 (cyclohexane/AcOEt 9:1 (v/v))): 1H NMR (300 MHz, CDCl3) δ 7.36–7.17 (m, 5H, H6–10), 5.12 (s, 1H, H3), 4.32 (dd, J = 7.2, 6.2 Hz, 1H, H2), 3.92 (ddd, J = 14.1, 9.3, 7.2 Hz, 1H, H20), 3.20 (dd, J = 14.1, 6.2 Hz, 1H, H4), 3.03 (ddd, J = 9.3, 7.2, 3.4 Hz, 1H, H20), 2.98 (dd, J = 13.8, 7.2 Hz, 1H, H4), 1.35–1.22 (m, 23H, H22–28, H17–19), 1.06 (s, 9H, H12–14), 0.87 (t, J = 6.7 Hz, 3H, H29). 13C NMR (75 MHz, CDCl3) δ 171.5 (C1), 156.0 (C15), 138.3 (C5), 129.7 (C6, C10), 128.4 (C7, C9), 126.5 (C8), 81.1 (C16), 79.2 (C3), 61.9 (C2), 42.8 (C20), 39.8 (C4), 37.4 (C11), 31.9 (CH2), 29.5 (CH2), 29.5 (CH2), 29.3 (CH2), 29.2 (CH2), 28.0 (C17–19), 27.0 (C12–14), 26.7 (CH2), 26.5 (C21), 22.7 (CH2), 14.1 (C29). IR (KBr) 2927, 2856, 1708, 1366, 1167, 786, 751, 699 cm−1. HRMS (DCI-CH4) Calculated for C29H49N2O3 473.3743 [M+H]+, found 473.3761.
- Compound 8 (Rf 0.35 (cyclohexane/AcOEt 9:1 (v/v))): 1H NMR (300 MHz, CDCl3) δ 7.33–7.14 (m, 5H, H6–10), 5.19 (s, 1H, H3), 4.52 (dd, J = 7.8, 5.7 Hz, 1H, H2), 4.24 (dt, J = 10.5, 6.5 Hz, 1H, H20), 4.03 (dt, J = 10.5, 6.6 Hz, 1H, H20), 3.07 (dd, J = 13.6, 5.7 Hz, 1H, H4), 2.93 (dd, J = 13.6, 7.8 Hz, 1H, H4), 1.68–1.54 (m, 2H, H21), 1.36 (s, 9H, H17–19), 1.32–1.22 (m, 14H, H22–28), 0.98 (s, 9H, H12–14), 0.88 (t, J = 6.7 Hz, 3H, H29). 13C NMR (75 MHz, CDCl3) δ 167.6 (C1), 156.1 (C15), 138.5 (C5), 129.6 (C6, C10), 128.2 (C7, C9), 126.3 (C8), 90.1 (C3), 80.3 (C16), 68.3 (C20), 63.9 (C2), 40.2 (C4), 36.6 (C11), 31.9 (CH2), 29.6 (CH2), 29.5 (CH2), 29.3 (CH2), 29.3 (CH2), 28.7 (C21), 28.3 (C17–19), 26.7 (C12–14), 25.9 (CH2), 22.7 (CH2), 14.1 (C29). IR (KBr) 3416, 2927, 2856, 1712, 1667, 1366, 1331, 1163, 786, 745, 699 cm−1. HRMS (DCI-CH4) Calculated for C29H49N2O3 473.3743 [M+H]+, found 473.3751.
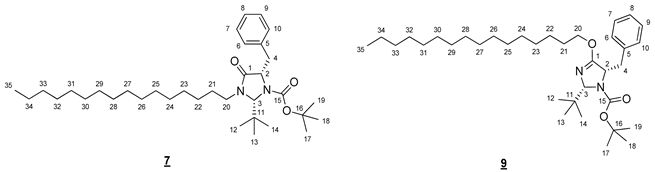
- tert-Butyl (2R,5S)-5-benzyl-2-(tert-butyl)-4-(hexadecyloxy)-2,5-dihydro-1H-imidazole-1-carboxylate (7) and tert-butyl (2R,5S)-5-benzyl-2-(tert-butyl)-3-hexadecyl-4-oxoimidazolidine-1-carboxylate (9)
To a solution of 1.000 g (3.02 mmol, 1 equiv) of 3 in 20 mL of DMF, 0.242 g (6.04 mmol, 2 equiv) of sodium hydride (60% in mineral oil) was added. The mixture obtained was stirred for 5 min. A solution of 0.967 g (3.02 mmol, 1 equiv) of 5 in 5 mL of DMF was then added and the medium was stirred at 60 °C for 6 h. It was cooled to room temperature and hydrolyzed with 20 mL of water. The resulting aqueous phase was extracted with 50 mL of ethyl acetate. The combined organic phases were washed five times with 20 mL of water, dried (MgSO4), and concentrated in vacuo. The crude product was purified by flash chromatography on silica gel (cyclohexane/AcOEt 95:5 (v/v)) to give 0.497 g (30%) of 7 and 0.596 g (35%) of 9 as colorless oils.
- Compound 7 (Rf 0.17 (cyclohexane/AcOEt 9:1 (v/v))): 1H NMR (300 MHz, CDCl3) δ 7.36–7.14 (m, 5H, H6–10), 5.12 (s, 1H, H3), 4.39–4.27 (m, 1H, H2), 3.92 (ddd, J = 14.2, 9.3, 7.3 Hz, 1H, H20), 3.20 (dd, J = 13.8, 6.0 Hz, 1H, H4), 3.03 (ddd, J = 9.3, 6.7, 3.4 Hz, 1H, H20), 2.98 (dd, J = 13.8, 7.2 Hz, 1H, H4), 1.67 (m, 2H, H21), 1.38–1.15 (m, 26H, H22–34), 1.26 (s, 9H, H17–19) 1.06 (s, 9H, H12–14), 0.88 (t, J = 6.7 Hz, 3H, H35). 13C NMR (75 MHz, CDCl3) δ 171.5 (C1), 156.0 (C15), 138.3 (C5), 129.7 (C6, C10), 128.3 (C7, C9), 126.5 (C8), 81.1 (C16), 79.2 (C3), 61.9 (C2), 42.7 (C20), 39.8 (C4), 37.4 (C11), 31.9 (CH2), 29.7 (3 * CH2), 29.7 (2 * CH2), 29.6 (CH2), 29.5 (2 * CH2), 29.4 (CH2), 29.2 (CH2), 28,0 (C17–19), 27.0 (C12–14), 26.7 (C22), 26.5 (C21), 22.7 (CH2), 14.1 (C35). IR (KBr) 2926, 2854, 1708, 1367, 1167, 786, 750, 698 cm−1. HRMS (DCI-CH4) Calculated for C35H61N2O3 557.4682 [M+H]+, found 557.4688.
- Compound 9 (Rf 0.38 (cyclohexane/AcOEt 9:1 (v/v))): 1H NMR (300 MHz, CDCl3) δ 7.35–7.13 (m, 5H, H6–10), 5.19 (s, 1H, H3), 4.53 (dd, J = 7.7, 5.8 Hz, 1H, H2), 4.24 (dt, J = 10.5, 6.5 Hz, 1H, H20), 4.03 (dt, J = 10.5, 6.6 Hz, 1H, H20), 3.07 (dd, J = 13.6, 5.6 Hz, 1H, H4), 2.93 (dd, J = 13.6, 7.9 Hz, 1H, H4), 1.68–1.55 (m, 2H, H21), 1.36 (s, 9H, H17–19), 1.32–1.20 (m, 26H, H22–34), 0.98 (s, 9H, H12–14), 0.88 (t, J = 6.7 Hz, 3H, H35). 13C NMR (75 MHz, CDCl3) δ 167.6 (C1), 156.1 (C15), 138.5 (C5), 129.6 (C6, C10), 128.1 (C7, C9), 126.3 (C8), 90.1 (C3), 80.2 (C16), 68.3 (C20), 63.9 (C2), 40.2 (C4), 36.6 (C11), 32.0 (C33), 29.7 (4 * CH2), 29.7 (2 * CH2), 29.6 (CH2), 29.6 (CH2), 29.4 (CH2), 29.3 (CH2), 28.7 (C21), 28.3 (C17–19), 26.7 (C12–14), 25.9 (C22), 22.7 (CH2), 14.1 (C35). IR (KBr) 2925, 2855, 1712, 1666, 1365, 1331, 1175, 786, 745, 698 cm−1. HRMS (DCI-CH4) Calculated for C35H61N2O3 557.4682 [M+H]+, found 557.4700.
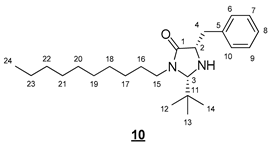
- (2S,5S)-5-benzyl-2-(tert-butyl)-3-decylimidazolidin-4-one (10)
To a solution of 0.403 g (0.85 mmol, 1 equiv) of 6 in 2.5 mL of DCM, 1.378 mL (17.09 mmol, 20 equiv) of trifluoroacetic acid was added. The resulting mixture was stirred at room temperature for 3 h. The volatile compounds were evaporated in a rotary evaporator. The medium was resolubilized in 3 mL of DCM and then treated with 1 mL of aqueous sodium hydroxide solution (1M). The organic phase was dried (MgSO4) and concentrated in vacuo. The crude product was purified by flash chromatography on silica gel (cyclohexane/EtOAc 7:3 (v/v)) to give 0.262 g of 10 (80%) as a yellow oil.
- Rf 0.33 (cyclohexane/AcOEt 7:3 (v/v)). 1H NMR (500 MHz, CDCl3) δ 7.31–7.19 (m, 5H, H6–10), 4.23 (s, 1H, H3), 3.77 (ddd, J = 14.0, 9.7, 7.0 Hz, 1H, H15), 3.72–3.65 (m, 1H, H2), 3.15 (dd, J = 13.7, 3.9 Hz, 1H, H4), 3.05 (ddd, J = 14.0, 9.4, 4.6 Hz, 1H, H15), 2.91 (dd, J = 13.7, 7.7 Hz, 1H, H4), 1.63–1.54 (m, 1H, H16), 1.52–1.44 (m, 1H, H16), 1.34–1.20 (m, 14H, H17–23), 0.88 (t, J = 7.0 Hz, 3H, H24), 0.84 (s, 9H, H12–14). 13C NMR (126 MHz, CDCl3) δ 175.3 (C1), 138.0 (C5), 129.6 (C6, C10), 128.5 (C7, C9), 126.6 (C8), 79.6 (C3), 59.3 (C2), 42.6 (C15), 38.2 (C4), 35.4 (C11), 31.9 (CH2), 29.6 (CH2), 29.5 (CH2), 29.3 (2 * CH2), 27.0 (CH2), 26.9 (C16), 25.5 (C12–14), 22.7 (CH2), 14.1 (C24). IR (KBr) 2927, 2855, 1698, 1455, 751, 721, 700 cm−1. HRMS (DCI-CH4) Calculated for C24H41N2O 373.3219 [M+H]+, found 373.3213.
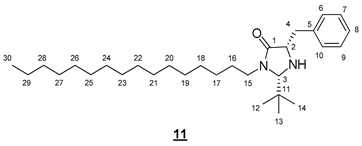
- (2S,5S)-5-benzyl-2-(tert-butyl)-3-hexadecylimidazolidin-4-one (11)
To a solution of 0.497 g (0.89 mmol, 1 equiv) of 7 in 2.5 mL of DCM, 1.378 mL (17.85 mmol, 20 equiv) of trifluoroacetic acid was added. The resulting mixture was stirred at room temperature for 3 h. The volatile compounds were evaporated in a rotary evaporator. The medium was resolubilized in 3 mL of DCM and then treated with 1 mL of aqueous sodium hydroxide solution (1M). The organic phase was dried (MgSO4) and concentrated in vacuo. The crude product was purified by flash chromatography on silica gel (cyclohexane/EtOAc 7:3 (v/v)) to give 0.270 g of 11 (66%) as a yellow oil.
- Rf 0.37 (cyclohexane/AcOEt 7:3 (v/v)). 1H NMR (500 MHz, CDCl3) δ 7.31–7.19 (m, 5H, H6–10), 4.23 (s, 1H, H3), 3.77 (ddd, J = 14.1, 9.7, 7.1 Hz, 1H, H15), 3.72–3.65 (m, 1H, H2), 3.15 (dd, J = 13.7, 3.9 Hz, 1H, H4), 3.05 (ddd, J = 14.1, 9.3, 4.5 Hz, 1H, H15), 2.91 (dd, J = 13.7, 7.7 Hz, 1H, H4), 1.63–1.54 (m, 1H, H16), 1.51–1.44 (m, 1H, H16), 1.34–1.19 (s, 26H, H17–29), 0.88 (t, J = 6.9 Hz, 3H, H30), 0.84 (s, 9H, H12–14). 13C NMR (126 MHz, CDCl3) δ 175.3 (C1), 138.1 (C5), 129.6 (C6, C10), 128.5 (C7, C9), 126.6 (C8), 79.6 (C3), 59.3 (C2), 42.6 (C15), 38.3 (C4), 35.4 (C11), 31.9 (CH2), 29.7 (3 * CH2), 29.7 (CH2), 29.7 (CH2), 29.7 (CH2), 29.6 (CH2), 29.6 (CH2), 29.4 (CH2), 29.3 (CH2), 27.0 (CH2), 26.9 (C16), 25.5 (C12–14), 22.7 (CH2), 14.1 (C30). IR (KBr) 2926, 2854, 1699, 1455, 699 cm−1. HRMS (DCI-CH4) Calculated for C30H53N2O 457.4158 [M+H]+, found 457.4177.
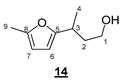
- 3-(5-Methylfuran-2-yl)butan-1-ol (14)
To a solution of 0.027 g (0.07 mmol, 0.1 equiv) of 10 in 2.5 mL of DCM, the following were added: 5.5 µL (0.07 mmol, 0.1 equiv) of trifluoroacetic acid, 329 µL (3.57 mmol, 5 equiv) of 2-methylfuran, and 58 µL (0.71 mmol, 1 equiv) of crotonaldehyde. The resulting mixture was stirred at room temperature for 6 h and then transferred into a vial containing 0.135 g (3.57 mmol, 5 equiv) of NaBH4 in 2.5 mL of EtOH. After 15 min of stirring at room temperature, 10 mL of a saturated aqueous NaHCO3 solution was added. The resulting medium was extracted three times with 10 mL of DCM. The combined organic phases were then washed with 10 mL of saturated aqueous NaHCO3 solution and 10 mL of brine. The organic phase was dried (MgSO4) and concentrated in vacuo. The crude product was purified by flash chromatography on silica gel (cyclohexane/EtOAc 9:1 (v/v)) to give 0.104 g of 14 (95%) as a yellow oil. The structure of 14 was confirmed by comparison with literature reported data.
- Rf 0.37 (cyclohexane/AcOEt 7:3 (v/v)). 1H NMR (500 MHz, CDCl3) δ 5.86 (d, J = 3.0 Hz, 1H, H6), 5.84 (d, J = 3.0 Hz, 1H, H7), 3.71–3.61 (m, 2H, H1), 2.94 (m, 1H, H3), 2.25 (s, 3H, H9), 1.89 (ddt, J = 14.0, 8.0, 6.2 Hz, 1H, H2), 1.77 (td, J = 14.0, 6.6 Hz, 1H, H2), 1.25 (d, J = 7.0 Hz, 3H, H4). 13C NMR (126 MHz, CDCl3) δ 158.1 (C5), 150.3 (C8), 105.6 (C6), 104.3 (C7), 61.0 (C1), 38.8 (C2), 29.9 (C3), 19.4 (C4), 13.5 (C9). HRMS (DCI-CH4) Calculated for C9H15O2 155.1072 [M+H]+, found 155.1073.
3.3. Surface Tension Analysis
The surface tension of the surfactants was determined at 25 °C with a GBX instrument using a platinum plate. A 1 g·L−1 solution of each surfactant was prepared in ultrapure water. The surface tension of this solution was measured. The solution was then diluted with ultrapure water. Each experimental sample was measured in triplicate and averaged. The relationship curves between surface tension (γ) and logarithmic concentration (Log C) were drawn. In addition, the surface tension of ultrapure water was measured to be 72.00 mN·m−1.
3.4. Dynamic Light Scattering Tests
The hydrodynamic diameter of surfactant aggregates in aqueous solution (3 g·L−1 and 1 g·L−1 for molecules 12 and 13, respectively) was determined with a Zetasizer Nano-ZS nanoparticle size potentiometer. The scattering angle is 173° using a helium–neon laser with a wavelength of 633 nm. The samples were kept at 25 °C for more than 5 h prior to the experiment. The aqueous solutions of the surfactants were filtered with a 0.45 mm filter head to remove any impurities. Each analysis was performed in triplicate to ensure the reproducibility of the measurement.
4. Conclusions
In conclusion, we have shown in this work that it is possible to synthesize new organocatalytic surfactants. These were obtained from as many biobased compounds as possible, in accordance with the 12 principles of Green Chemistry. The properties of these molecules were studied in water: we were able to demonstrate that they present critical aggregation concentrations, surface tensions at the CACw, and surface adsorption parameters close to those of conventional surfactants. DLS analyses also confirmed that these molecules self-assemble to form vesicles in water. Finally, we tested the asymmetric catalytic activity of these organocatalytic surfactants in Diels–Alder reactions carried out in dichloromethane. The results were encouraging, as the desired molecules were obtained in good yields and enantioselectivities. However, the enantioselectivity decreased when the reaction was carried out in water. Work is currently underway to improve these results, as this methodology would eventually allow the synthesis of asymmetric molecules with high added value in water, which is a current challenge for many chemical sectors.
Author Contributions
Conceptualization, C.C., P.d.C., S.T.-R. and E.V.; methodology, C.C., P.d.C., E.V. and E.C.; validation, C.C., P.d.C., S.T.-R., E.V. and E.C.; formal analysis, E.C.; investigation, E.C.; resources, C.C., P.d.C. and E.V.; writing—original draft preparation, C.C., P.d.C. and E.V.; writing—review and editing, C.C., P.d.C. and E.V.; visualization, C.C., P.d.C. and E.V.; supervision, C.C., P.d.C. and E.V.; project administration, C.C.; funding acquisition, C.C. and S.T.-R. All authors have read and agreed to the published version of the manuscript.
Funding
The authors gratefully thank the ANR Sargassum program for “SarTrib: Tribological and electrochemical recovery of Sargassum pyrolysis residues” (Joint call «Research, development and innovation», SARGASSUM, 2019), supported by ADEME (20GAC0011) and Region Guadeloupe FEDER GP0026401 (2020-FED-15).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are contained within the article.
Acknowledgments
This work was certified EUR BioEco by the French National Research Agency “Investissement d’avenir” Program (N°ANR-18-EURE-0021) and has benefited from the financial support of the French Higher Education and Research Minister (MESR).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Pace, V.; Hoyos, P.; Castoldi, L.; Dominguez De Maria, P.; Alcantara, A. 2-Methyltetrahydrofuran (2-MeTHF): A Biomass-Derived Solvent with Broad Application in Organic Chemistry. ChemSusChem 2012, 5, 1369–1379. [Google Scholar] [CrossRef] [PubMed]
- Anastas, P.; Warner, J. Green Chemistry: Theory and Practice; Oxford University Press: Oxford, UK, 1998. [Google Scholar]
- Lelais, G.; MacMillan, D. History and Perspective of Chiral Organic Catalysts; John Wiley & Sons: Hoboken, NJ, USA, 2007. [Google Scholar]
- La Sorella, G.; Strukul, G.; Scarso, A. Recent advances in catalysis in micellar media. Green Chem. 2015, 17, 644–683. [Google Scholar] [CrossRef]
- Aratake, S.; Itoh, T.; Okano, T.; Nagae, N.; Sumiya, T.; Shoi, M.; Hayashi, Y. Highly Diastereo- and Enantioselective Direct Aldol Reactions of Aldehydes and Ketones Catalyzed by Siloxyproline in the Presence of Water. Chem. A Eur. J. 2007, 13, 10246–10256. [Google Scholar] [CrossRef] [PubMed]
- Hu, F.; Guo, C.-S.; Xie, J.; Zhu, H.-L.; Huang, Z.-Z. Efficient and Highly Enantioselective Michael Addition of Aldehydes to Nitroalkenes Catalyzed by a Surfactant-type Organocatalyst in the Presence of Water. Chem. Lett. 2010, 39, 412–414. [Google Scholar] [CrossRef]
- Li, J.; Liu, Y.; Liu, L. Efficient Enantioselective Michael Addition of Nitroalkenes Catalyzed by a Surfactant-Type Bifunctional Thiourea Organocatalyst in the Presence of Water. Lett. Org. Chem. 2012, 9, 51–55. [Google Scholar] [CrossRef]
- Wu, C.; Fu, X.; Ma, X.; Li, S.; Li, C. Threonine-surfactant organocatalysts for the highly diastereo- and enantioselective direct anti-Mannich reactions of hydroxyacetone. Tetrahedron Lett. 2010, 51, 5775–5777. [Google Scholar] [CrossRef]
- Wu, C.; Fu, X.; Ma, X.; Li, S. One-step, efficient synthesis of combined threonine–surfactant organocatalysts for the highly enantioselective direct aldol reactions of cyclic ketones with aromatic aldehydes in the presence of water. Tetrahedron Asymmetry 2010, 21, 2465–2470. [Google Scholar] [CrossRef]
- Kagan, H.B.; Tabart, M. Chiralité et synthèse asymétrique en chimie thérapeutique. L'actual. Chim. 2015, 393, 31–38. [Google Scholar]
- Mac Millan, D. The advent and development of organocatalysis. Nature 2008, 455, 304–308. [Google Scholar] [CrossRef] [PubMed]
- Bauta, J.; Calbrix, E.; Capblancq, S.; Cecutti, C.; Peydecastaing, J.; Delgado Raynaud, C.; Rouilly, A.; Simon, V.; Vaca-Medina, G.; Vandenbosshe, V.; et al. Global chemical characterization of sargassum spp. seaweeds from different locations on Caribbean islands: A screening of organic compounds and heavy metals contents. Phycology 2024, 4, 190–212. [Google Scholar] [CrossRef]
- Giry, C.; Bertrand, D.; Cecutti, C.; Brossard, C.; Moreau, E.; Thiébaud-Roux, S.; Vaca-Garcia, C.; Vedrenne, E. Green Optimization of the First Steps for the Synthesis of a Novel Surfactant: Towards the Elimination of CMR Solvents and the Drastic Reduction of the Used Solvent Volume. ChemistrySelect 2019, 4, 8621–8625. [Google Scholar] [CrossRef]
- Giry, C.; Bertrand, D.; Pierret, A.; Vedrenne, E.; Lacaze-Dufaure, C.; Fabre, J.-F.; Thiébaud-Roux, S.; Vaca-Garcia, C.; Cecutti, C. Synthesis and Characterization of a New Organocatalytic Biosourced Surfactant. Sustain. Chem. 2021, 2, 335–342. [Google Scholar] [CrossRef]
- Paras, N.A.; MacMillan, D.W.C. The Enantioselective Organocatalytic 1,4-Addition of Electron-Rich Benzenes to α,β-Unsaturated Aldehydes. J. Am. Chem. Soc. 2002, 124, 7894–7895. [Google Scholar] [CrossRef] [PubMed]
- Shendage, D.M.; Fröhlich, R.; Haufe, G. Highly Efficient Stereoconservative Amidation and Deamidation of α-Amino Acids. Org. Lett. 2004, 6, 3675–3678. [Google Scholar] [CrossRef] [PubMed]
- Taveira, A.F.; Hyaric, M.L.; Reis, E.F.C.; Araújo, D.P.; Ferreira, A.P.; de Souza, M.A.; Alves, L.L.; Lourenço, M.C.S.; Vicente, F.R.C.; de Almeida, M.V. Preparation and Antitubercular Activities of Alkylated Amino Alcohols and Their Glycosylated Derivatives. Bioorg. Med. Chem. 2007, 15, 7789–7794. [Google Scholar] [CrossRef] [PubMed]
- Francavilla, C.; Turtle, E.D.; Kim, B.; O’Mahony, D.J.R.; Shiau, T.P.; Low, E.; Alvarez, N.J.; Celeri, C.E.; D’Lima, L.; Friedman, L.C.; et al. Novel N-Chloroheterocyclic Antimicrobials. Bioorg. Med. Chem. Lett. 2011, 21, 3029–3033. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Liu, H.; Bi, J.; Zhang, C.; Zhang, H.; Bai, C.; Hu, Y.; Zhang, X. Pyridine–Oxazoline and Quinoline–Oxazoline Ligated Cobalt Complexes: Synthesis, Characterization, and 1,3-Butadiene Polymerization Behaviors. Inorg. Chim. Acta 2015, 435, 305–312. [Google Scholar] [CrossRef]
- Ashworth, I.W.; Cox, B.G.; Meyrick, B. Kinetics and Mechanism of N-Boc Cleavage: Evidence of a Second-Order Dependence upon Acid Concentration. J. Org. Chem. 2010, 75, 8117–8125. [Google Scholar] [CrossRef]
- Teunissen, H.P. Hexadecylamine (Cetylamine). Recl. Trav. Chim. Pays-Bas 1927, 46, 208–211. [Google Scholar] [CrossRef]
- El-Dossoki, F.I.; Gomaa, E.A.; Hamza, O.K. Solvation Thermodynamic Parameters for Alkyl Benzyl Dimethyl Ammonium Chloride and Cetyl Trimethyl Ammonium Chloride Surfactants in Water and Alcoholic-Water Solvents. J. Chem. Eng. Data 2019, 64, 4482–4492. [Google Scholar] [CrossRef]
- Ralston, A.W.; Hoffman, E.J.; Hoerr, C.W.; Selby, W.M. Studies on High Molecular Weight Aliphatic Amines and their Salts. I. Behavior of the Hydrochlorides of Dodecylamine and Octadecylamine in Water. J. Am. Chem. Soc. 1941, 63, 1598–1601. [Google Scholar] [CrossRef]
- Church, J.; Willner, M.R.; Renfro, B.R.; Chen, Y.; Diaz, D.; Lee, W.H.; Dutcher, C.S.; Lundin, J.G.; Paynter, D.M. Impact of Interfacial Tension and Critical Micelle Concentration on Bilgewater Oil Separation. J. Water Process Eng. 2021, 39, 101684. [Google Scholar] [CrossRef]
- Blachechen, L.S.; Silva, J.O.; Barbosa, L.R.S.; Itri, R.; Petri, D.F.S. Hofmeister effects on the colloidal stability of poly(ethylene glycol)-decorated nanoparticles. Colloid Polym. Sci. 2012, 290, 1537–1546. [Google Scholar] [CrossRef]
- El-Aila Hisham, J.Y. Interaction of Nonionic Surfactant Triton-X-100 with Ionic Surfactants. J. Dispers. Sci. Technol. 2009, 30, 1277–1280. [Google Scholar] [CrossRef]
- Szymczyk, K.; Zdziennicka, A.; Jańczuk, B. Adsorption and Aggregation Properties of Some Polysorbates at Different Temperatures. J. Solut. Chem. 2018, 47, 1824–1840. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.K.; Bhattarai, A. Interfacial and Micellization Behavior of Cetyltrimethylammonium Bromide (CTAB) in Water and methanol-Water Mixture at 298.15 to 323.15 K. J. Chem. 2020, 280, 4653092. [Google Scholar] [CrossRef]
- Geng, F.; Yu, L.; Lu, T.; Li, Z.; Zheng, L.; Li, G. Studies on the Effects of Additional Components on Micellization of CTAB via Surface Tension Measurements. J. Dispers. Sci. Technol. 2008, 29, 1209–1213. [Google Scholar] [CrossRef]
- Man, Z.; Wu, W. Study on the Synthesis, Surface Activity, and Self-Assembly Behaviour of Anionic Non-Ionic Gemini Surfactants. Molecules 2024, 29, 1725. [Google Scholar] [CrossRef]
- Saito, T.; Hayamizu, K.; Yanagisawa, M.; Yamamoto, O.; Wasada, N.; Kinugasa, S.; Tanabe, K.; Tamura, T. Integrated Spectral Data Base System of Organic Compounds; National Institute of Advanced Industrial Science and Technology: Tokyo, Japan, 2004.
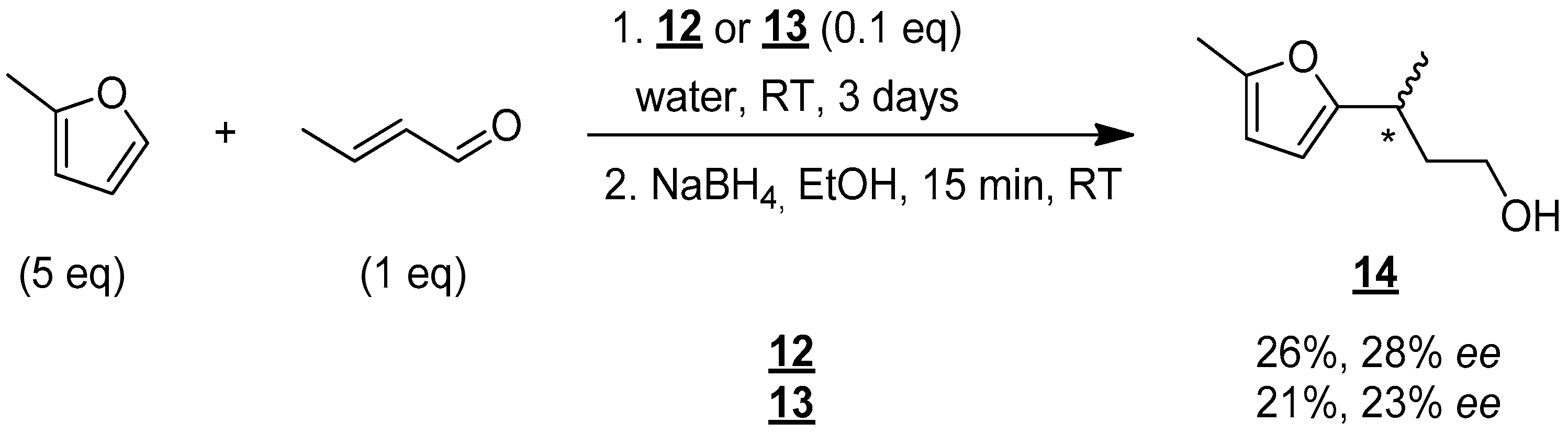
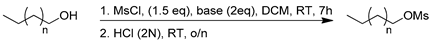
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).