
Synthetic and Functional Engineering of Bacteriophages: Approaches for Tailored Bactericidal, Diagnostic, and Delivery Platforms

 , , , , , , and
, , , , , , and 
Abstract
1. Introduction
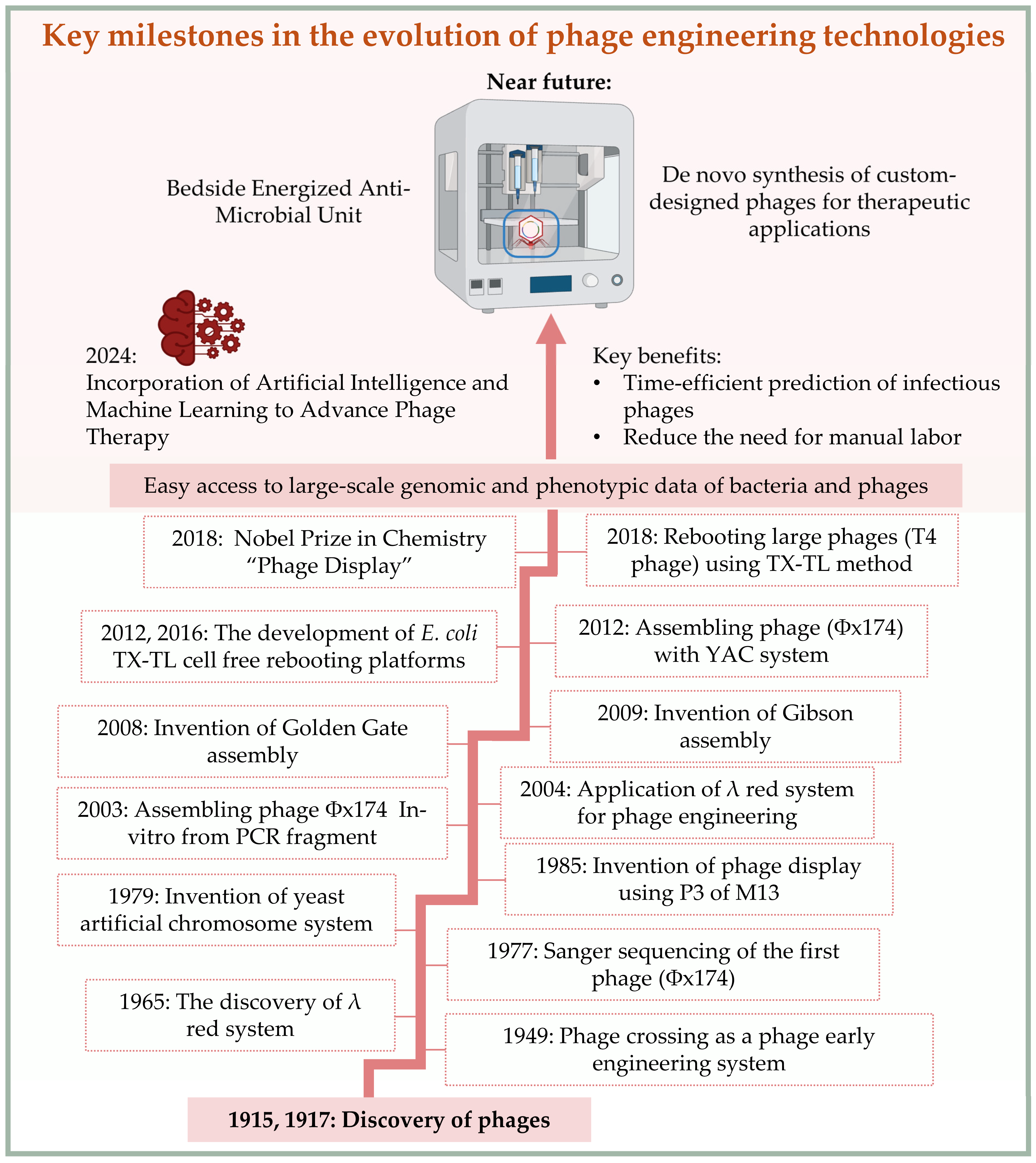
1.1. Historical Background of Bacteriophage Research
1.2. Modern Applications and the Need for Phage Engineering
2. Biological Foundations of Phage Engineering
2.1. Phage Structure and Morphology
2.2. Phage Life Cycles and Genome Packaging Mechanisms
2.3. Host Recognition and Engineering of Receptor-Binding Proteins

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phage Name | Phage Type | Original RBPs/Host | New RBPs/Host | Introduced Modification | Purpose and Application | References |
|---|---|---|---|---|---|---|
| λ | Temperate | LamB, OmpC | OmpC and LPS | P2-STF | Genetic engineering of bacteria in the mouse gut/Transduction capsid/dCas9 base editor | [22] |
| λ | Temperate | LamB, OmpC | OmpC and O-antigen (O157) | STF tail fiber that recognize O-antigen (O157) | Eliminate STEC/Transduction capsid/Cas12a | [23] |
| P2 | Temperate | LPS | Shigilla flexneri M90T, Escherichia coli O-antigen O157 | Hybrid long tail fiber genes (gpHG or gpH only) P1-S’ and P1-U’, or PhiV10 tail spike protein, respectively | Eliminate STEC/Transduction capsid/Cas9 | [35] |
| P2vir1 | Lytic | LPS | Salmonella (OmpC) | P2-gpH and S16-gp37 | Expand the Host range | [24] |
| T7 | Lytic | Rough LPS | - | Selective mutations in HRDRs of gp17 | Expand host range/Transduction capsid | [61] |
| T7 | Lytic | Rough LPS | Klebsiella | Swapping gp11, gp12, and gp17 with those of phage K11 | Expand host range | [76] |
| T3 | Lytic | - | Escherichia coli (BW25113) | Swapping T3-gp17 with T7-gp17 | Expand host range | [78] |
| α15 | Lytic | LPS | Tsx | Knock-In of gp38 from phage α17 | Expand host range/Cas delivery | [25] |
| Others | R-type pyocin | Pseudomonas | Escherichia coli O-antigen (O157) | Utilize the C-terminal of the phiV10 tail spike protein | Expand host range/Bacteria killing agent | [77] |
| Others | Nisin-nanoparticles | - | MRSA | RBPs of Staphylococcal phage Sb-1 | Expand host range/Bacteria killing agent | [42] |
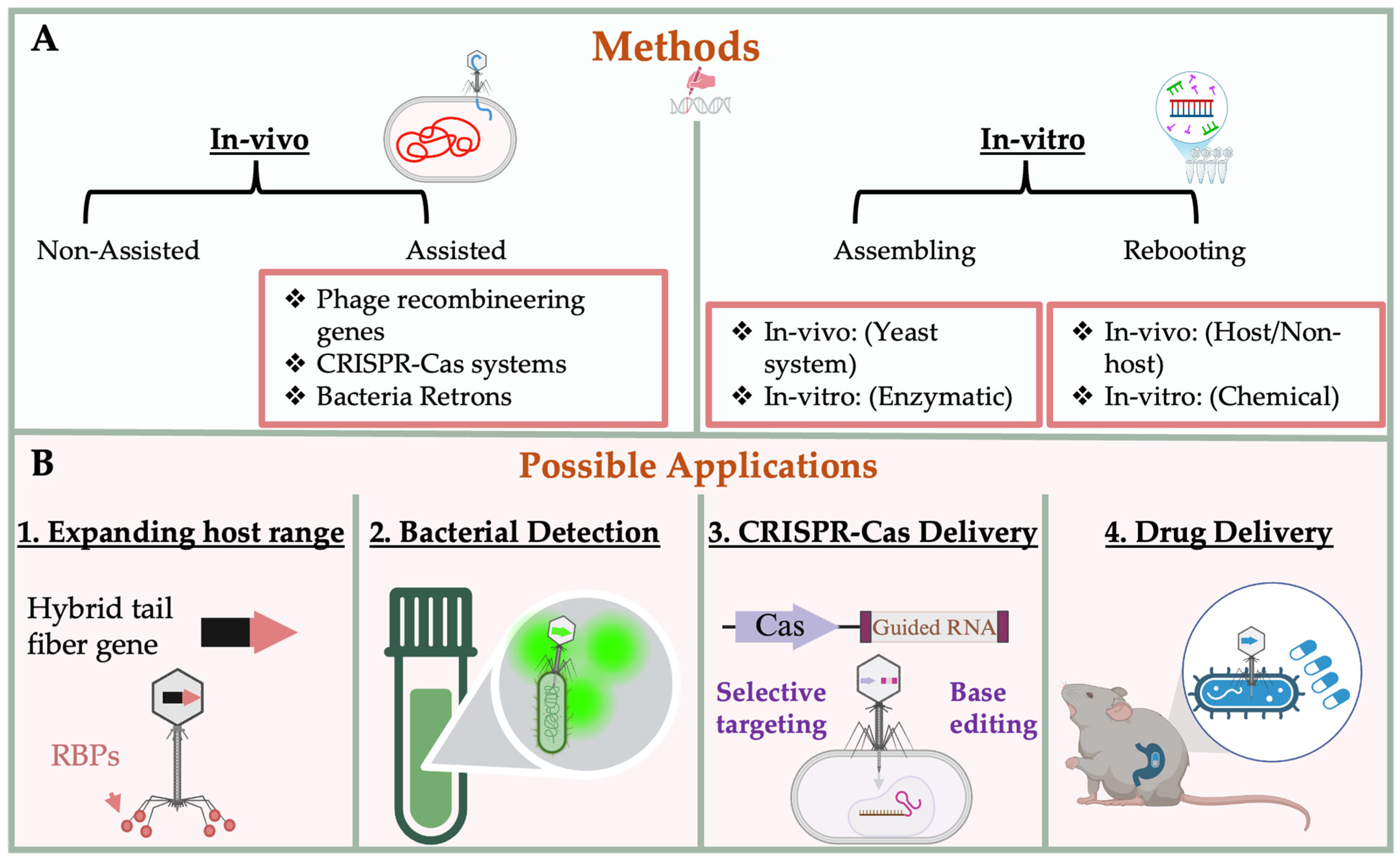
3. Engineering Strategies for Bacteriophages
3.1. Genetic Engineering Approaches
3.1.1. Phage Recombineering System
3.1.2. CRISPR-Cas-Assisted Phage Editing
3.1.3. Retron-Mediated Genome Modification
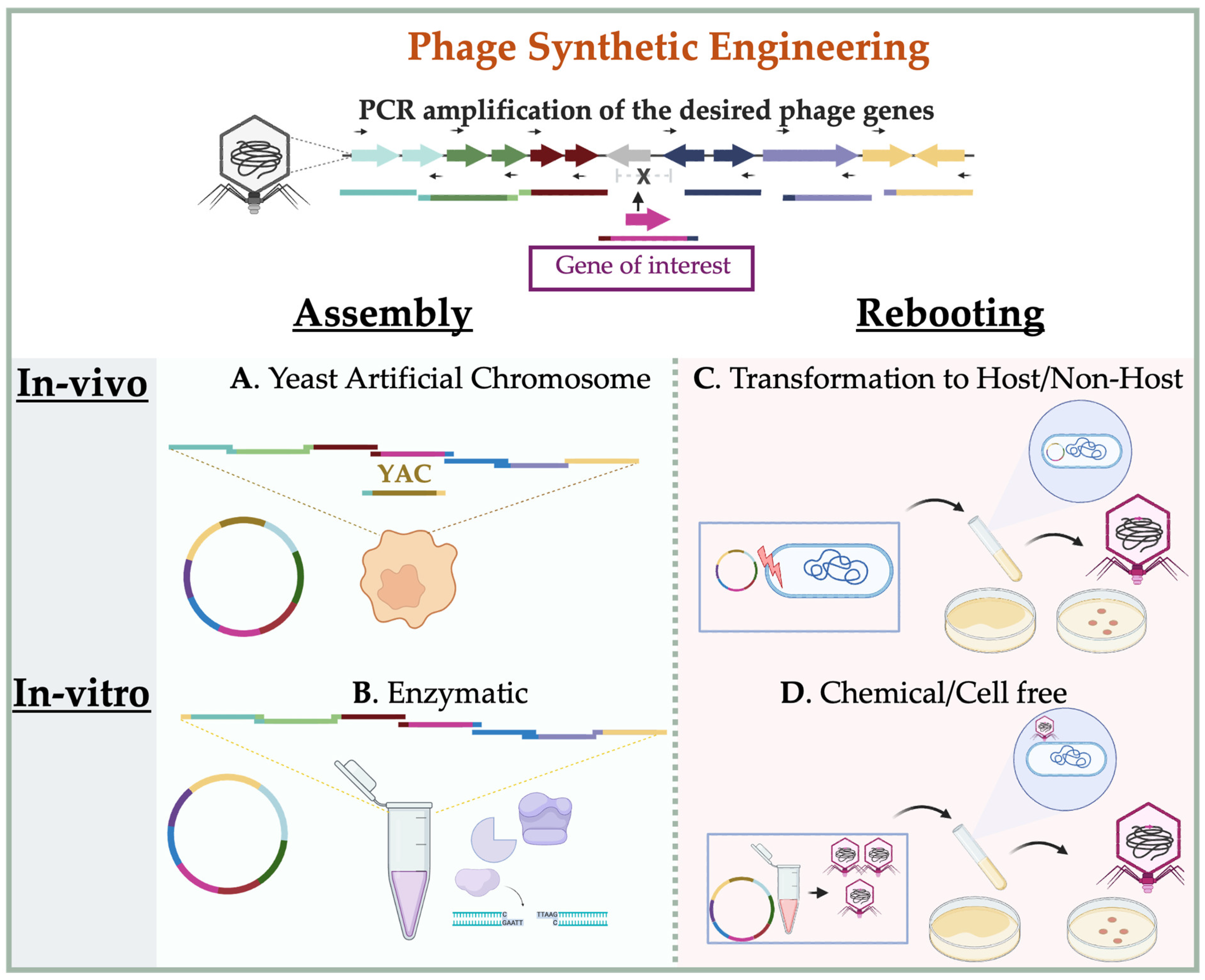
3.2. In-Vitro Synthetic Engineering Platforms
3.2.1. Synthetic Assembly of Phage Genomes
3.2.2. Rebooting Engineered Phage
4. Applications and Innovations in Engineered Phage Platforms
4.1. Host Range Expansion and Targeting
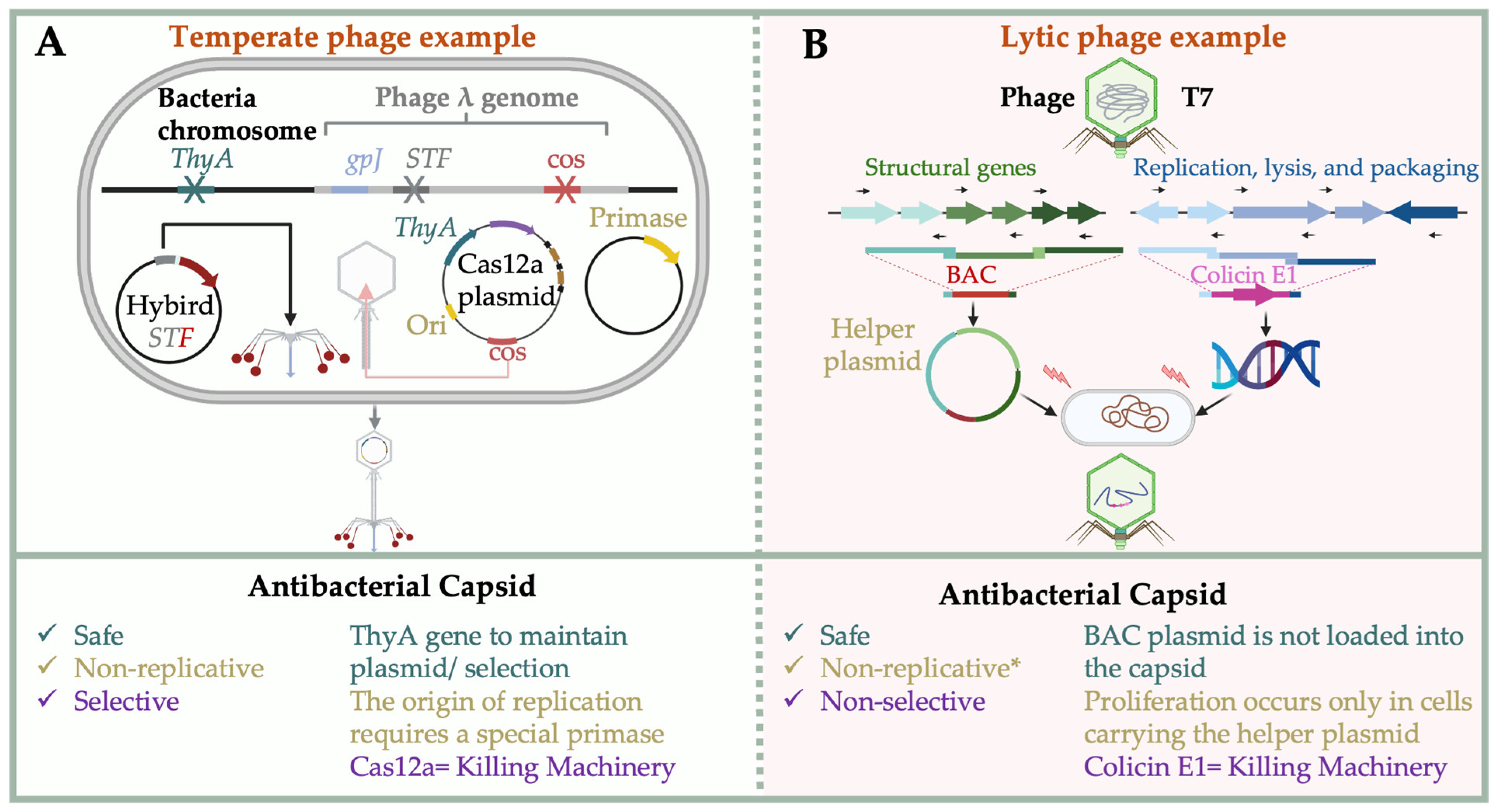
4.2. CRISPR-Cas Delivery via Phage Capsids
4.3. Biocontained Non-Replicative Phage Therapeutics
4.4. Diagnostic and Antimicrobial Payload Delivery
5. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hankin, M.E. L’action Bactéricide Des Eaux de La et Du Gange Sur Le Vibrion Du Choléra. Ann. Inst. Pasteur 1896, 10, 511–523. [Google Scholar] [CrossRef]
- Sulakvelidze, A.; Alavidze, Z.; Morris, J. Bacteriophage Therapy. Antimicrob. Agents Chemother. 2001, 45, 649–659. [Google Scholar] [CrossRef]
- Twort, F.W. An investigation on the nature of ultra-microscopic viruses. Lancet 1915, 186, 1241–1243. [Google Scholar] [CrossRef]
- D’Herelle, F. On an invisible microbe antagonistic toward dysenteric bacilli: Brief note by Mr. F. D’Herelle, presented by Mr. Roux. 1917. Res. Microbiol. 2007, 158, 553–554. [Google Scholar] [CrossRef]
- Wollman, E. The phenomenon of twort-d’hérelle and its significance. Lancet 1935, 226, 1312–1314. [Google Scholar] [CrossRef]
- Chanishvili, N. Phage Therapy—History from Twort and d’Herelle Through Soviet Experience to Current Approaches. Adv. Virus Res. 2012, 83, 3–40. [Google Scholar] [CrossRef] [PubMed]
- Peitzman, S.J. Félix d’Hérelle and the Origins of Molecular Biology (Review). Bull. Hist. Med. 2001, 75, 159–161. [Google Scholar] [CrossRef]
- Summers, W.C. The Strange History of Phage Therapy. Bacteriophage 2012, 2, 130–133. [Google Scholar] [CrossRef]
- Hershey, A.D.; Chase, M.M. Independent Functions of Viral Protein and Nucleic Acid in Growth of Bacteriophage. J. Gen. Physiol. 1952, 36, 39–56. [Google Scholar] [CrossRef]
- Van Valen, D.; Wu, D.; Chen, Y.J.; Tuson, H.; Wiggins, P.; Phillips, R. A Single-Molecule Hershey-Chase Experiment. Curr. Biol. 2012, 22, 1339–1343. [Google Scholar] [CrossRef]
- Hershey, A.D.; Rotman, R. Genetic Recombination between Host-Range and Plaque-Type Mutants of Bacteriophage in Single Bacterial Cells. Genetics 1949, 34, 44. [Google Scholar] [CrossRef]
- Harshey, R.M. The Mu Story: How a Maverick Phage Moved the Field Forward. Mob. DNA 2012, 3, 21. [Google Scholar] [CrossRef] [PubMed]
- Pingoud, A.; Wilson, G.G.; Wende, W. Type II Restriction Endonucleases—A Historical Perspective and More. Nucleic Acids Res. 2014, 42, 7489–7527. [Google Scholar] [CrossRef] [PubMed]
- Rossi, R.; Montecucco, A.; Ciarrocchi, G.; Biamonti, G. Functional Characterization of the T4 DNA Ligase: A New Insight into the Mechanism of Action; Oxford University Press: Oxford, UK, 1997; Volume 25. [Google Scholar] [CrossRef]
- Sanger, F.; Air, G.M.; Barrell, B.G.; Brown, N.L.; Coulson, A.R.; Fiddes, J.C.; Hutchison, C.A.; Slocombe, P.M.; Smith, M. Nucleotide Sequence of Bacteriophage Φx174 DNA. Nature 1977, 265, 687–695. [Google Scholar] [CrossRef] [PubMed]
- Navarro, F.; Muniesa, M. Phages in the Human Body. Front. Microbiol. 2017, 8, 566. [Google Scholar] [CrossRef]
- Breitbart, M.; Rohwer, F. Here a Virus, There a Virus, Everywhere the Same Virus? Trends Microbiol. 2005, 13, 278–284. [Google Scholar] [CrossRef]
- Dion, M.B.; Oechslin, F.; Moineau, S. Phage Diversity, Genomics and Phylogeny. Nat. Rev. Microbiol. 2020, 18, 125–138. [Google Scholar] [CrossRef]
- Uyttebroek, S.; Chen, B.; Onsea, J.; Ruythooren, F.; Debaveye, Y.; Devolder, D.; Spriet, I.; Depypere, M.; Wagemans, J.; Lavigne, R.; et al. Safety and Efficacy of Phage Therapy in Difficult-to-Treat Infections: A Systematic Review. Lancet Infect. Dis. 2022, 22, e208–e220. [Google Scholar] [CrossRef]
- Azam, A.H.; Tan, X.E.; Veeranarayanan, S.; Kiga, K.; Cui, L. Bacteriophage Technology and Modern Medicine. Antibiotics 2021, 10, 999. [Google Scholar] [CrossRef]
- Pirnay, J.P.; Djebara, S.; Steurs, G.; Griselain, J.; Cochez, C.; De Soir, S.; Glonti, T.; Spiessens, A.; Vanden Berghe, E.; Green, S.; et al. Personalized Bacteriophage Therapy Outcomes for 100 Consecutive Cases: A Multicentre, Multinational, Retrospective Observational Study. Nat. Microbiol. 2024, 9, 1434–1453. [Google Scholar] [CrossRef]
- Brödel, A.K.; Charpenay, L.H.; Galtier, M.; Fuche, F.J.; Terrasse, R.; Poquet, C.; Havránek, J.; Pignotti, S.; Krawczyk, A.; Arraou, M.; et al. In Situ Targeted Base Editing of Bacteria in the Mouse Gut. Nature 2024, 632, 877–884. [Google Scholar] [CrossRef]
- Galtier, M.; Krawczyk, A.; Fuche, F.J.; Charpenay, L.H.; Stzepourginski, I.; Pignotti, S.; Arraou, M.; Terrasse, R.; Brödel, A.K.; Poquet, C.; et al. Treatment of STEC Infection via CRISPR-Cas Targeted Cleavage of the Shiga Toxin Gene in Animal Models. bioRxiv 2025. [Google Scholar] [CrossRef]
- Cunliffe, T.G.; Parker, A.L.; Jaramillo, A. Pseudotyping Bacteriophage P2 Tail Fibers to Extend the Host Range for Biomedical Applications. ACS Synth. Biol. 2022, 11, 3207–3215. [Google Scholar] [CrossRef] [PubMed]
- Gencay, Y.E.; Jasinskytė, D.; Robert, C.; Semsey, S.; Martínez, V.; Petersen, A.Ø.; Brunner, K.; de Santiago Torio, A.; Salazar, A.; Turcu, I.C.; et al. Engineered Phage with Antibacterial CRISPR–Cas Selectively Reduce E. coli Burden in Mice. Nat. Biotechnol. 2023, 42, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.K.; Collins, J.J. Dispersing Biofilms with Engineered Enzymatic Bacteriophage. Proc. Natl. Acad. Sci. USA 2007, 104, 11197–11202. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Deng, Z.; Tao, H.; Song, W.; Xing, B.; Liu, W.; Kong, L.; Yuan, S.; Ma, Y.; Wu, Y.; et al. Harnessing Stepping-Stone Hosts to Engineer, Select, and Reboot Synthetic Bacteriophages in One Pot. Cell Rep. Methods 2022, 2, 100217. [Google Scholar] [CrossRef]
- Eghbalpoor, F.; Gorji, M.; Alavigeh, M.Z.; Moghadam, M.T. Genetically Engineered Phages and Engineered Phage-Derived Enzymes to Destroy Biofilms of Antibiotics Resistance Bacteria. Heliyon 2024, 10, e35666. [Google Scholar] [CrossRef]
- Meile, S.; Du, J.; Staubli, S.; Grossmann, S.; Koliwer-Brandl, H.; Piffaretti, P.; Leitner, L.; Matter, C.I.; Baggenstos, J.; Hunold, L.; et al. Engineered Reporter Phages for Detection of Escherichia Coli, Enterococcus, and Klebsiella in Urine. Nat. Commun. 2023, 14, 4336. [Google Scholar] [CrossRef]
- Kim, J.; Kim, M.; Kim, S.; Ryu, S. Sensitive Detection of Viable Escherichia coli O157:H7 from Foods Using a Luciferase-Reporter Phage PhiV10lux. Int. J. Food Microbiol. 2017, 254, 11–17. [Google Scholar] [CrossRef]
- Tamura, A.; Azam, A.H.; Nakamura, T.; Lee, K.; Iyoda, S.; Kondo, K.; Ojima, S.; Chihara, K.; Yamashita, W.; Cui, L.; et al. Synthetic Phage-Based Approach for Sensitive and Specific Detection of Escherichia coli O157. Commun. Biol. 2024, 7, 535. [Google Scholar] [CrossRef]
- Pulkkinen, E.M.; Hinkley, T.C.; Nugen, S.R. Utilizing In Vitro DNA Assembly to Engineer a Synthetic T7 Nanoluc Reporter Phage for Escherichia coli Detection. Integr. Biol. 2019, 11, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Baker, Z.R.; Zhang, Y.; Zhang, H.; Franklin, H.C.; Serpa, P.B.S.; Southard, T.; Li, L.; Hsu, B.B. Sustained in Situ Protein Production and Release in the Mammalian Gut by an Engineered Bacteriophage. Nat. Biotechnol. 2025, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Dhasmana, N.; Ram, G.; McAllister, K.N.; Chupalova, Y.; Lopez, P.; Ross, H.F.; Novick, R.P. Dynamics of Antibacterial Drone Establishment in Staphylococcus Aureus: Unexpected Effects of Antibiotic Resistance Genes. mBio 2021, 12, e0208321. [Google Scholar] [CrossRef] [PubMed]
- Fa-Arun, J.; Huan, Y.W.; Darmon, E.; Wang, B. Tail-Engineered Phage P2 Enables Delivery of Antimicrobials into Multiple Gut Pathogens. ACS Synth. Biol. 2023, 12, 596–607. [Google Scholar] [CrossRef]
- Kiga, K.; Tan, X.E.; Ibarra-Chávez, R.; Watanabe, S.; Aiba, Y.; Sato’o, Y.; Li, F.Y.; Sasahara, T.; Cui, B.; Kawauchi, M.; et al. Development of CRISPR-Cas13a-Based Antimicrobials Capable of Sequence-Specific Killing of Target Bacteria. Nat. Commun. 2020, 11, 2934. [Google Scholar] [CrossRef]
- Shimamori, Y.; Tan, X.E.; Li, F.Y.; Nishikawa, Y.; Watanabe, S.; Sasahara, T.; Miyanaga, K.; Aiba, Y.; Veeranarayanan, S.; Thitiananpakorn, K.; et al. Efficient Synthesis of CRISPR-Cas13a-Antimicrobial Capsids against MRSA Facilitated by Silent Mutation Incorporation. Sci. Rep. 2024, 14, 16225. [Google Scholar] [CrossRef]
- Li, F.Y.; Tan, X.E.; Shimamori, Y.; Kiga, K.; Veeranarayanan, S.; Watanabe, S.; Nishikawa, Y.; Aiba, Y.; Sato’o, Y.; Miyanaga, K.; et al. Phagemid-Based Capsid System for CRISPR-Cas13a Antimicrobials Targeting Methicillin-Resistant Staphylococcus Aureus. Commun. Biol. 2024, 7, 1–11. [Google Scholar] [CrossRef]
- Mcgillin, M.; Tokman, J.I.; Hsu, E.; Alcaine, S.D.; Denes, T.G. Assessment of Resistance to Colicinogenic Synthetic Phage Antimicrobial System. Microbiol. Spectr. 2024, 12, e0079324. [Google Scholar] [CrossRef]
- Kiga, K.; Sato’o, Y.; Tan, X.-E.; Miyanaga, K.; Nguyen, H.M.; Li, F.-Y.; Azam, A.H.; Veeranarayanan, S.; Watanabe, S.; Aiba, Y.; et al. Development of a Non-Replicative Phage-Based DNA Delivery System and Its Application to Antimicrobial Therapies. PNAS Nexus 2025, 4, pgaf176. [Google Scholar] [CrossRef]
- Zhu, J.; Batra, H.; Ananthaswamy, N.; Mahalingam, M.; Tao, P.; Wu, X.; Guo, W.; Fokine, A.; Rao, V.B. Design of Bacteriophage T4-Based Artificial Viral Vectors for Human Genome Remodeling. Nat. Commun. 2023, 14, 2928. [Google Scholar] [CrossRef]
- Wan, H.; Zhong, X.; Yang, S.; Deng, J.; Song, X.; Liu, Y.; Li, Y.; Yin, Z.; Zhao, X. Enhancing the Therapeutic Potential of Peptide Antibiotics Using Bacteriophage Mimicry Strategies. Adv. Sci. 2024, 12, 2411753. [Google Scholar] [CrossRef]
- He, Y.; Chen, J. CRISPR/Cas9-Mediated Genome Editing of T4 Bacteriophage for High-Throughput Antimicrobial Susceptibility Testing. Anal. Chem. 2024, 96, 18301–18310. [Google Scholar] [CrossRef]
- Huan, Y.W.; Torraca, V.; Brown, R.; Fa-Arun, J.; Miles, S.L.; Oyarzún, D.A.; Mostowy, S.; Wang, B. P1 Bacteriophage-Enabled Delivery of CRISPR-Cas9 Antimicrobial Activity Against Shigella flexneri. ACS Synth. Biol. 2023, 12, 709–721. [Google Scholar] [CrossRef]
- Valencia-Toxqui, G.; Ramsey, J. How to Introduce a New Bacteriophage on the Block: A Short Guide to Phage Classification. J. Virol. 2024, 98, e0182123. [Google Scholar] [CrossRef]
- Ackermann, H.W. 5500 Phages Examined in the Electron Microscope. Arch. Virol. 2007, 152, 227–243. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.M.; Watanabe, S.; Sharmin, S.; Kawaguchi, T.; Tan, X.E.; Wannigama, D.L.; Cui, L. RNA and Single-Stranded DNA Phages: Unveiling the Promise from the Underexplored World of Viruses. Int. J. Mol. Sci. 2023, 24, 17029. [Google Scholar] [CrossRef] [PubMed]
- Albrycht, K.; Rynkiewicz, A.A.; Harasymczuk, M.; Barylski, J.; Zielezinski, A. Daily Reports on Phage-Host Interactions. Front. Microbiol. 2022, 13, 946070. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Gao, M. Jumbo Bacteriophages: An Overview. Front. Microbiol. 2017, 8, 403. [Google Scholar] [CrossRef]
- Michniewski, S.; Rihtman, B.; Cook, R.; Jones, M.A.; Wilson, W.H.; Scanlan, D.J.; Millard, A. A New Family of “Megaphages” Abundant in the Marine Environment. ISME Commun. 2021, 1, 58. [Google Scholar] [CrossRef]
- Zrelovs, N.; Dislers, A.; Kazaks, A. Motley Crew: Overview of the Currently Available Phage Diversity. Front. Microbiol. 2020, 11, 579452. [Google Scholar] [CrossRef]
- Olszak, T.; Latka, A.; Roszniowski, B.; Valvano, M.A.; Drulis-Kawa, Z. Phage Life Cycles Behind Bacterial Biodiversity. Curr. Med. Chem. 2017, 24, 3987–4001. [Google Scholar] [CrossRef]
- Shkoporov, A.N.; Khokhlova, E.V.; Stephens, N.; Hueston, C.; Seymour, S.; Hryckowian, A.J.; Scholz, D.; Ross, R.P.; Hill, C. Long-Term Persistence of CrAss-like Phage CrAss001 Is Associated with Phase Variation in Bacteroides Intestinalis. BMC Biol. 2021, 19, 163. [Google Scholar] [CrossRef] [PubMed]
- Dougherty, P.E.; Bernard, C.; Carstens, A.B.; Stanford, K.; McAllister, T.A.; Rocha, E.P.C.; Hansen, L.H. Persistent Virulent Phages Exist in Bacterial Isolates. bioRxiv 2025. [Google Scholar] [CrossRef]
- Łobocka, M.B.; Rose, D.J.; Plunkett, G.; Rusin, M.; Samojedny, A.; Lehnherr, H.; Yarmolinsky, M.B.; Blattner, F.R. Genome of Bacteriophage P1. J. Bacteriol. 2004, 186, 7032. [Google Scholar] [CrossRef] [PubMed]
- Marvin, D.A.; Symmons, M.F.; Straus, S.K. Structure and Assembly of Filamentous Bacteriophages. Prog. Biophys. Mol. Biol. 2014, 114, 80–122. [Google Scholar] [CrossRef] [PubMed]
- Fujisawa, H.; Morita, M. Phage DNA Packaging. Genes Cells 1997, 2, 537–545. [Google Scholar] [CrossRef]
- Casjens, S.R.; Gilcrease, E.B. Determining DNA Packaging Strategy by Analysis of the Termini of the Chromosomes in Tailed-Bacteriophage Virions. Methods Mol. Biol. 2009, 502, 91–111. [Google Scholar] [CrossRef]
- Merrill, B.D.; Ward, A.T.; Grose, J.H.; Hope, S. Software-Based Analysis of Bacteriophage Genomes, Physical Ends, and Packaging Strategies. BMC Genom. 2016, 17, 679. [Google Scholar] [CrossRef]
- Garneau, J.R.; Depardieu, F.; Fortier, L.C.; Bikard, D.; Monot, M. PhageTerm: A Tool for Fast and Accurate Determination of Phage Termini and Packaging Mechanism Using next-Generation Sequencing Data. Sci. Rep. 2017, 7, 8292. [Google Scholar] [CrossRef]
- Apjok, G.; Számel, M.; Christodoulou, C.; Seregi, V.; Vásárhelyi, B.M.; Stirling, T.; Eszenyi, B.; Sári, T.; Vidovics, F.; Nagrand, E.; et al. Characterization of Antibiotic Resistomes by Reprogrammed Bacteriophage-Enabled Functional Metagenomics in Clinical Strains. Nat. Microbiol. 2023, 8, 410–423. [Google Scholar] [CrossRef]
- Dunne, M.; Prokhorov, N.S.; Loessner, M.J.; Leiman, P.G. Reprogramming Bacteriophage Host Range: Design Principles and Strategies for Engineering Receptor Binding Proteins. Curr. Opin. Biotechnol. 2021, 68, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Taslem Mourosi, J.; Awe, A.; Guo, W.; Batra, H.; Ganesh, H.; Wu, X.; Zhu, J. Understanding Bacteriophage Tail Fiber Interaction with Host Surface Receptor: The Key “Blueprint” for Reprogramming Phage Host Range. Int. J. Mol. Sci. 2022, 23, 12146. [Google Scholar] [CrossRef] [PubMed]
- Leprince, A.; Mahillon, J. Phage Adsorption to Gram-Positive Bacteria. Viruses 2023, 15, 196. [Google Scholar] [CrossRef] [PubMed]
- Bennett, N.J.; Gagic, D.; Sutherland-Smith, A.J.; Rakonjac, J. Characterization of a Dual-Function Domain That Mediates Membrane Insertion and Excision of Ff Filamentous Bacteriophage. J. Mol. Biol. 2011, 411, 972–985. [Google Scholar] [CrossRef]
- Kleinbeck, F.; Kuhn, A. Membrane Insertion of the M13 Minor Coat Protein G3p Is Dependent on Yidc and the Secayeg Translocase. Viruses 2021, 13, 1414. [Google Scholar] [CrossRef]
- Chang, C.; Guo, W.; Yu, X.; Guo, C.; Zhou, N.; Guo, X.; Huang, R.L.; Li, Q.; Zhu, Y. Engineered M13 Phage as a Novel Therapeutic Bionanomaterial for Clinical Applications: From Tissue Regeneration to Cancer Therapy. Mater. Today Bio 2023, 20, 100612. [Google Scholar] [CrossRef]
- Raetz, C.R.H.; Whitfield, C. Lipopolysaccharide Endotoxins. Annu. Rev. Biochem. 2002, 71, 635–700. [Google Scholar] [CrossRef]
- Washizaki, A.; Yonesaki, T.; Otsuka, Y. Characterization of the Interactions between Escherichia coli Receptors, LPS and OmpC, and Bacteriophage T4 Long Tail Fibers. Microbiologyopen 2016, 5, 1003–1015. [Google Scholar] [CrossRef]
- Montag, D.; Schwarz, H.; Henning, U. A Component of the Side Tail Fiber of Escherichia Coli Bacteriophage Lambda Can Functionally Replace the Receptor-Recognizing Part of a Long Tail Fiber Protein of the Unrelated Bacteriophage T4. J. Bacteriol. 1989, 171, 4378–4384. [Google Scholar] [CrossRef]
- González-García, V.A.; Pulido-Cid, M.; Garcia-Doval, C.; Bocanegra, R.; Van Raaij, M.J.; Martín-Benito, J.; Cuervo, A.; Carrascosa, J.L. Conformational Changes Leading to T7 DNA Delivery upon Interaction with the Bacterial Receptor. J. Biol. Chem. 2015, 290, 10038–10044. [Google Scholar] [CrossRef]
- Witte, S.; Zinsli, L.V.; Gonzalez-Serrano, R.; Matter, C.I.; Loessner, M.J.; van Mierlo, J.T.; Dunne, M. Structural and Functional Characterization of the Receptor Binding Proteins of Escherichia coli O157 Phages EP75 and EP335. Comput. Struct. Biotechnol. J. 2021, 19, 3416–3426. [Google Scholar] [CrossRef]
- Oats, M.F.; Coronel-Aguilera, C.P.; Applegate, B.M.; Csonka, L.N.; Bhunia, A.K.; Gehring, A.G.; Paoli, G.C. Determination of the Infection Dynamics of Escherichia Coli O157:H7 by Bacteriophage ΦV10. Foods 2025, 14, 617. [Google Scholar] [CrossRef]
- Marti, R.; Zurfluh, K.; Hagens, S.; Pianezzi, J.; Klumpp, J.; Loessner, M.J. Long Tail Fibres of the Novel Broad-Host-Range T-Even Bacteriophage S16 Specifically Recognize Salmonella OmpC. Mol. Microbiol. 2013, 87, 818–834. [Google Scholar] [CrossRef]
- Baxa, U.; Steinbacher, S.; Miller, S.; Weintraub, A.; Huber, R.; Seckler, R. Interactions of Phage P22 Tails with Their Cellular Receptor, Salmonella O-Antigen Polysaccharide. Biophys. J. 1996, 71, 2040–2048. [Google Scholar] [CrossRef] [PubMed]
- Ando, H.; Lemire, S.; Pires, D.P.; Lu, T.K. Engineering Modular Viral Scaffolds for Targeted Bacterial Population Editing. Cell Syst. 2015, 1, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Scholl, D.; Cooley, M.; Williams, S.R.; Gebhart, D.; Martin, D.; Bates, A.; Mandrell, R. An Engineered R-Type Pyocin Is a Highly Specific and Sensitive Bactericidal Agent for the Food-Borne Pathogen Escherichia coli O157:H7. Antimicrob. Agents Chemother. 2009, 53, 3074–3080. [Google Scholar] [CrossRef] [PubMed]
- Mitsunaka, S.; Yamazaki, K.; Pramono, A.K.; Ikeuchi, M.; Kitao, T.; Ohara, N.; Kubori, T.; Nagai, H.; Ando, H. Synthetic Engineering and Biological Containment of Bacteriophages. Proc. Natl. Acad. Sci. USA 2022, 119, e2206739119. [Google Scholar] [CrossRef]
- Luria, S.E.; Dulbecco, R. Genetic Recombinations Leading to Production of Active Bacteriophage from Ultraviolet Inactivated Bacteriophage Particles. Genetics 1949, 34, 93. [Google Scholar] [CrossRef]
- Duong, M.M.; Carmody, C.M.; Ma, Q.; Peters, J.E.; Nugen, S.R. Optimization of T4 Phage Engineering via CRISPR/Cas9. Sci. Rep. 2020, 10, 18229. [Google Scholar] [CrossRef]
- Filsinger, G.T.; Wannier, T.M.; Pedersen, F.B.; Lutz, I.D.; Zhang, J.; Stork, D.A.; Debnath, A.; Gozzi, K.; Kuchwara, H.; Volf, V.; et al. Characterizing the Portability of Phage-Encoded Homologous Recombination Proteins. Nat. Chem. Biol. 2021, 17, 394–402. [Google Scholar] [CrossRef]
- Fishman, C.B.; Crawford, K.D.; Bhattarai-Kline, S.; Poola, D.; Zhang, K.; González-Delgado, A.; Rojas-Montero, M.; Shipman, S.L. Continuous Multiplexed Phage Genome Editing Using Recombitrons. Nat. Biotechnol. 2024, 1–12. [Google Scholar] [CrossRef]
- Marinelli, L.J.; Piuri, M.; Swigoňová, Z.; Balachandran, A.; Oldfield, L.M.; van Kessel, J.C.; Hatfull, G.F. BRED: A Simple and Powerful Tool for Constructing Mutant and Recombinant Bacteriophage Genomes. PLoS ONE 2008, 3, e3957. [Google Scholar] [CrossRef]
- Poteete, A.R. What Makes the Bacteriophage λ Red System Useful for Genetic Engineering: Molecular Mechanism and Biological Function. FEMS Microbiol. Lett. 2001, 201, 9–14. [Google Scholar] [CrossRef]
- Clark, A.J.; Margulies, A.D. Isolation and characterization of recombination-deficient mutants of Escherichia coli K12. Proc. Natl. Acad. Sci. USA 1965, 53, 451–459. [Google Scholar] [CrossRef]
- Maresca, M.; Erler, A.; Fu, J.; Friedrich, A.; Zhang, Y.; Stewart, A.F. Single-Stranded Heteroduplex Intermediates in λ Red Homologous Recombination. BMC Mol. Biol. 2010, 11, 54. [Google Scholar] [CrossRef]
- Marinelli, L.J.; Hatfull, G.F.; Piuri, M. Recombineering. Bacteriophage 2012, 2, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, K.; Rutvisuttinunt, W.; Scott, W.; Myers, R.S. The Enzymatic Basis of Processivity in Lambda Exonuclease. Nucleic Acids Res. 2003, 31, 1585–1596. [Google Scholar] [CrossRef] [PubMed]
- Erler, A.; Wegmann, S.; Elie-Caille, C.; Bradshaw, C.R.; Maresca, M.; Seidel, R.; Habermann, B.; Muller, D.J.; Stewart, A.F. Conformational Adaptability of Redβ during DNA Annealing and Implications for Its Structural Relationship with Rad52. J. Mol. Biol. 2009, 391, 586–598. [Google Scholar] [CrossRef] [PubMed]
- Court, R.; Cook, N.; Saikrishnan, K.; Wigley, D. The Crystal Structure of Lambda-Gam Protein Suggests a Model for RecBCD Inhibition. J. Mol. Biol. 2007, 371, 25–33. [Google Scholar] [CrossRef]
- Datsenko, K.A.; Wanner, B.L. One-Step Inactivation of Chromosomal Genes in Escherichia coli K-12 Using PCR Products. Proc. Natl. Acad. Sci. USA 2000, 97, 6640–6645. [Google Scholar] [CrossRef]
- Karlinsey, J.E. Λ-Red Genetic Engineering in Salmonella Enterica Serovar Typhimurium. Methods Enzymol. 2007, 421, 199–209. [Google Scholar] [CrossRef]
- Zhang, Y.; Buchholz, F.; Muyrers, J.P.P.; Francis Stewart, A. A New Logic for DNA Engineering Using Recombination in Escherichia coli. Nat. Genet. 1998, 20, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Oppenheim, A.B.; Rattray, A.J.; Bubunenko, M.; Thomason, L.C.; Court, D.L. In Vivo Recombineering of Bacteriophage λ by PCR Fragments and Single-Strand Oligonucleotides. Virology 2004, 319, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Serra-Moreno, R.; Acosta, S.; Hernalsteens, J.P.; Jofre, J.; Muniesa, M. Use of the Lambda Red Recombinase System to Produce Recombinant Prophages Carrying Antibiotic Resistance Genes. BMC Mol. Biol. 2006, 7, 31. [Google Scholar] [CrossRef] [PubMed]
- Fehér, T.; Karcagi, I.; Blattner, F.R.; Pósfai, G. Bacteriophage Recombineering in the Lytic State Using the Lambda Red Recombinases. Microb. Biotechnol. 2012, 5, 466–476. [Google Scholar] [CrossRef]
- Thomason, L.C.; Sawitzke, J.A.; Li, X.; Costantino, N.; Court, D.L. Recombineering: Genetic Engineering in Bacteria Using Homologous Recombination. Curr. Protoc. Mol. Biol. 2014, 106, 1.16.1–1.16.39. [Google Scholar] [CrossRef]
- Isaev, A.; Andriianov, A.; Znobishcheva, E.; Zorin, E.; Morozova, N.; Severinov, K. Editing of Phage Genomes—Recombineering-Assisted SpCas9 Modification of Model Coliphages T7, T5, and T3. Mol. Biol. 2022, 56, 801–815. [Google Scholar] [CrossRef]
- Derbise, A.; Lesic, B.; Dacheux, D.; Ghigo, J.M.; Carniel, E. A Rapid and Simple Method for Inactivating Chromosomal Genes in Yersinia. FEMS Immunol. Med. Microbiol. 2003, 38, 113–116. [Google Scholar] [CrossRef]
- Lesic, B.; Rahme, L.G. Use of the Lambda Red Recombinase System to Rapidly Generate Mutants in Pseudomonas Aeruginosa. BMC Mol. Biol. 2008, 9, 20. [Google Scholar] [CrossRef]
- Muyrers, J.P.P.; Zhang, Y.; Buchholz, F.; Stewart, A.F. RecE/RecT and Redα/Redβ Initiate Double-Stranded Break Repair by Specifically Interacting with Their Respective Partners. Genes Dev. 2000, 14, 1971. [Google Scholar] [CrossRef]
- Muyrers, J.P.P.; Zhang, Y.; Stewart, A.F. Techniques: Recombinogenic Engineering–New Options for Cloning and Manipulating DNA. Trends Biochem. Sci. 2001, 26, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Van Kessel, J.C.; Hatfull, G.F. Recombineering in Mycobacterium Tuberculosis. Nat. Methods 2006, 4, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Van Kessel, J.C.; Hatfull, G.F. Efficient Point Mutagenesis in Mycobacteria Using Single-Stranded DNA Recombineering: Characterization of Antimycobacterial Drug Targets. Mol. Microbiol. 2008, 67, 1094–1107. [Google Scholar] [CrossRef] [PubMed]
- Lopes, A.; Amarir-Bouhram, J.; Faure, G.; Petit, M.A.; Guerois, R. Detection of Novel Recombinases in Bacteriophage Genomes Unveils Rad52, Rad51 and Gp2.5 Remote Homologs. Nucleic Acids Res. 2010, 38, 3952–3962. [Google Scholar] [CrossRef]
- Swingle, B.; Bao, Z.; Markel, E.; Chambers, A.; Cartinhour, S. Recombineering Using RecTE from Pseudomonas Syringae. Appl. Environ. Microbiol. 2010, 76, 4960–4968. [Google Scholar] [CrossRef]
- Jacobs, W.R.; Snapper, S.B.; Tuckinan, M.; Bloom, B.R. Mycobacteriophage Vector Systems. Rev. Infect. Dis. 1989, 11, S404–S410. [Google Scholar] [CrossRef]
- Pearson, R.E.; Jurgensen, S.; Sarkis, G.J.; Hatfull, G.F.; Jacobs, W.R. Construction of D29 Shuttle Phasmids and Luciferase Reporter Phages for Detection of Mycobacteria. Gene 1996, 183, 129–136. [Google Scholar] [CrossRef]
- Sarkis, G.J.; Jacobs, W.R.; Hatfulll, G.F. L5 Luciferase Reporter Mycobacteriophages: A Sensitive Tool for the Detection and Assay of Live Mycobacteria. Mol. Microbiol. 1995, 15, 1055–1067. [Google Scholar] [CrossRef]
- Wetzel, K.S.; Guerrero-Bustamante, C.A.; Dedrick, R.M.; Ko, C.C.; Freeman, K.G.; Aull, H.G.; Divens, A.M.; Rock, J.M.; Zack, K.M.; Hatfull, G.F. CRISPY-BRED and CRISPY-BRIP: Efficient Bacteriophage Engineering. Sci. Rep. 2021, 11, 6796. [Google Scholar] [CrossRef]
- Yin, J.; Zheng, W.; Gao, Y.; Jiang, C.; Shi, H.; Diao, X.; Li, S.; Chen, H.; Wang, H.; Li, R.; et al. Single-Stranded DNA-Binding Protein and Exogenous RecBCD Inhibitors Enhance Phage-Derived Homologous Recombination in Pseudomonas. iScience 2019, 14, 1. [Google Scholar] [CrossRef]
- Bao, Z.; Cartinhour, S.; Swingle, B. Substrate and Target Sequence Length Influence RecTEPsy Recombineering Efficiency in Pseudomonas Syringae. PLoS ONE 2012, 7, e50617. [Google Scholar] [CrossRef]
- Zheng, W.; Xia, Y.; Wang, X.; Gao, S.; Zhou, D.; Fu, J.; Li, R.; Yin, J. Cascade-Cas3 Facilitates High-Accuracy Genome Engineering in Pseudomonas Using Phage-Encoded Homologous Recombination. Eng. Microbiol. 2022, 2, 100046. [Google Scholar] [CrossRef]
- Liang, R.; Liu, J. Scarless and Sequential Gene Modification in Pseudomonas Using PCR Product Flanked by Short Homology Regions. BMC Microbiol. 2010, 10, 209. [Google Scholar] [CrossRef]
- Chen, Y.; Yan, B.; Chen, W.; Zhang, X.; Liu, Z.; Zhang, Q.; Li, L.; Hu, M.; Zhao, X.; Xu, X.; et al. Development of the CRISPR-Cas12a System for Editing of Pseudomonas Aeruginosa Phages. iScience 2024, 27, 110210. [Google Scholar] [CrossRef] [PubMed]
- Yosef, I.; Goren, M.G.; Globus, R.; Molshanski-Mor, S.; Qimron, U. Extending the Host Range of Bacteriophage Particles for DNA Transduction. Mol. Cell 2017, 66, 721–728.e3. [Google Scholar] [CrossRef] [PubMed]
- Hatoum-Aslan, A. Phage Genetic Engineering Using CRISPR–Cas Systems. Viruses 2018, 10, 335. [Google Scholar] [CrossRef] [PubMed]
- Mahler, M.; Costa, A.R.; van Beljouw, S.P.B.; Fineran, P.C.; Brouns, S.J.J. Approaches for Bacteriophage Genome Engineering. Trends Biotechnol. 2023, 41, 669–685. [Google Scholar] [CrossRef]
- Hoshiga, F.; Yoshizaki, K.; Takao, N.; Miyanaga, K.; Tanji, Y. Modification of T2 Phage Infectivity toward Escherichia coli O157:H7 via Using CRISPR/Cas9. FEMS Microbiol. Lett. 2019, 366, 41. [Google Scholar] [CrossRef]
- Dionne, E.N.; Cornely, K. An Application of Mycobacteriophage Genome Engineering Using Bacteriophage Recombineering with Electroporated DNA (BRED) and CRISPR Cas-9 Systems. FASEB J. 2022, 36. [Google Scholar] [CrossRef]
- Choi, S.Y.; Romero-Calle, D.X.; Cho, H.G.; Bae, H.W.; Cho, Y.H. Use of Cas9 Targeting and Red Recombination for Designer Phage Engineering. J. Microbiol. 2024, 62, 1–10. [Google Scholar] [CrossRef]
- Heler, R.; Samai, P.; Modell, J.W.; Weiner, C.; Goldberg, G.W.; Bikard, D.; Marraffini, L.A. Cas9 Specifies Functional Viral Targets during CRISPR-Cas Adaptation. Nature 2015, 519, 199. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, P.; Jakimo, N.; Lee, J.; Amrani, N.; Rodríguez, T.; Koseki, S.R.T.; Tysinger, E.; Qing, R.; Hao, S.; Sontheimer, E.J.; et al. An Engineered ScCas9 with Broad PAM Range and High Specificity and Activity. Nat. Biotechnol. 2020, 38, 1154–1158. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Koseki, S.R.T.; Silverstein, R.A.; Amrani, N.; Peng, C.; Kramme, C.; Savic, N.; Pacesa, M.; Rodríguez, T.C.; Stan, T.; et al. PAM-Flexible Genome Editing with an Engineered Chimeric Cas9. Nat. Commun. 2023, 14, 6175. [Google Scholar] [CrossRef] [PubMed]
- Walton, R.T.; Christie, K.A.; Whittaker, M.N.; Kleinstiver, B.P. Unconstrained Genome Targeting with Near-PAMless Engineered CRISPR-Cas9 Variants. Science 2020, 368, 290–296. [Google Scholar] [CrossRef]
- Hibshman, G.N.; Bravo, J.P.K.; Hooper, M.M.; Dangerfield, T.L.; Zhang, H.; Finkelstein, I.J.; Johnson, K.A.; Taylor, D.W. Unraveling the Mechanisms of PAMless DNA Interrogation by SpRY-Cas9. Nat. Commun. 2024, 15, 3663. [Google Scholar] [CrossRef]
- Tao, P.; Wu, X.; Tang, W.C.; Zhu, J.; Rao, V. Engineering of Bacteriophage T4 Genome Using CRISPR-Cas9. ACS Synth. Biol. 2017, 6, 1952. [Google Scholar] [CrossRef]
- Fernbach, J.; Baggenstos, J.; Riedo, J.; McCallin, S.; Loessner, M.J.; Kilcher, S. CRISPR-Cas9 Enables Efficient Genome Engineering of the Strictly Lytic, Broad Host-Range Staphylococcal Bacteriophage K. bioRxiv 2024. [Google Scholar] [CrossRef]
- Shen, J.; Zhou, J.; Chen, G.-Q.; Xiu, Z.-L. Efficient Genome Engineering of a Virulent Klebsiella Bacteriophage Using CRISPR-Cas9. J. Virol. 2018, 92, e0053418. [Google Scholar] [CrossRef]
- Zetsche, B.; Gootenberg, J.S.; Abudayyeh, O.O.; Slaymaker, I.M.; Makarova, K.S.; Essletzbichler, P.; Volz, S.E.; Joung, J.; Van Der Oost, J.; Regev, A.; et al. Cpf1 Is a Single RNA-Guided Endonuclease of a Class 2 CRISPR-Cas System. Cell 2015, 163, 759–771. [Google Scholar] [CrossRef]
- Liu, Y.; Tao, P. Genetic Engineering of Bacteriophage Using CRISPR-Cas12a; Humana: New York, NY, USA, 2025; pp. 43–53. [Google Scholar] [CrossRef]
- Yuan, S.; Li, Y.; Kou, C.; Sun, Y.C.; Ma, Y. CRISPR/Cas12a-Based Genome Editing for Cyanophage of Anabeana sp. Synth. Syst. Biotechnol. 2025, 10, 140–147. [Google Scholar] [CrossRef]
- Abudayyeh, O.O.; Gootenberg, J.S.; Konermann, S.; Joung, J.; Slaymaker, I.M.; Cox, D.B.T.; Shmakov, S.; Makarova, K.S.; Semenova, E.; Minakhin, L.; et al. C2c2 Is a Single-Component Programmable RNA-Guided RNA-Targeting CRISPR Effector. Science 2016, 353, aaf5573. [Google Scholar] [CrossRef]
- Shmakov, S.; Abudayyeh, O.O.; Makarova, K.S.; Wolf, Y.I.; Gootenberg, J.S.; Semenova, E.; Minakhin, L.; Joung, J.; Konermann, S.; Severinov, K.; et al. Discovery and Functional Characterization of Diverse Class 2 CRISPR-Cas Systems. Mol. Cell 2015, 60, 385–397. [Google Scholar] [CrossRef] [PubMed]
- East-Seletsky, A.; O’Connell, M.R.; Knight, S.C.; Burstein, D.; Cate, J.H.D.; Tjian, R.; Doudna, J.A. Two Distinct RNase Activities of CRISPR-C2c2 Enable Guide-RNA Processing and RNA Detection. Nature 2016, 538, 270–273. [Google Scholar] [CrossRef] [PubMed]
- Adler, B.A.; Hessler, T.; Cress, B.F.; Lahiri, A.; Mutalik, V.K.; Barrangou, R.; Banfield, J.; Doudna, J.A. Broad-Spectrum CRISPR-Cas13a Enables Efficient Phage Genome Editing. Nat. Microbiol. 2022, 7, 1967–1979. [Google Scholar] [CrossRef] [PubMed]
- Guan, J.; Oromí-Bosch, A.; Mendoza, S.D.; Karambelkar, S.; Berry, J.D.; Bondy-Denomy, J. Bacteriophage Genome Engineering with CRISPR–Cas13a. Nat. Microbiol. 2022, 7, 1956–1966. [Google Scholar] [CrossRef]
- Yee, T.; Furuichi, T.; Inouye, S.; Inouye, M. Multicopy Single-Stranded DNA Isolated from a Gram-Negative Bacterium, Myxococcus Xanthus. Cell 1984, 38, 203–209. [Google Scholar] [CrossRef]
- Dhundale, A.; Lampson, B.; Furuichi, T.; Inouye, M.; Inouye, S. Structure of MsDNA from Myxococcus Xanthus: Evidence for a Long, Self-Annealing RNA Precursor for the Covalently Linked, Branched RNA. Cell 1987, 51, 1105–1112. [Google Scholar] [CrossRef]
- Hsu, M.Y.; Inouye, S.; Inouye, M. Structural Requirements of the RNA Precursor for the Biosynthesis of the Branched RNA-Linked Multicopy Single-Stranded DNA of Myxococcus Xanthus. J. Biol. Chem. 1989, 264, 6214–6219. [Google Scholar] [CrossRef]
- Lampson, B.C.; Inouye, M.; Inouye, S. Reverse Transcriptase with Concomitant Ribonuclease H Activity in the Cell-Free Synthesis of Branched RNA-Linked MsDNA of Myxococcus Xanthus. Cell 1989, 56, 701–707. [Google Scholar] [CrossRef]
- Lampson, B.; Inouye, M.; Inouye, S. The MsDNAs of Bacteria. Prog. Nucleic Acid. Res. Mol. Biol. 2001, 67, 65–91. [Google Scholar] [CrossRef]
- Simon, A.J.; Ellington, A.D.; Finkelstein, I.J. Retrons and Their Applications in Genome Engineering. Nucleic Acids Res. 2019, 47, 11007–11019. [Google Scholar] [CrossRef]
- Millman, A.; Bernheim, A.; Stokar-Avihail, A.; Fedorenko, T.; Voichek, M.; Leavitt, A.; Oppenheimer-Shaanan, Y.; Sorek, R. Bacterial Retrons Function In Anti-Phage Defense. Cell 2020, 183, 1551–1561.e12. [Google Scholar] [CrossRef] [PubMed]
- Lopez, S.C.; Crawford, K.D.; Lear, S.K.; Bhattarai-Kline, S.; Shipman, S.L. Precise Genome Editing across Kingdoms of Life Using Retron-Derived DNA. Nat. Chem. Biol. 2022, 18, 199–206. [Google Scholar] [CrossRef] [PubMed]
- González-Delgado, A.; Lopez, S.C.; Rojas-Montero, M.; Fishman, C.B.; Shipman, S.L. Simultaneous Multi-Site Editing of Individual Genomes Using Retron Arrays. Nat. Chem. Biol. 2024, 20, 1482–1492. [Google Scholar] [CrossRef] [PubMed]
- Jaschke, P.R.; Lieberman, E.K.; Rodriguez, J.; Sierra, A.; Endy, D. A Fully Decompressed Synthetic Bacteriophage ØX174 Genome Assembled and Archived in Yeast. Virology 2012, 434, 278–284. [Google Scholar] [CrossRef]
- Gibson, D.G.; Young, L.; Chuang, R.Y.; Venter, J.C.; Hutchison, C.A.; Smith, H.O. Enzymatic Assembly of DNA Molecules up to Several Hundred Kilobases. Nat. Methods 2009, 6, 343–345. [Google Scholar] [CrossRef]
- Engler, C.; Kandzia, R.; Marillonnet, S. A One Pot, One Step, Precision Cloning Method with High Throughput Capability. PLoS ONE 2008, 3, e3647. [Google Scholar] [CrossRef]
- Nozaki, S. Rapid and Accurate Assembly of Large DNA Assisted by In Vitro Packaging of Bacteriophage. ACS Synth. Biol. 2022, 11, 4113–4122. [Google Scholar] [CrossRef]
- Garenne, D.; Haines, M.C.; Romantseva, E.F.; Freemont, P.; Strychalski, E.A.; Noireaux, V. Cell-Free Gene Expression. Nat. Rev. Methods Primers 2021, 1, 49. [Google Scholar] [CrossRef]
- Shin, J.; Jardine, P.; Noireaux, V. Genome Replication, Synthesis, and Assembly of the Bacteriophage T7 in a Single Cell-Free Reaction. ACS Synth. Biol. 2012, 1, 408–413. [Google Scholar] [CrossRef]
- Rustad, M.; Eastlund, A.; Jardine, P.; Noireaux, V. Cell-Free TXTL Synthesis of Infectious Bacteriophage T4 in a Single Test Tube Reaction. Synth. Biol. 2018, 3, ysy002. [Google Scholar] [CrossRef]
- Burke, D.T.; Carle, G.F.; Olson, M.V. Cloning of Large Segments of Exogenous DNA into Yeast by Means of Artificial Chromosome Vectors. Science 1987, 236, 806–812. [Google Scholar] [CrossRef] [PubMed]
- Frazer, L.A.N.; O’Keefe, R.T. A New Series of Yeast Shuttle Vectors for the Recovery and Identification of Multiple Plasmids from Saccharomyces Cerevisiae. Yeast 2007, 24, 777–789. [Google Scholar] [CrossRef] [PubMed]
- Gibson, D.G.; Benders, G.A.; Axelrod, K.C.; Zaveri, J.; Algire, M.A.; Moodie, M.; Montague, M.G.; Venter, J.C.; Smith, H.O.; Hutchison, C.A. One-Step Assembly in Yeast of 25 Overlapping DNA Fragments to Form a Complete Synthetic Mycoplasma Genitalium Genome. Proc. Natl. Acad. Sci. USA 2008, 105, 20404–20409. [Google Scholar] [CrossRef] [PubMed]
- Brownstein, B.H.; Silverman, G.A.; Little, R.D.; Burke, D.T.; Korsmeyer, S.J.; Schlessinger, D.; Olson, M.V. Isolation of Single-Copy Human Genes from a Library of Yeast Artificial Chromosome Clones. Science 1989, 244, 1348–1351. [Google Scholar] [CrossRef]
- Ipoutcha, T.; Racharaks, R.; Huttelmaier, S.; Wilson, C.J.; Ozer, E.A.; Hartmann, E.M. A Synthetic Biology Approach to Assemble and Reboot Clinically Relevant Pseudomonas Aeruginosa Tailed Phages. Microbiol. Spectr. 2024, 12, e0289723. [Google Scholar] [CrossRef]
- Kristensen, C.S.; Petersen, A.Ø.; Kilstrup, M.; van der Helm, E.; Takos, A. Cell-Free Synthesis of Infective Phages from in Vitro Assembled Phage Genomes for Efficient Phage Engineering and Production of Large Phage Libraries. Synth. Biol. 2024, 9, 12. [Google Scholar] [CrossRef]
- Lu, C.; He, L.; Guo, Y.; Wang, T.; Ye, Y.; Lin, Z. Synthesis of Headful Packaging Phages Through Yeast Transformation-Associated Recombination. Viruses 2025, 17, 45. [Google Scholar] [CrossRef]
- Assad-Garcia, N.; D’Souza, R.; Buzzeo, R.; Tripathi, A.; Oldfield, L.M.; Vashee, S.; Fouts, D.E. Cross-Genus “Boot-Up” of Synthetic Bacteriophage in Staphylococcus Aureus by Using a New and Efficient DNA Transformation Method. Appl. Environ. Microbiol. 2022, 88, e0148621. [Google Scholar] [CrossRef]
- Gibson, D.G. Enzymatic Assembly of Overlapping DNA Fragments. Methods Enzymol. 2011, 498, 349–361. [Google Scholar] [CrossRef]
- Lin, Z.; Li, H.; He, L.; Jing, Y.; Pistolozzi, M.; Wang, T.; Ye, Y. Efficient Genome Editing for Pseudomonas Aeruginosa Using CRISPR-Cas12a. Gene 2021, 790, 145693. [Google Scholar] [CrossRef] [PubMed]
- Levrier, A.; Karpathakis, I.; Nash, B.; Bowden, S.D.; Lindner, A.B.; Noireaux, V. PHEIGES: All-Cell-Free Phage Synthesis and Selection from Engineered Genomes. Nat. Commun. 2024, 15, 2223. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.J.; Ripley, L.S. The Spectrum of Acridine Resistant Mutants of Bacteriophage T4 Reveals Cryptic Effects of the TsL141 DNA Polymerase Allele on Spontaneous Mutagenesis. Genetics 1998, 148, 1655. [Google Scholar] [CrossRef] [PubMed]
- Pryor, J.M.; Potapov, V.; Bilotti, K.; Pokhrel, N.; Lohman, G.J.S. Rapid 40 Kb Genome Construction from 52 Parts through Data-Optimized Assembly Design. ACS Synth. Biol. 2022, 11, 2036–2042. [Google Scholar] [CrossRef]
- Kilcher, S.; Studer, P.; Muessner, C.; Klumpp, J.; Loessner, M.J.; Adhya, S. Cross-Genus Rebooting of Custom-Made, Synthetic Bacteriophage Genomes in L-Form Bacteria. Proc. Natl. Acad. Sci. USA 2018, 115, 567–572. [Google Scholar] [CrossRef]
- Nirenberg, M.W.; Matthaei, J.H. The Dependence of Cell-Free Protein Synthesis in E. Coli upon Naturally Occurring or Synthetic Polyribonucleotides. Proc. Natl. Acad. Sci. USA 1961, 47, 1588–1602. [Google Scholar] [CrossRef]
- Carlson, E.D.; Gan, R.; Hodgman, C.E.; Jewett, M.C. Cell-Free Protein Synthesis: Applications Come of Age. Biotechnol. Adv. 2012, 30, 1185–1194. [Google Scholar] [CrossRef]
- Gregorio, N.E.; Levine, M.Z.; Oza, J.P. A User’s Guide to Cell-Free Protein Synthesis. Methods Protoc. 2019, 2, 24. [Google Scholar] [CrossRef]
- Pizarro-Bauerle, J.; Ando, H. Engineered Bacteriophages for Practical Applications. Biol. Pharm. Bull. 2020, 43, 240–249. [Google Scholar] [CrossRef]
- Kortright, K.E.; Chan, B.K.; Koff, J.L.; Turner, P.E. Phage Therapy: A Renewed Approach to Combat Antibiotic-Resistant Bacteria. Cell Host Microbe 2019, 25, 219–232. [Google Scholar] [CrossRef]
- Borin, J.M.; Avrani, S.; Barrick, J.E.; Petrie, K.L.; Meyer, J.R. Coevolutionary Phage Training Leads to Greater Bacterial Suppression and Delays the Evolution of Phage Resistance. Proc. Natl. Acad. Sci. USA 2021, 118, e2104592118. [Google Scholar] [CrossRef] [PubMed]
- Hodyra-Stefaniak, K.; Lahutta, K.; Majewska, J.; Kaźmierczak, Z.; Lecion, D.; Harhala, M.; Kęska, W.; Owczarek, B.; Jończyk-Matysiak, E.; Kłopot, A.; et al. Bacteriophages Engineered to Display Foreign Peptides May Become Short-Circulating Phages. Microb. Biotechnol. 2019, 12, 730–741. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.P.; Cha, J.D.; Jang, E.H.; Klumpp, J.; Hagens, S.; Hardt, W.D.; Lee, K.Y.; Loessner, M.J. PEGylation of Bacteriophages Increases Blood Circulation Time and Reduces T-Helper Type 1 Immune Response. Microb. Biotechnol. 2008, 1, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Dąbrowska, K.; Abedon, S.T. Pharmacologically Aware Phage Therapy: Pharmacodynamic and Pharmacokinetic Obstacles to Phage Antibacterial Action in Animal and Human Bodies. Microbiol. Mol. Biol. Rev. 2019, 83, e0001219. [Google Scholar] [CrossRef]
- Doud, M.B.; Robertson, J.M.; Strathdee, S.A. Optimizing Phage Therapy with Artificial Intelligence: A Perspective. Front. Cell. Infect. Microbiol. 2025, 15, 1611857. [Google Scholar] [CrossRef]
- Boeckaerts, D.; Stock, M.; Ferriol-González, C.; Oteo-Iglesias, J.; Sanjuán, R.; Domingo-Calap, P.; De Baets, B.; Briers, Y. Prediction of Klebsiella Phage-Host Specificity at the Strain Level. Nat. Commun. 2024, 15, 4355. [Google Scholar] [CrossRef]
- Gaborieau, B.; Vaysset, H.; Tesson, F.; Charachon, I.; Dib, N.; Bernier, J.; Dequidt, T.; Georjon, H.; Clermont, O.; Hersen, P.; et al. Prediction of Strain Level Phage–Host Interactions across the Escherichia Genus Using Only Genomic Information. Nat. Microbiol. 2024, 9, 2847–2861. [Google Scholar] [CrossRef]
- Pirnay, J.P. Phage Therapy in the Year 2035. Front. Microbiol. 2020, 11, 1171. [Google Scholar] [CrossRef]
- Cao, X.; Yu, T.; Sun, Z.; Chen, M.; Xie, W.; Pang, Q.; Deng, H. Engineered Phages in Anti-Infection and Anti-Tumor Fields: A Review. Microb. Pathog. 2025, 198, 107052. [Google Scholar] [CrossRef]
- Peng, H.; Chen, I.A.; Qimron, U. Engineering Phages to Fight Multidrug-Resistant Bacteria. Chem. Rev. 2025, 125, 933–971. [Google Scholar] [CrossRef]
- Woudstra, C.; Sørensen, A.N.; Sørensen, M.C.H.; Brøndsted, L. Strategies for Developing Phages into Novel Antimicrobial Tailocins. Trends Microbiol. 2024, 32, 996–1006. [Google Scholar] [CrossRef]
- Cui, L.; Watanabe, S.; Miyanaga, K.; Kiga, K.; Sasahara, T.; Aiba, Y.; Tan, X.-E.; Veeranarayanan, S.; Thitiananpakorn, K.; Nguyen, H.M.; et al. A Comprehensive Review on Phage Therapy and Phage-Based Drug Development. Antibiotics 2024, 13, 870. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Veeranarayanan, S.; Thitiananpakorn, K.; Wannigama, D.L. Bacteriophage Bioengineering: A Transformative Approach for Targeted Drug Discovery and Beyond. Pathogens 2023, 12, 1179. [Google Scholar] [CrossRef] [PubMed]
- Veeranarayanan, S.; Azam, A.H.; Kiga, K.; Watanabe, S.; Cui, L. Bacteriophages as Solid Tumor Theragnostic Agents. Int. J. Mol. Sci. 2021, 23, 402. [Google Scholar] [CrossRef] [PubMed]
- Pirnay, J.P.; Kutter, E. Bacteriophages: It’s a Medicine, Jim, but Not as We Know It. Lancet Infect. Dis. 2021, 21, 309–311. [Google Scholar] [CrossRef]
- Pirnay, J.P.; Blasdel, B.G.; Bretaudeau, L.; Buckling, A.; Chanishvili, N.; Clark, J.R.; Corte-Real, S.; Debarbieux, L.; Dublanchet, A.; De Vos, D.; et al. Quality and Safety Requirements for Sustainable Phage Therapy Products. Pharm. Res. 2015, 32, 2173–2179. [Google Scholar] [CrossRef]
- Cui, L.; Kiga, K.; Kondabagil, K.; Węgrzyn, A. Current and Future Directions in Bacteriophage Research for Developing Therapeutic Innovations. Sci. Rep. 2024, 14, 24404. [Google Scholar] [CrossRef]
- Smith, G.P. Filamentous Fusion Phage: Novel Expression Vectors That Display Cloned Antigens on the Virion Surface. Science 1985, 228, 1315–1317. [Google Scholar] [CrossRef]
- Smith, H.O.; Hutchison, C.A.; Pfannkoch, C.; Venter, J.C. Generating a Synthetic Genome by Whole Genome Assembly: ΦX174 Bacteriophage from Synthetic Oligonucleotides. Proc. Natl. Acad. Sci. USA 2003, 100, 15440–15445. [Google Scholar] [CrossRef]
- Garamella, J.; Marshall, R.; Rustad, M.; Noireaux, V. The All E. coli TX-TL Toolbox 2.0: A Platform for Cell-Free Synthetic Biology. ACS Synth. Biol. 2016, 5, 344–355. [Google Scholar] [CrossRef]

| Function | Phage Name | Cargo | Targeted Host | Phage Feature | References |
|---|---|---|---|---|---|
| Antibiofilm | CPB0329 | Dispersin B | Klebsiella pneumoniae | Replicative phage particles | [27] |
| Antimicrobial agent | T7 | Colicin E1 and Colicin M | Escherichia coli | Replicative phage particles | [39] |
| Antimicrobial agent | T7 | Colicin E1 | Escherichia coli | Non-replicative particles * | [40] |
| Antimicrobial agent | T-even like phages | Cas (type I-E) | Pathogenic Escherichia coli | Replicative phage particles | [25] |
| Bacteria detection | T4 | LacZα | Escherichia coli | Replicative phage particles | [43] |
| Bacteria detection | T7 | NanoLuc luciferse | Escherichia coli | Replicative phage particles | [32] |
| Bacteria detection | PhiV10 | luxCDABE | Escherichia coli (Food samples) | Replicative phage particles | [30] |
| Bacteria detection | E2, E4, EfS3, EfS7, K1and K4 | NanoLuc luciferse | Escherichia coli, Enterococcus spp., and Klebsiella spp. (Urine samples) | Replicative phage particles | [29] |
| Bacteria detection | vB_Eco4M-7 | HiBiT | STEC | Replicative phage particles | [31] |
| Bacteria genetic engineering | λ | Base editor dCas9 | Escherichia coli (β-lactamase) | Non-replicative particles | [22] |
| Delivery of antimicrobial peptide | Sb-1 | Nisin | MRSA | Phage structural components (Tail) | [42] |
| Drug delivery system for mammalian gut | T4 | Serpine B1a, Chaperone protein clpB | Nonpathogenic Escherichia coli | Replicative phage particles | [33] |
| Gene delivery system for the human cells | T4 | Gene editing, in situ protein expression and others | - | Phage structural components (Head) | [41] |
| Selective antimicrobial agent | λ | Cas12a | STEC | Non-replicative particles | [23] |
| Selective antimicrobial agent | P1 | Cas9 | Escherichia coli | Non-replicative particles | [44] |
| Selective antimicrobial agent | Phi80, M13, 80α and Tan2 | Cas13a | Escherichia coli and Staphylococcus aureus | Non-replicative particles | [36,37,38] |
| Selective antimicrobial agent | P2 | Cas9 | STEC and Shigella flexneri | Non-replicative particles | [35] |
| Phage General Information. | Phage Engineering Method. | In-Vivo Engineering Conditions. | In-Vitro (Synthetic) Engineering Conditions. | Engineering Purpose. | Reference | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Phage Host. | Phage Name. | Recombineering Genes. | Counter Selection Method. | Assembly Method. | Rebooting Method. | Rebooting Host. | Intermediate Host. | Phage Genome Size Kbp. | Modification. | Purpose. | ||
| Escherichia coli | Phage α15 | In-vivo | -. | - | - | - | - | - | - | Gene replacement of ~7 kp | Load Cas (type I-E) and gene for Tsx-binding adhesin | [25] |
| Escherichia coli | T7 | In-vivo | Flippase | Induced Phenotype *1 | - | - | BW25113ΔtrxA | - | - | Gene replacement | Tail fiber modification | [116] |
| Escherichia coli | T4 | In-vivo | - | Induced Phenotype *2 | - | - | - | - | - | Gene replacement | In situ protein expression within mammalian cells | [33] |
| Escherichia coli | T4 | In-vivo | - | CRISPR/Cas9 | - | - | - | - | - | NanoLuc luciferase. | Reporter gene | [80] |
| Escherichia coli | T4 | In-vivo *3 | - | CRISPR/Cas9 or Cas12 | - | - | - | - | - | Eliminate phage DNA packaging to create an empty head. | Load various cargoes to human cells | [41] |
| Escherichia coli | T3, T7, and T5 | In-vivo | λ-red | CRISPR/Cas9 | - | - | - | - | - | Point substitutions, insertions, or deletions. | Tail fiber modification | [98] |
| Escherichia coli | P1 | In-vivo | λ-red | Selection Marker | - | - | - | - | - | Deletion of packaging region Δpac of plasmid phage P1 | Phage capsid construction | [44] |
| Escherichia coli | λ. | In-vivo | λ-red | CRISPR/Cas9 | - | - | - | - | - | Deletion | Tail fiber modification | [22] |
| Klebsiella pneumonia. | T7 family and non-family Klebsiella pneumoniae phages. | In-vivo | λ-red | CRISPR/Cas9 | - | In-vivo | Escherichia coli DH10B | Yes | 41 to 46 | Either gene replacement of a non-essential ligase gene with dispersin B (DspB) or just gene insertion of the mentioned gene DspB | Distribute biofilm | [27] |
| Anabaena | Cyanophage A-1(L) and A-4(L) | In-vivo | - | CRISPR/Cas12a | - | - | - | - | - | Deletion | Minimize genome reduction of 2.4 kbp | [132] |
| Pseudomonas aeruginosa | -. | In-vivo | λ-red | CRISPR/Cas12a | - | - | - | - | - | Deletion | 15 kbp deletion | [163] |
| Pseudomonas aeruginosa. | KZ | In-vivo | - | CRISPR/Cas13a and acrVIA1 | - | - | - | - | - | Insertion, deletion and fluorescent tagging | - | [137] |
| Escherichia coli. | T4, T7 and EdH4 | In-vivo. | - | CRISPR/Cas13a | - | - | - | - | - | Multi gene deletion and single base modification | - | [136] |
| Escherichia coli. | T7 | In-vivo. | - | Recombitrons | - | - | - | - | - | Amino acid substitutions in gp17 gene | Expand host range | [82] |
| Escherichia coli. | T7 | Synthetic | - | - | NEBuilder HiFi DNA | In-vivo | Escherichia coli 10G | No | 39.937 + 0.977 | Insertion of NanoLuc luciferase | Reporter gene | [32] |
| Escherichia coli, Klebsiella and Yersinia. | T7 family | Synthetic | - | - | YAC | In-vivo | Escherichia cloni 10G | Yes | 37 to 45 | Modify Tail fiber | Expand host range | [76] |
| Escherichia coli. | T7 | Synthetic | - | - | Exonuclease only | TXTL | - | - | 39 | Modify Tail fiber | Expand host range | [164] |
| Salmonella. | P22 | Synthetic | - | - | Gibson | In-vivo | Salmonella Typhimurium strain LT2 | - | - | Deletion of lytic cycle repressor c2 | Modify lifestyle | [78] |
| Mycobacterium. | D29 | Synthetic | - | - | Gibson | In-vivo | M. smegmatis mc2155 | - | - | Gene replacement and insertion of NanoLuc luciferase gene | Reporter | |
| Escherichia coli | T7 | Synthetic | - | - | Gibson | TXTL | - | - | - | Gene replacement and insertion of LacZ operon | Reporter | |
| Salmonella | SP6 | Synthetic | - | - | Gibson | In-vivo | Salmonella Typhimurium strain LT2 | - | - | Deletion of phage head | Biocontained Phages | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alessa, O.; Aiba, Y.; Arbaah, M.; Hidaka, Y.; Watanabe, S.; Miyanaga, K.; Wannigama, D.L.; Cui, L. Synthetic and Functional Engineering of Bacteriophages: Approaches for Tailored Bactericidal, Diagnostic, and Delivery Platforms. Molecules 2025, 30, 3132. https://doi.org/10.3390/molecules30153132
Alessa O, Aiba Y, Arbaah M, Hidaka Y, Watanabe S, Miyanaga K, Wannigama DL, Cui L. Synthetic and Functional Engineering of Bacteriophages: Approaches for Tailored Bactericidal, Diagnostic, and Delivery Platforms. Molecules. 2025; 30(15):3132. https://doi.org/10.3390/molecules30153132
Chicago/Turabian StyleAlessa, Ola, Yoshifumi Aiba, Mahmoud Arbaah, Yuya Hidaka, Shinya Watanabe, Kazuhiko Miyanaga, Dhammika Leshan Wannigama, and Longzhu Cui. 2025. "Synthetic and Functional Engineering of Bacteriophages: Approaches for Tailored Bactericidal, Diagnostic, and Delivery Platforms" Molecules 30, no. 15: 3132. https://doi.org/10.3390/molecules30153132
APA StyleAlessa, O., Aiba, Y., Arbaah, M., Hidaka, Y., Watanabe, S., Miyanaga, K., Wannigama, D. L., & Cui, L. (2025). Synthetic and Functional Engineering of Bacteriophages: Approaches for Tailored Bactericidal, Diagnostic, and Delivery Platforms. Molecules, 30(15), 3132. https://doi.org/10.3390/molecules30153132