
Synthesis, Characterization, HSA/DNA Binding, and Cytotoxic Activity of [RuCl2(η6-p-cymene)(bph-κN)] Complex

 ,
,  ,
,  , ,
, ,  , , ,
, , ,  and
and 
Abstract
1. Introduction
2. Results and Discussion
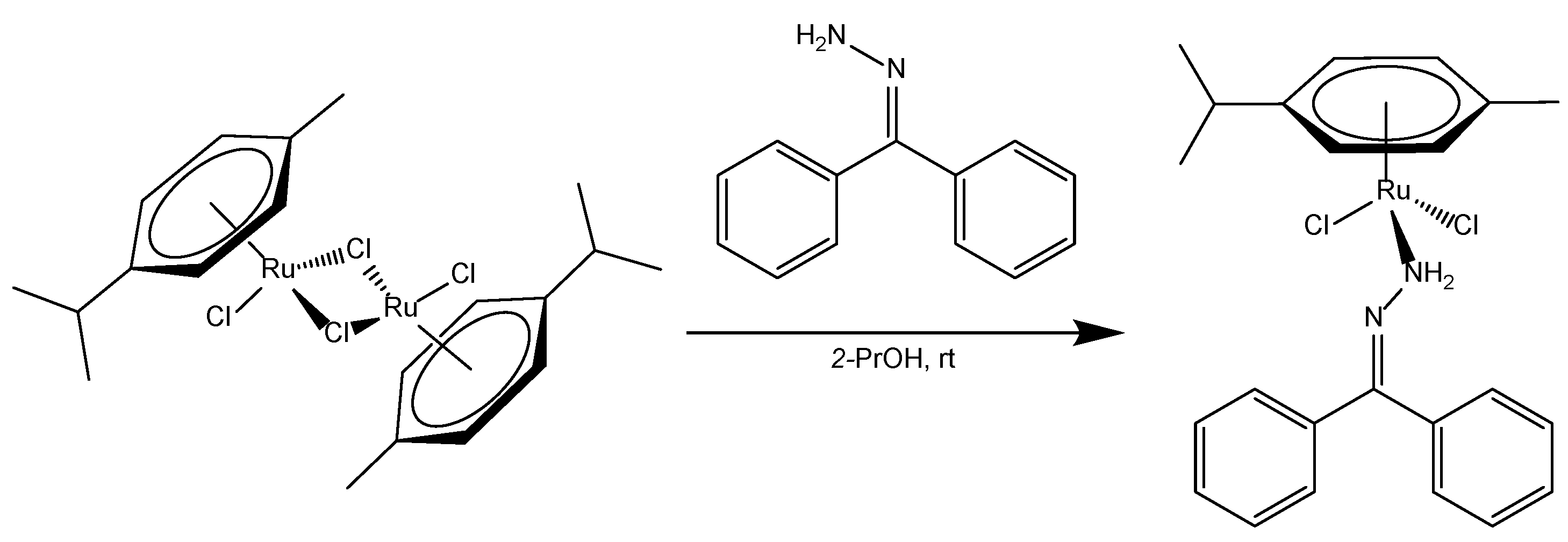
2.1. Synthetic Procedure
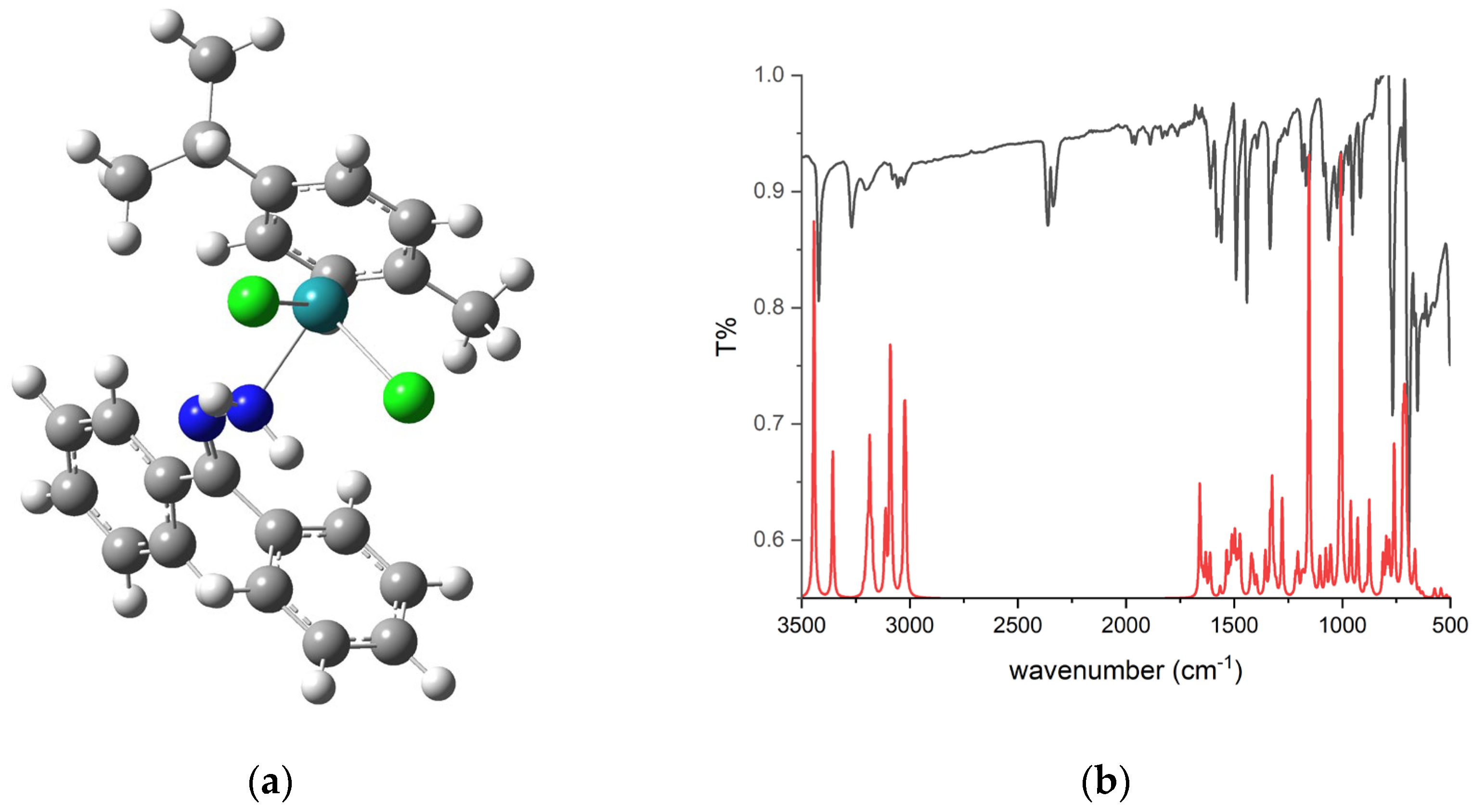
2.2. Optimization of Structure
2.3. Comparison of Experimental and Theoretical FTIR Spectra
2.4. Comparison of Experimental and Theoretical NMR Spectra
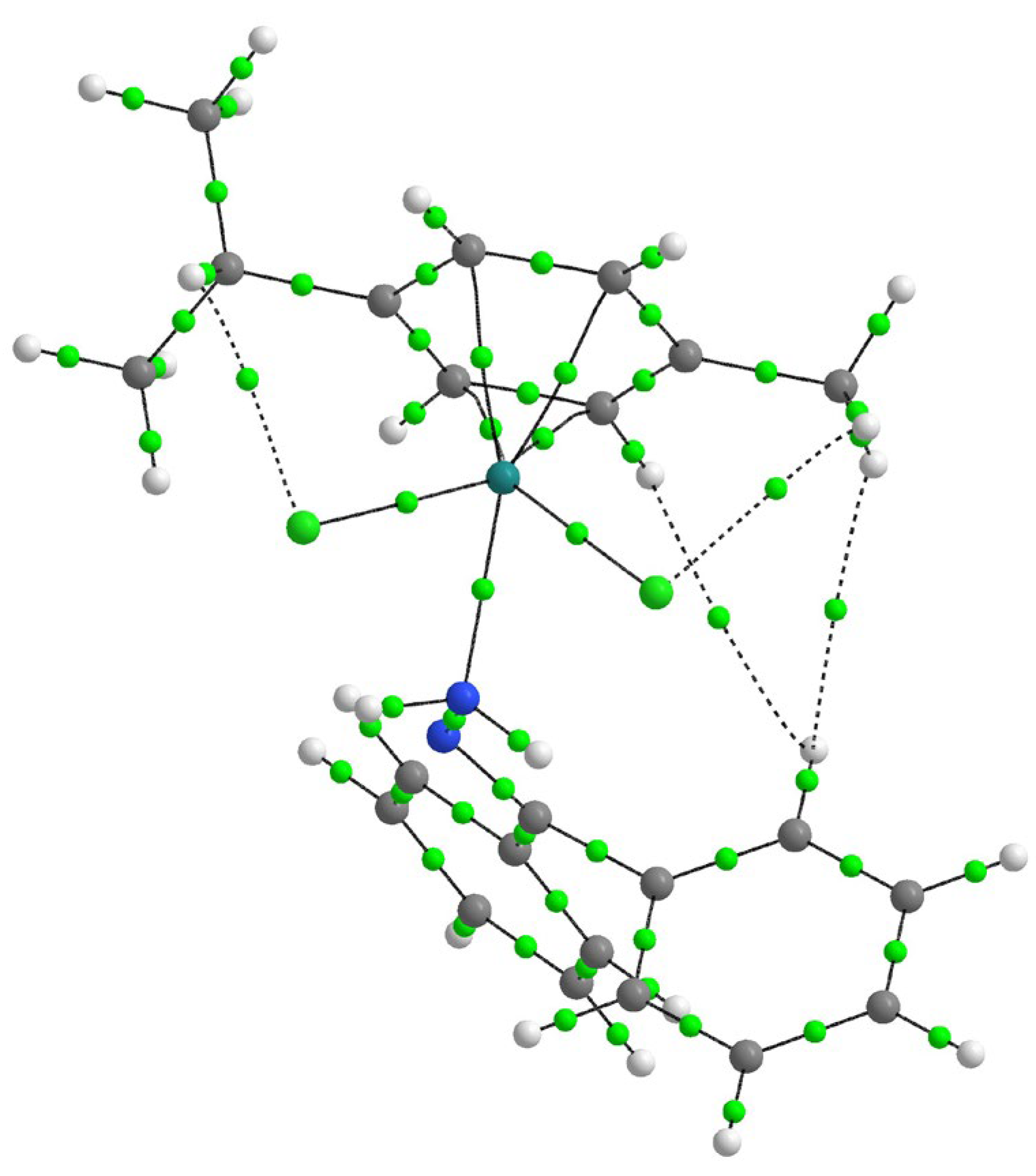
2.5. QTAIM Analysis of Intramolecular Interactions
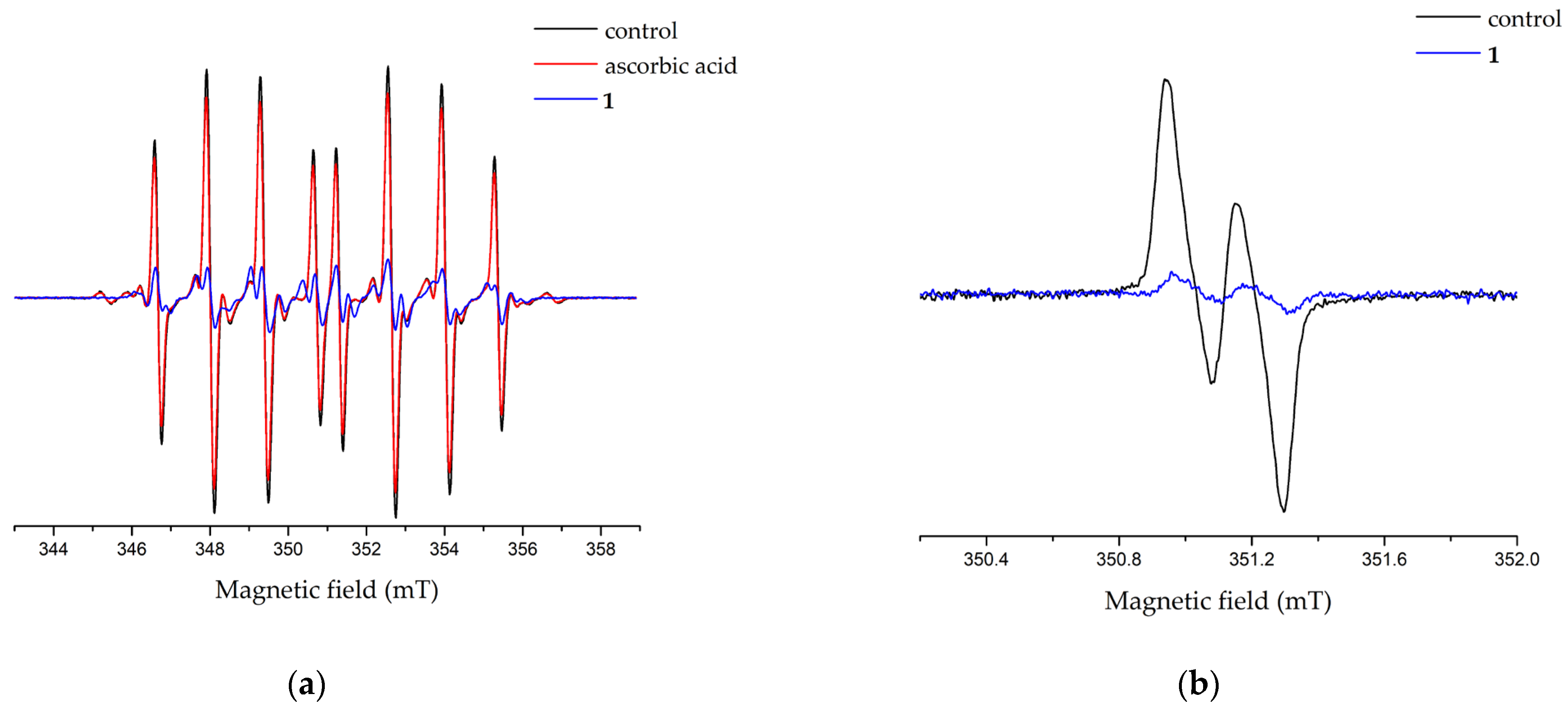
2.6. Antioxidant Activity
2.7. Experimental Assessment of Protein Binding Affinity
2.8. Experimental Assessment of DNA Binding Affinity
2.8.1. Spectrofluorimetric Titration of 1 by CT-DNA
2.8.2. Ethidium Bromide Displacement Studies
2.9. Molecular Docking Simulation of HSA/DNA Binding
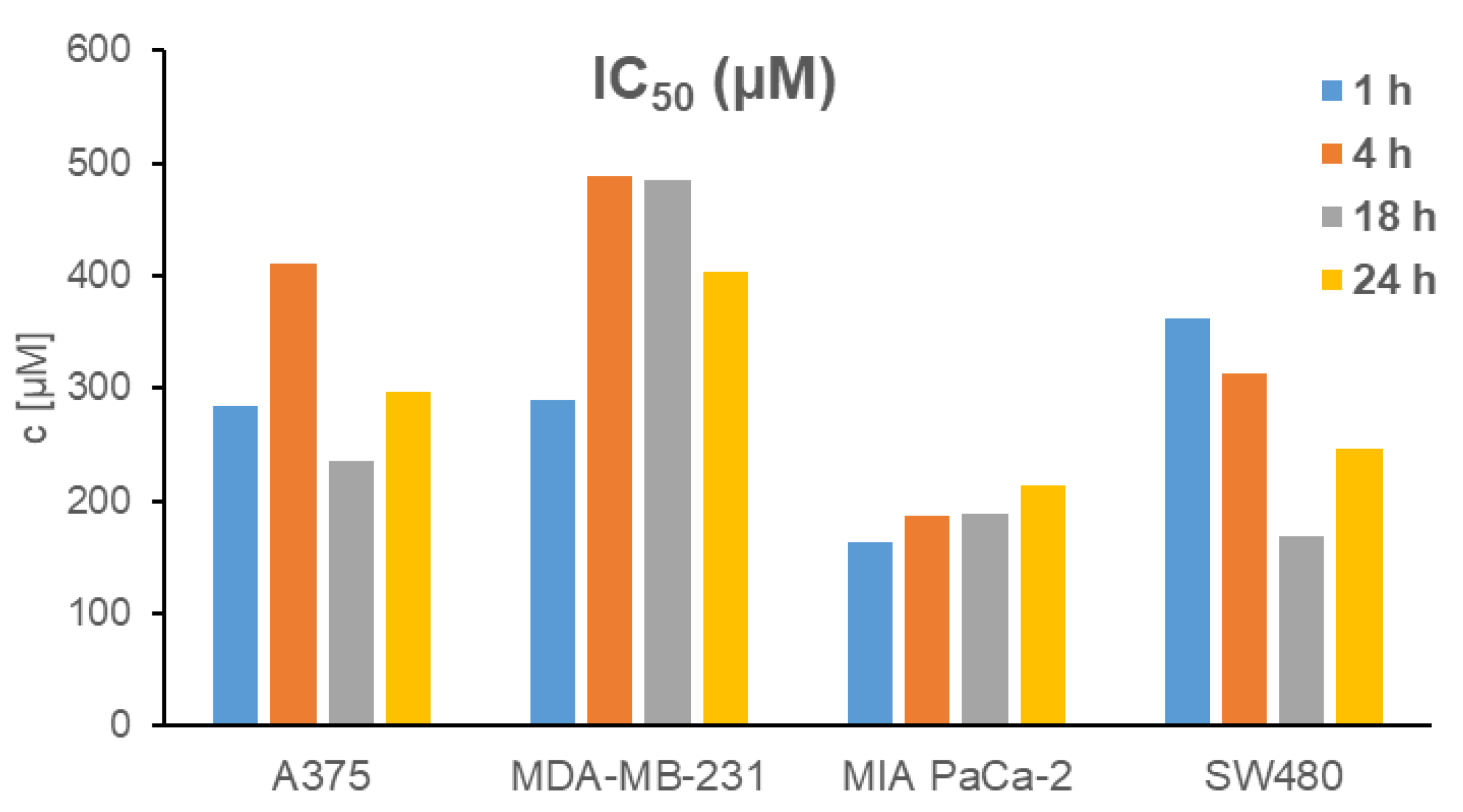
2.10. In Vitro Activity
2.10.1. MTT Test
2.10.2. Malondialdehyde (MDA) and Protein Carbonyl (PC)
3. Materials and Methods
3.1. Materials and Instrumentation
3.2. Synthesis of Dichlorido((diphenylmethylidene)hydrazine-κN)(η6-p-cymene)ruthenium(II) Complex
3.3. Theoretical Calculations
3.4. ESR Spectroscopy
3.4.1. ESR Detection of Anti-Hydroxyl Radical (HO•) Scavenging Activity
3.4.2. ESR Detection of Anti-Ascorbyl Radical (•Asc) Scavenging Activity
3.5. Spectrofluorimetric Determination of HSA Binding Affinity
3.6. Spectrofluorimetric Determination of DNA Binding Affinity
3.7. Molecular Docking
3.8. In Vitro Activity
3.8.1. Reagents and Chemicals
3.8.2. Cell Culture and Experimental Design
3.8.3. MTT Assay
3.8.4. Determination of Malondialdehyde and Protein Carbonyl Levels
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Catalano, A.; Iacopetta, D.; Ceramella, J.; Scumaci, D.; Giuzio, F.; Saturnino, C.; Aquaro, S.; Rosano, C.; Sinicropi, M.S. Multidrug Resistance (MDR): A Widespread Phenomenon in Pharmacological Therapies. Molecules 2022, 27, 616. [Google Scholar] [CrossRef]
- Wang, J.; Seebacher, N.; Shi, H.; Kan, Q.; Duan, Z. Novel Strategies to Prevent the Development of Multidrug Resistance (MDR) in Cancer. Oncotarget 2017, 8, 84559–84571. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Lan, X.; Lin, H.; Xi, Q.; Wang, M.; Quan, X.; Yao, G.; Yu, Z.; Wang, Y.; Yu, M. Tumor Microenvironment-regulating Nanomedicine Design to Fight Multi-drug Resistant Tumors. WIREs Nanomed. Nanobiotechnol. 2023, 15, e1842. [Google Scholar] [CrossRef] [PubMed]
- Duan, C.; Yu, M.; Xu, J.; Li, B.-Y.; Zhao, Y.; Kankala, R.K. Overcoming Cancer Multi-Drug Resistance (MDR): Reasons, Mechanisms, Nanotherapeutic Solutions, and Challenges. Biomed. Pharmacother. 2023, 162, 114643. [Google Scholar] [CrossRef] [PubMed]
- Szakács, G.; Paterson, J.K.; Ludwig, J.A.; Booth-Genthe, C.; Gottesman, M.M. Targeting Multidrug Resistance in Cancer. Nat. Rev. Drug Discov. 2006, 5, 219–234. [Google Scholar] [CrossRef]
- Podolski-Renić, A.; Čipak Gašparović, A.; Valente, A.; López, Ó.; Bormio Nunes, J.H.; Kowol, C.R.; Heffeter, P.; Filipović, N.R. Schiff Bases and Their Metal Complexes to Target and Overcome (Multidrug) Resistance in Cancer. Eur. J. Med. Chem. 2024, 270, 116363. [Google Scholar] [CrossRef]
- Salmain, M.; Gaschard, M.; Baroud, M.; Lepeltier, E.; Jaouen, G.; Passirani, C.; Vessières, A. Thioredoxin Reductase and Organometallic Complexes: A Pivotal System to Tackle Multidrug Resistant Tumors? Cancers 2023, 15, 4448. [Google Scholar] [CrossRef]
- Yu, Y.; Zhang, L.; Qin, Z.; Karges, J.; Xiao, H.; Su, X. Unraveling and Overcoming Platinum Drug-Resistant Cancer Tumors with DNA Nanostructures. Adv. Funct. Mater. 2023, 33, 2208797. [Google Scholar] [CrossRef]
- Zhong, T.; Yu, J.; Pan, Y.; Zhang, N.; Qi, Y.; Huang, Y. Recent Advances of Platinum-Based Anticancer Complexes in Combinational Multimodal Therapy. Adv. Healthc. Mater. 2023, 12, 2300253. [Google Scholar] [CrossRef]
- Cao, X.; Li, R.; Xiong, H.; Su, J.; Guo, C.; An, T.; Zong, H.; Zhao, R. Novel Pt(IV) Complexes to Overcome Multidrug Resistance in Gastric Cancer by Targeting P-Glycoprotein. Eur. J. Med. Chem. 2021, 221, 113520. [Google Scholar] [CrossRef]
- Opačak, S.; Kovač, M.P.; Landais, C.; Debeljak, Ž.; Golding, T.M.; Smith, G.S.; Brozovic, A.; Kirin, S.I. Dissimilar Effect of Organometallic Ruthenium Complexes on the Viability of MDR and Non-MDR Experimental Models. J. Inorg. Biochem. 2024, 257, 112614. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, R.G.; Salaroglio, I.C.; Oliveira, N.F.B.; Sequeira, J.G.N.; Fontrodona, X.; Romero, I.; Machuqueiro, M.; Tomaz, A.I.; Garcia, M.H.; Riganti, C.; et al. Fighting Multidrug Resistance with Ruthenium–Cyclopentadienyl Compounds: Unveiling the Mechanism of P-Gp Inhibition. J. Med. Chem. 2023, 66, 14080–14094. [Google Scholar] [CrossRef] [PubMed]
- Vock, C.A.; Ang, W.H.; Scolaro, C.; Phillips, A.D.; Lagopoulos, L.; Juillerat-Jeanneret, L.; Sava, G.; Scopelliti, R.; Dyson, P.J. Development of Ruthenium Antitumor Drugs That Overcome Multidrug Resistance Mechanisms. J. Med. Chem. 2007, 50, 2166–2175. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Li, J.; Zhang, C.; Zhang, Y.-K.; Zhang, W.; Huang, J.; Ashby, C.R.; Chen, Z.-S.; Chao, H. An Organoruthenium Complex Overcomes ABCG2-Mediated Multidrug Resistance via Multiple Mechanisms. Chem. Commun. 2019, 55, 3833–3836. [Google Scholar] [CrossRef]
- Kawano, M.; Hoshino, C.; Matsumoto, K. Synthesis, Properties, and Crystal Structure of a Novel.Mu.-Hydrazine-Bridged Mixed-Valence Ruthenium(II,III) Complex Stabilized by Hydrazine Hydrogen Bonds, [RuCl(TMP)2]2(.Mu.-Cl)(.Mu.-N2H4)(.Mu.-S2) (TMP = Trimethyl Phosphite). Inorg. Chem. 1992, 31, 5158–5159. [Google Scholar] [CrossRef]
- Rozenel, S.S.; Arnold, J. Bimetallic Ruthenium PNP Pincer Complex As a Platform to Model Proposed Intermediates in Dinitrogen Reduction to Ammonia. Inorg. Chem. 2012, 51, 9730–9739. [Google Scholar] [CrossRef]
- Matsumoto, K.; Uemura, H.; Kawano, M. A Novel Diruthenium (II, III) Complex,({Ru (AN)(TMP) 2} 2 (. MU.-S2)(. MU.-NH2NH2) 2)(CF3SO3) 3Et2O, with Two Hydrazine Bridges (AN= acetonitrile, TMP= P (OMe) 3). Chem. Lett. 1994, 23, 1215–1218. [Google Scholar] [CrossRef]
- Mashima, K.; Kaneko, S.; Tani, K.; Kaneyoshi, H.; Nakamura, A. Synthesis and Reactions of Coordinatively Unsaturated 16-Electron Chalcogenolate Complexes, Ru(EAr) 2(H6-Arene) and Cationic Binuclear Chalcogenolate Complexes, [(H6-Arene) Ru(μ-EPh)3 Ru(H6-Arene)]PF6. J. Organomet. Chem. 1997, 545–546, 345–356. [Google Scholar] [CrossRef]
- Eichhorn, T.; Kolbe, F.; Mišić, S.; Dimić, D.; Morgan, I.; Saoud, M.; Milenković, D.; Marković, Z.; Rüffer, T.; Marković, J.D.; et al. Synthesis, Crystallographic Structure, Theoretical Analysis, Molecular Docking Studies, and Biological Activity Evaluation of Binuclear Ru(II)-1-Naphthylhydrazine Complex. Int. J. Mol. Sci. 2023, 24, 689. [Google Scholar] [CrossRef]
- Marković, J.D.; Dimić, D.; Eichhorn, T.; Milenković, D.; Pavićević, A.; Đikić, D.; Živković, E.; Čokić, V.; Rüffer, T.; Kaluđerović, G.N. Ru(II) Complexes with 3,4-Dimethylphenylhydrazine: Exploring In Vitro Anticancer Activity and Protein Affinities. Biomolecules 2025, 15, 350. [Google Scholar] [CrossRef]
- Eichhorn, T.; Đošić, M.; Dimić, D.; Morgan, I.; Milenković, D.; Rennert, R.; Amić, A.; Marković, Z.; Kaluđerović, G.N.; Marković, J.D. Ru(II)-Nitrophenylhydrazine/Chlorophenylhydrazine Complexes: Nanoarchitectonics, Biological Evaluation and In Silico Study. Eur. J. Inorg. Chem. 2024, 27, e202300683. [Google Scholar] [CrossRef]
- Dasmahapatra, U.; Maiti, B.; Alam, M.M.; Chanda, K. Anti-Cancer Property and DNA Binding Interaction of First Row Transition Metal Complexes: A Decade Update. Eur. J. Med. Chem. 2024, 275, 116603. [Google Scholar] [CrossRef] [PubMed]
- Arakelyan, J.; Rusanov, D.A.; Chang, M.R.; Asaturova, A.V.; Kireeva, G.S.; Alshehri, M.; Ang, W.H.; Babak, M.V. Targeting Emerging Cancer Hallmarks by Transition Metal Complexes: Epigenetic Reprogramming and Epitherapies. Part II. Coord. Chem. Rev. 2023, 475, 214899. [Google Scholar] [CrossRef]
- Khan, H.Y.; Ansari, M.F.; Tabassum, S.; Arjmand, F. A Review on the Recent Advances of Interaction Studies of Anticancer Metal-Based Drugs with Therapeutic Targets, DNA and RNAs. Drug Discov. Today 2024, 29, 104055. [Google Scholar] [CrossRef]
- Sharfalddin, A.A.; Emwas, A.-H.; Jaremko, M.; Hussien, M.A. Synthesis and Theoretical Calculations of Metal-Antibiotic Chelation with Thiamphenicol: In Vitro DNA and HSA Binding, Molecular Docking, and Cytotoxicity Studies. New J. Chem. 2021, 45, 9598–9613. [Google Scholar] [CrossRef]
- Sharmin, S.; Rahaman, M.; Martorell, M.; Sastre-Serra, J.; Sharifi-Rad, J.; Butnariu, M.; Bagiu, I.C.; Bagiu, R.V.; Islam, M.T. Cytotoxicity of Synthetic Derivatives against Breast Cancer and Multi-Drug Resistant Breast Cancer Cell Lines: A Literature-Based Perspective Study. Cancer Cell Int. 2021, 21, 612. [Google Scholar] [CrossRef]
- Lalrempuia, R.; Kollipara, M.R.; Carroll, P.J. Syntheses and Characterization of Arene Ruthenium (II) Complexes Containing N,N′-Donor Schiff Base Ligands. Crystal and Molecular Structure of [(H6-C6Me6)Ru(C5H4N-2-CH N C6H4-p-NO2)]PF6. Polyhedron 2003, 22, 605–609. [Google Scholar] [CrossRef]
- Gulaczyk, I.; Kręglewski, M.; Valentin, A. The N–N Stretching Band of Hydrazine. J. Mol. Spectrosc. 2003, 220, 132–136. [Google Scholar] [CrossRef]
- Durig, J.R.; McAllister, W.A.; Mercer, E.E. On the I.R. Spectra of the Isomers of Trichlorotriaquoruthenium(III). J. Inorg. Nucl. Chem. 1967, 29, 1441–1447. [Google Scholar] [CrossRef]
- Arlt, S.; Petković, V.; Ludwig, G.; Eichhorn, T.; Lang, H.; Rüffer, T.; Mijatović, S.; Maksimović-Ivanić, D.; Kaluđerović, G.N. Arene Ruthenium(II) Complexes Bearing the κ-P or κ-P,κ-S Ph2P(CH2)3SPh Ligand. Molecules 2021, 26, 1860. [Google Scholar] [CrossRef]
- Pallewela, G.N.; Bettens, R.P.A. A DFT, TDDFT and QTAIM Study of the Acridine Pincer Ligand-Based Ru(II) and Rh(III) Complexes: Detailed Analysis of the Metal-F Bonding. Chem. Pap. 2023, 77, 47–61. [Google Scholar] [CrossRef]
- Sánchez-Coronilla, A.; Sánchez-Márquez, J.; Zorrilla, D.; Martín, E.I.; de los Santos, D.M.; Navas, J.; Fernández-Lorenzo, C.; Alcántara, R.; Martín-Calleja, J. Convergent Study of Ru–Ligand Interactions through QTAIM, ELF, NBO Molecular Descriptors and TDDFT Analysis of Organometallic Dyes. Mol. Phys. 2014, 112, 2063–2077. [Google Scholar] [CrossRef]
- Matveevskaya, V.V.; Pavlov, D.I.; Samsonenko, D.G.; Bonfili, L.; Cuccioloni, M.; Benassi, E.; Pettinari, R.; Potapov, A.S. Arene-Ruthenium(II) Complexes with Tetracyclic Oxime Derivatives: Synthesis, Structure and Antiproliferative Activity against Human Breast Cancer Cells. Inorganica Chim. Acta 2022, 535, 120879. [Google Scholar] [CrossRef]
- Kasalović, M.P.; Jelača, S.; Milanović, Ž.; Maksimović-Ivanić, D.; Mijatović, S.; Lađarević, J.; Božić, B.; Marković, Z.; Dunđerović, D.; Rüffer, T.; et al. Novel Triphenyltin (iv) Compounds with Carboxylato N-Functionalized 2-Quinolones as Promising Potential Anticancer Drug Candidates: In Vitro and in Vivo Evaluation. Dalton Trans. 2024, 53, 8298–8314. [Google Scholar] [CrossRef]
- Espinosa, E.; Molins, E.; Lecomte, C. Hydrogen Bond Strengths Revealed by Topological Analyses of Experimentally Observed Electron Densities. Chem. Phys. Lett. 1998, 285, 170–173. [Google Scholar] [CrossRef]
- Bianchi, R.; Gervasio, G.; Marabello, D. Experimental Electron Density Analysis of Mn 2 (CO) 10: Metal−Metal and Metal−Ligand Bond Characterization. Inorg. Chem. 2000, 39, 2360–2366. [Google Scholar] [CrossRef]
- Soliman, S.M.; Albering, J.; Abu-Youssef, M.A.M. Structural Analyses of Two New Highly Distorted Octahedral Copper(II) Complexes with Quinoline-Type Ligands; Hirshfeld, AIM and NBO Studies. Polyhedron 2017, 127, 36–50. [Google Scholar] [CrossRef]
- Lepetit, C.; Vabre, B.; Canac, Y.; Alikhani, M.E.; Zargarian, D. Pentacoordinated, Square Pyramidal Cationic PCP Ni(II) Pincer Complexes: ELF and QTAIM Topological Analyses of Nickel–Triflate Interactions. Theor. Chem. Acc. 2018, 137, 141. [Google Scholar] [CrossRef]
- Galano, A.; Mazzone, G.; Alvarez-Diduk, R.; Marino, T.; Alvarez-Idaboy, J.R.; Russo, N. Food Antioxidants: Chemical Insights at the Molecular Level. Annu. Rev. Food Sci. Technol. 2016, 7, 335–352. [Google Scholar] [CrossRef]
- Kompany-Zareh, M.; Akbarian, S.; Najafpour, M.M. Unsupervised Recognition of Components from the Interaction of BSA with Fe Cluster in Different Conditions Utilizing 2D Fluorescence Spectroscopy. Sci. Rep. 2022, 12, 16875. [Google Scholar] [CrossRef]
- Szymańska, M.; Pospieszna-Markiewicz, I.; Mańka, M.; Insińska-Rak, M.; Dutkiewicz, G.; Patroniak, V.; Fik-Jaskółka, M.A. Synthesis and Spectroscopic Investigations of Schiff Base Ligand and Its Bimetallic Ag(I) Complex as DNA and BSA Binders. Biomolecules 2021, 11, 1449. [Google Scholar] [CrossRef]
- Cao, X.; He, Y.; Liu, D.; He, Y.; Hou, X.; Cheng, Y.; Liu, J. Characterization of Interaction between Scoparone and Bovine Serum Albumin: Spectroscopic and Molecular Docking Methods. RSC Adv. 2018, 8, 25519–25525. [Google Scholar] [CrossRef]
- Abdalla, E.M.; Al-Sulami, A.I.; Aly, S.A.; Abd-Allah, M.T.; Nasr, G.M.; Albohy, S.A.H.; Hosny, S. Synthesis, Characterization, DNA Binding, DFT, Anticancer, Antibacterial, and the Effect of Gamma Irradiation of Novel Co(II), Ag (I), and Cd (II) Complexes with Hydrazone Derivatives. J. Saudi Chem. Soc. 2023, 27, 101770. [Google Scholar] [CrossRef]
- Palchaudhuri, R.; Hergenrother, P.J. DNA as a Target for Anticancer Compounds: Methods to Determine the Mode of Binding and the Mechanism of Action. Curr. Opin. Biotechnol. 2007, 18, 497–503. [Google Scholar] [CrossRef]
- Shahabadi, N.; Kashanian, S.; Mahdavi, M.; Sourinejad, N. DNA Interaction and DNA Cleavage Studies of a New Platinum(II) Complex Containing Aliphatic and Aromatic Dinitrogen Ligands. Bioinorg. Chem. Appl. 2011, 2011, 525794. [Google Scholar] [CrossRef] [PubMed]
- Shahabadi, N.; Kashanian, S.; Khosravi, M.; Mahdavi, M. Multispectroscopic DNA Interaction Studies of a Water-Soluble Nickel(II) Complex Containing Different Dinitrogen Aromatic Ligands. Transit. Met. Chem. 2010, 35, 699–705. [Google Scholar] [CrossRef]
- Lincoln, P.; Nordén, B. DNA Binding Geometries of Ruthenium(II) Complexes with 1,10-Phenanthroline and 2,2‘-Bipyridine Ligands Studied with Linear Dichroism Spectroscopy. Borderline Cases of Intercalation. J. Phys. Chem. B 1998, 102, 9583–9594. [Google Scholar] [CrossRef]
- Mahmood, K.; Akhter, Z.; Perveen, F.; Aisha; Bibi, M.; Ismail, H.; Tabassum, N.; Yousuf, S.; Ashraf, A.R.; Qayyum, M.A. Synthesis, DNA Binding and Biological Evaluation of Benzimidazole Schiff Base Ligands and Their Metal(ii) Complexes. RSC Adv. 2023, 13, 11982–11999. [Google Scholar] [CrossRef]
- Pushkaran, A.C.; Arabi, A.A. Accurate Prediction of DNA-Intercalator Binding Energies: Ensemble of Short or Long Molecular Dynamics Simulations? Int. J. Biol. Macromol. 2025, 306, 141408. [Google Scholar] [CrossRef]
- Muralisankar, M.; Dheepika, R.; Haribabu, J.; Balachandran, C.; Aoki, S.; Bhuvanesh, N.S.P.; Nagarajan, S. Design, Synthesis, DNA/HSA Binding, and Cytotoxic Activity of Half-Sandwich Ru(II)-Arene Complexes Containing Triarylamine–Thiosemicarbazone Hybrids. ACS Omega 2019, 4, 11712–11723. [Google Scholar] [CrossRef]
- Colina-Vegas, L.; Luna-Dulcey, L.; Plutín, A.M.; Castellano, E.E.; Cominetti, M.R.; Batista, A.A. Half Sandwich Ru(ii)-Acylthiourea Complexes: DNA/HSA-Binding, Anti-Migration and Cell Death in a Human Breast Tumor Cell Line. Dalton Trans. 2017, 46, 12865–12875. [Google Scholar] [CrossRef]
- Hikisz, P.; Namiecińska, E.; Paneth, P.; Budzisz, E. Mechanistic Studies of Arene–Ruthenium(II) Complexes with Carbothioamidopyrazoles as Alternative Cancer Drugs. Molecules 2023, 28, 3969. [Google Scholar] [CrossRef] [PubMed]
- Palakkeezhillam, V.N.V.; Haribabu, J.; Kumar, V.S.; Santibanez, J.F.; Manakkadan, V.; Rasin, P.; Garg, M.; Bhuvanesh, N.; Sreekanth, A. Unraveling the Anticancer Efficacy and Biomolecular Properties of Ru(II)-Arene Complexes of Pyrene-Based Thiosemicarbazone Ligands: A Comprehensive In Silico/In Vitro Exploration. Organometallics 2024, 43, 242–260. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Becke, A.D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648. [Google Scholar] [CrossRef]
- Dunning, T.H. Gaussian Basis Sets for Use in Correlated Molecular Calculations. I. The Atoms Boron through Neon and Hydrogen. J. Chem. Phys. 1989, 90, 1007. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab Initio Effective Core Potentials for Molecular Calculations. Potentials for the Transition Metal Atoms Sc to Hg. J. Chem. Phys. 1985, 82, 270–283. [Google Scholar] [CrossRef]
- Hay, P.J.; Wadt, W.R. Ab Initio Effective Core Potentials for Molecular Calculations. Potentials for K to Au Including the Outermost Core Orbitale. J. Chem. Phys. 1985, 82, 299–310. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Bohmann, J.A.; Weinhold, F.; Farrar, T.C. Natural Chemical Shielding Analysis of Nuclear Magnetic Resonance Shielding Tensors from Gauge-Including Atomic Orbital Calculations. J. Chem. Phys. 1997, 107, 1173. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules. Acc. Chem. Res. 1985, 18, 9–15. [Google Scholar] [CrossRef]
- Bader, R.F.W.; Slee, T.S.; Cremer, D.; Kraka, E. Description of Conjugation and Hyperconjugation in Terms of Electron Distributions. J. Am. Chem. Soc. 1983, 105, 5061–5068. [Google Scholar] [CrossRef]
- Keith, T.A. AIMAl l, version 19.10.12; TK Gristmill Software: Overland Park, KS, USA, 2019.
- Frejaville, C.; Karoui, H.; Tuccio, B.; Moigne, F.L.; Culcasi, M.; Pietri, S.; Lauricella, R.; Tordo, P. 5-(Diethoxyphosphoryl)-5-Methyl-1-Pyrroline N-Oxide: A New Efficient Phosphorylated Nitrone for the in Vitro and in Vivo Spin Trapping of Oxygen-Centered Radicals. J. Med. Chem. 1995, 38, 258–265. [Google Scholar] [CrossRef]
- Vasić, J.; Dimić, D.; Antonijević, M.; Avdović, E.H.; Milenković, D.; Nakarada, Đ.; Marković, J.D.; Molnar, M.; Lončarić, M.; Bešlo, D.; et al. The Electronic Effects of 3-Methoxycarbonylcoumarin Substituents on Spectral, Antioxidant, and Protein Binding Properties. Int. J. Mol. Sci. 2023, 24, 11820. [Google Scholar] [CrossRef] [PubMed]
- Nakarada, Đ.; Pejin, B.; Tommonaro, G.; Mojović, M. Liposomal Integration Method for Assessing Antioxidative Activity of Water Insoluble Compounds towards Biologically Relevant Free Radicals: Example of Avarol. J. Liposome Res. 2020, 30, 218–226. [Google Scholar] [CrossRef] [PubMed]
- Petitpas, I.; Bhattacharya, A.A.; Twine, S.; East, M.; Curry, S. Crystal Structure Analysis of Warfarin Binding to Human Serum Albumin. Anatomy of Drug Site I. J. Biol. Chem. 2001, 276, 22804–22809. [Google Scholar] [CrossRef] [PubMed]
- Mandal, P.K.; Venkadesh, S.; Gautham, N. Structure of the Tetradecanucleotide d(CCCCGGTACCGGGG)2 as an A-DNA Duplex. Acta Crystallogr. F Struct. Biol. Cryst. Commun. 2012, 68, 393–399. [Google Scholar] [CrossRef]
- Ha, S.C.; Lowenhaupt, K.; Rich, A.; Kim, Y.-G.; Kim, K.K. Crystal Structure of a Junction between B-DNA and Z-DNA Reveals Two Extruded Bases. Nature 2005, 437, 1183–1186. [Google Scholar] [CrossRef]
- Ng, H.-L.; Kopka, M.L.; Dickerson, R.E. The Structure of a Stable Intermediate in the A ↔ B DNA Helix Transition. Proc. Natl. Acad. Sci. USA 2000, 97, 2035–2039. [Google Scholar] [CrossRef]
- Barton, J.K.; Rees, D.C.; Kielkopf, C.L.; Erkkila, K.E.; Hudson, B.P. Structure of a Photoactive Rhodium Complex Intercalated into DNA. Nat. Struct. Biol. 2000, 7, 117–121. [Google Scholar] [CrossRef]
- Di Pietro, M.L.; La Ganga, G.; Nastasi, F.; Puntoriero, F. Ru(II)-Dppz Derivatives and Their Interactions with DNA: Thirty Years and Counting. Appl. Sci. 2021, 11, 3038. [Google Scholar] [CrossRef]
- Dassault Systèmes BIOVIA. Discovery Studio Modeling Environment (Release 2017); Dassault Systèmes BIOVIA: San Diego, CA, USA, 2016. [Google Scholar]
- Mosmann, T. Rapid Colorimetric Assay for Cellular Growth and Survival: Application to Proliferation and Cytotoxicity Assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Baskić, D.D.; Radosavljević, G.; Čokanović, V.; Jeftić, I.; Zelen, I.; Popović, S.; Pavlović, S.; Arsenijević, N.N. Serumski Nivoi NO, IL-18 i MDA Kod Pacijenata Sa Karcinomom Dojke. Medicus 2005, 6, 62–65. [Google Scholar]
- Levine, R.L.; Garland, D.; Oliver, C.N.; Amici, A.; Climent, I.; Lenz, A.-G.; Ahn, B.-W.; Shaltiel, S.; Stadtman, E.R. Determination of Carbonyl Content in Oxidatively Modified Proteins. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 1990; pp. 464–478. [Google Scholar]
- Neidle, S. Principles of Nucleic Acid Structure, 1st ed.; Elsevier: Oxford, UK, 2008; pp. 38–67. [Google Scholar]
- Porschke, D. Boundary conditions for free A-DNA in solution and the relation of local to global DNA structures at reduced water activity. Eur. Biophys. J. 2016, 45, 413–421. [Google Scholar] [CrossRef]
- Hangan, A.C.; Oprean, L.S.; Dican, L.; Procopciuc, L.M.; Sevastre, B.; Lucaciu, R.L. Metal-Based Drug–DNA Interactions and Analytical Determination Methods. Molecules 2024, 29, 4361. [Google Scholar] [CrossRef] [PubMed]
- Krall, J.B.; Nichols, P.J.; Henen, M.A.; Vicens, Q.; Vögeli, B. Structure and Formation of Z-DNA and Z-RNA. Molecules 2023, 28, 843. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bond | ρ(r) [a.u.] | ∇2ρ(r) [a.u.] | G(r) [kJ mol−1] | V(r) [kJ mol−1] | H(r) [kJ mol−1] | −G(r)/V(r) | Ebond [kJ mol−1] |
|---|---|---|---|---|---|---|---|
| Ru−C1 | 0.082 | 0.240 | 208.9 | −260.1 | −51.1 | 0.8 | −130.0 |
| Ru−C2 | 0.083 | 0.241 | 209.9 | −261.5 | −51.7 | 0.8 | −130.8 |
| Ru−C3 | 0.086 | 0.239 | 214.6 | −272.7 | −58.1 | 0.8 | −136.4 |
| Ru−C4 | 0.086 | 0.240 | 215.0 | −272.4 | −57.4 | 0.8 | −136.2 |
| Ru−Cl1 | 0.069 | 0.184 | 154.8 | −188.8 | −34.0 | 0.8 | −94.4 |
| Ru-Cl2 | 0.067 | 0.181 | 151.6 | −184.2 | −32.6 | 0.8 | −92.1 |
| Ru−N | 0.077 | 0.293 | 226.0 | −259.9 | −33.9 | 0.9 | −130.0 |
| Cl1−H | 0.008 | 0.027 | 14.6 | −11.2 | 3.3 | 1.3 | −5.6 |
| Cl2−H | 0.007 | 0.025 | 13.4 | −10.4 | 3.1 | 1.3 | −5.2 |
| H−Hp-cym | 0.001 | 0.005 | 2.5 | −1.6 | 0.9 | 1.6 | −0.8 |
| H−Hp-cym | 0.002 | 0.005 | 2.6 | −1.7 | 0.9 | 1.5 | −0.9 |
| T [K] | KSV [M−1] | Kb [M−1] | n | ∆Hb [kJ mol−1] | ∆Sb [J mol−1 K−1] | ∆Gb [kJ mol−1] | R2 |
|---|---|---|---|---|---|---|---|
| 300 | 2.85 × 104 | 1.65 × 104 | 0.95 | −24.3 | |||
| 305 | 3.17 × 104 | 3.62 × 104 | 1.01 | 104.149 | 482.179 | −26.4 | 0.992 |
| 310 | 3.23 × 104 | 6.34 × 104 | 1.06 | −28.6 |
| T [K] | KSV [M−1] | Kb [M−1] | n | ∆Hb [kJ mol−1] | ∆Sb [J mol−1 K−1] | ∆Gb [kJ mol−1] | R2 |
|---|---|---|---|---|---|---|---|
| 300 | 1.86 × 104 | 5.34 × 103 | 0.88 | −21.7 | |||
| 305 | 1.95 × 104 | 1.67 × 104 | 0.98 | 124.635 | 487.683 | −24.1 | 0.949 |
| 310 | 1.98 × 104 | 2.67 × 104 | 1.03 | −26.5 |
| Target | ∆Gexp [kJ mol−1] at 27 °C | ∆Gcal [kJ mol−1] at 25 °C |
|---|---|---|
| HSA | −24.3 | −23.5 |
| A-DNA | −21.7 | −23.0 |
| B/A-DNA | −21.2 | |
| B-DNA | −25.1 | |
| Z-DNA | −26.8 | |
| DNA, intercalation site | −35.8 | −34.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perendija, S.; Dimić, D.; Eichhorn, T.; Rakić, A.; Saso, L.; Nakarada, Đ.; Đikić, D.; Dragojević, T.; Dimitrić Marković, J.; Kaluđerović, G.N. Synthesis, Characterization, HSA/DNA Binding, and Cytotoxic Activity of [RuCl2(η6-p-cymene)(bph-κN)] Complex. Molecules 2025, 30, 3088. https://doi.org/10.3390/molecules30153088
Perendija S, Dimić D, Eichhorn T, Rakić A, Saso L, Nakarada Đ, Đikić D, Dragojević T, Dimitrić Marković J, Kaluđerović GN. Synthesis, Characterization, HSA/DNA Binding, and Cytotoxic Activity of [RuCl2(η6-p-cymene)(bph-κN)] Complex. Molecules. 2025; 30(15):3088. https://doi.org/10.3390/molecules30153088
Chicago/Turabian StylePerendija, Stefan, Dušan Dimić, Thomas Eichhorn, Aleksandra Rakić, Luciano Saso, Đura Nakarada, Dragoslava Đikić, Teodora Dragojević, Jasmina Dimitrić Marković, and Goran N. Kaluđerović. 2025. "Synthesis, Characterization, HSA/DNA Binding, and Cytotoxic Activity of [RuCl2(η6-p-cymene)(bph-κN)] Complex" Molecules 30, no. 15: 3088. https://doi.org/10.3390/molecules30153088
APA StylePerendija, S., Dimić, D., Eichhorn, T., Rakić, A., Saso, L., Nakarada, Đ., Đikić, D., Dragojević, T., Dimitrić Marković, J., & Kaluđerović, G. N. (2025). Synthesis, Characterization, HSA/DNA Binding, and Cytotoxic Activity of [RuCl2(η6-p-cymene)(bph-κN)] Complex. Molecules, 30(15), 3088. https://doi.org/10.3390/molecules30153088