Ferroptosis and Metabolic Dysregulation: Emerging Chemical Targets in Cancer and Infection




Abstract
1. Introduction
1.1. Ferroptosis: Definition and Molecular Hallmarks
1.2. Overview: Relevance of Ferroptosis in Cancer and Infection
1.3. Aims of the Review
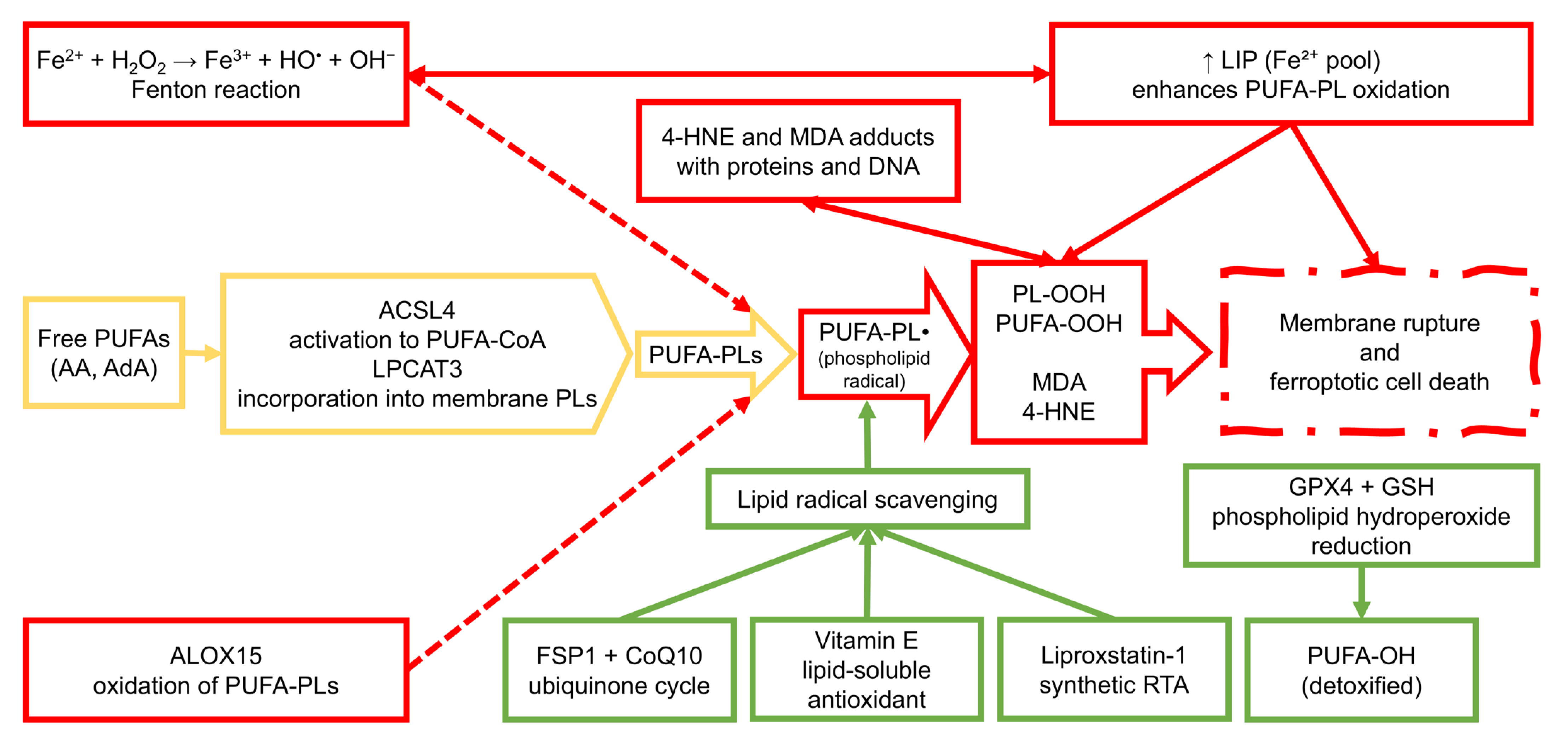
2. Chemical Basis of Ferroptosis
2.1. Lipid Peroxidation Pathways
2.2. Iron Metabolism and ROS Generation
2.3. Glutathione and GPX4 System
2.4. Iron-Independent Ferroptosis-like Mechanisms
3. Chemical Modulators of Ferroptosis
3.1. Ferroptosis Inducers
3.2. Ferroptosis Inhibitors
3.3. Natural Compounds Targeting Ferroptosis
3.4. Therapeutic Relevance of Ferroptosis-Modulating Compounds in Disease Contexts
4. Metabolic Dysregulation and Ferroptosis Sensitivity
4.1. Lipid Metabolism Reprogramming
4.2. Iron Dysregulation in Disease Context
4.3. Metabolic Ferroptosis Vulnerabilities Across Cancer Types
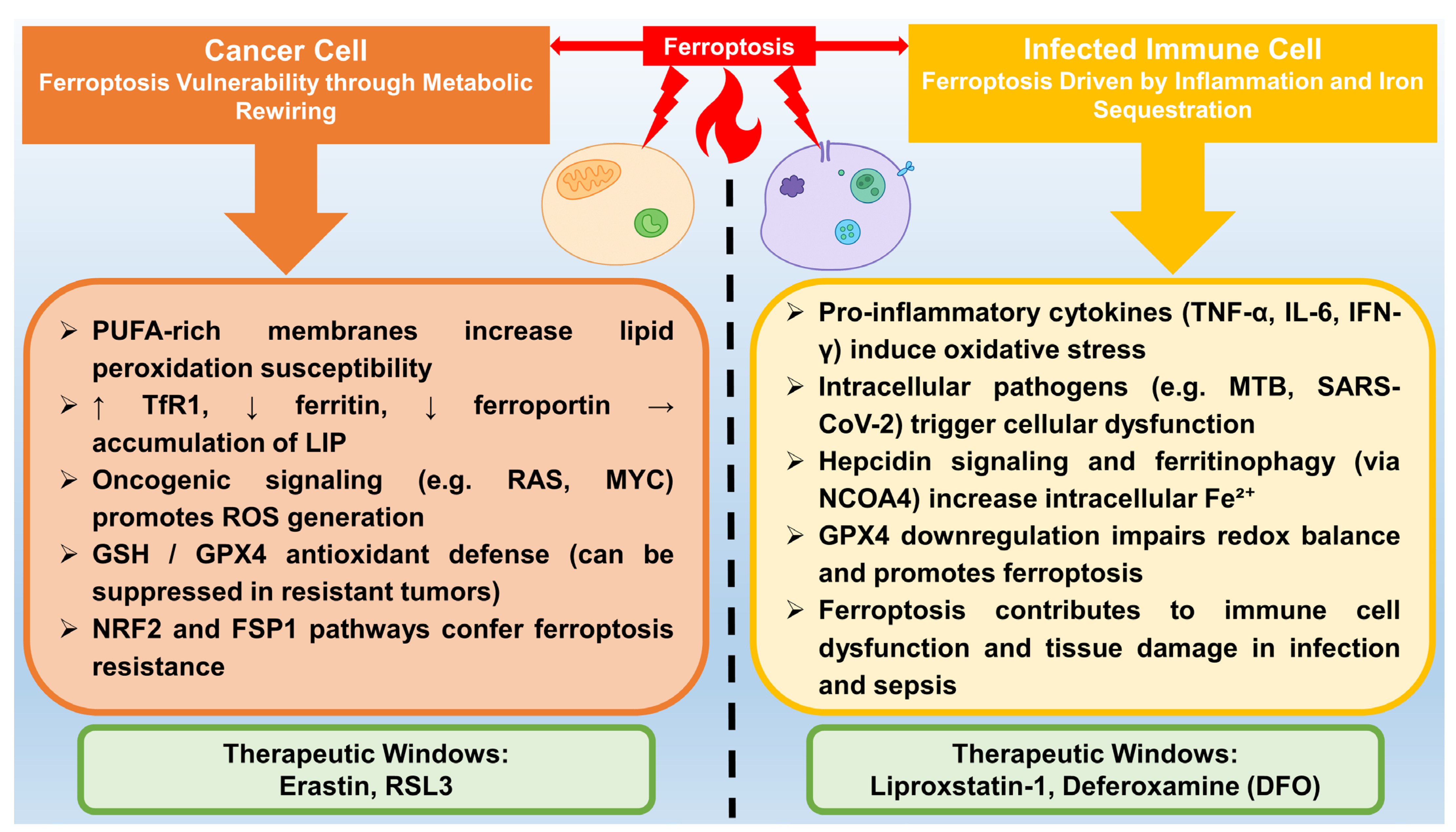
5. Ferroptosis in Disease Contexts
5.1. Cancer
5.2. Infection
5.3. Neurodegenerative Diseases
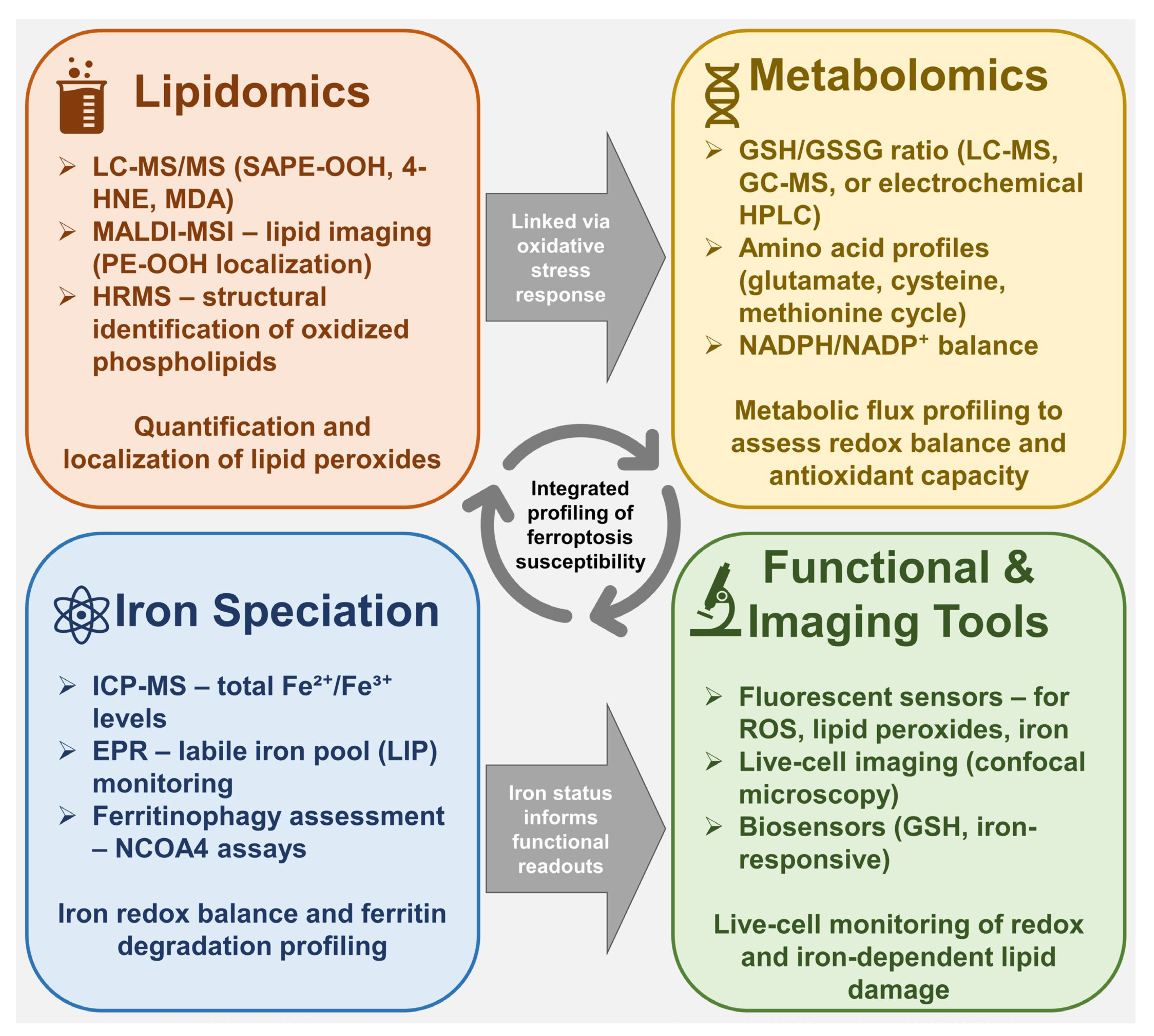
6. Analytical Tools in Ferroptosis Research
6.1. Lipidomics and Detection of Lipid Peroxidation Products
6.2. Metabolomics Approaches
7. Future Directions and Therapeutic Perspectives
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wang, B.; Wang, Y.; Zhang, J.; Hu, C.; Jiang, J.; Li, Y.; Peng, Z.Y. ROS-Induced Lipid Peroxidation Modulates Cell Death Outcome: Mechanisms behind Apoptosis, Autophagy, and Ferroptosis. Arch. Toxicol. 2023, 97, 1439–1451. [Google Scholar] [CrossRef]
- Liu, J.; Kang, R.; Tang, D. Signaling Pathways and Defense Mechanisms of Ferroptosis. FEBS J. 2022, 289, 7038–7050. [Google Scholar] [CrossRef]
- Zhang, T.; Deng, W.; Deng, Y.; Liu, Y.; Xiao, S.; Luo, Y.; Xiang, W.; He, Q. Mechanisms of Ferroptosis Regulating Oxidative Stress and Energy Metabolism in Myocardial Ischemia-Reperfusion Injury and a Novel Perspective of Natural Plant Active Ingredients for Its Treatment. Biomed. Pharmacother. 2023, 165, 114706. [Google Scholar] [CrossRef]
- Dai, Y.; Cui, C.; Jiao, D.; Zhu, X. JAK/STAT Signaling as a Key Regulator of Ferroptosis: Mechanisms and Therapeutic Potentials in Cancer and Diseases. Cancer Cell Int. 2025, 25, 83. [Google Scholar] [CrossRef]
- Liu, M.; Kong, X.-Y.; Yao, Y.; Wang, X.-A.; Yang, W.; Wu, H.; Li, S.; Ding, J.-W.; Yang, J. The Critical Role and Molecular Mechanisms of Ferroptosis in Antioxidant Systems: A Narrative Review. Ann. Transl. Med. 2022, 10, 368. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Sun, L.; Zhang, Y.; Wang, Y.; Zheng, J. Imbalanced GSH/ROS and Sequential Cell Death. J. Biochem. Mol. Toxicol. 2022, 36, e22942. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Kang, R.; Klionsky, D.J.; Tang, D. GPX4 in Cell Death, Autophagy, and Disease. Autophagy 2023, 19, 2621–2638. [Google Scholar] [CrossRef]
- Jiang, X.; Stockwell, B.R.; Conrad, M.; Program, B.; Sloan, M.; Cancer, K.; Death, C. Ferroptosis: Mechanisms, Biology, and Role in Disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef] [PubMed]
- Mustafa, M.; Ahmad, R.; Tantry, I.Q.; Ahmad, W.; Siddiqui, S.; Alam, M.; Abbas, K.; Moinuddin; Hassan, M.I.; Habib, S.; et al. Apoptosis: A Comprehensive Overview of Signaling Pathways, Morphological Changes, and Physiological Significance and Therapeutic Implications. Cells 2024, 13, 1838. [Google Scholar] [CrossRef]
- Kang, A.; Qiao, Y.; Pan, S.; Yan, F.; Chen, H.; Bai, Y. From RIPK1 to Necroptosis: Pathogenic Mechanisms in Neurodegenerative Diseases. Neurochem. Res. 2025, 50, 194. [Google Scholar] [CrossRef]
- Hu, J.J.; Liu, X.; Xia, S.; Zhang, Z.; Zhang, Y.; Zhao, J.; Ruan, J.; Luo, X.; Lou, X.; Bai, Y.; et al. FDA-Approved Disulfiram Inhibits Pyroptosis by Blocking Gasdermin D Pore Formation. Nat. Immunol. 2020, 21, 736–745. [Google Scholar] [CrossRef]
- Riegman, M.; Sagie, L.; Galed, C.; Levin, T.; Steinberg, N.; Dixon, S.J.; Wiesner, U.; Bradbury, M.S.; Niethammer, P.; Zaritsky, A.; et al. Ferroptosis Occurs through an Osmotic Mechanism and Propagates Independently of Cell Rupture. Nat. Cell Biol. 2020, 22, 1042–1048. [Google Scholar] [CrossRef]
- Sharma, A.; Flora, S.J.S. Positive and Negative Regulation of Ferroptosis and Its Role in Maintaining Metabolic and Redox Homeostasis. Oxid. Med. Cell. Longev. 2021, 2021, 9074206. [Google Scholar] [CrossRef]
- Tang, D.; Chen, X.; Kang, R.; Kroemer, G. Ferroptosis: Molecular Mechanisms and Health Implications. Cell Res. 2021, 31, 107–125. [Google Scholar] [CrossRef]
- Yan, H.F.; Zou, T.; Tuo, Q.Z.; Xu, S.; Li, H.; Belaidi, A.A.; Lei, P. Ferroptosis: Mechanisms and Links with Diseases. Signal Transduct. Target. Ther. 2021, 6, 49. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, C.; Wang, J.; Hu, W.; Feng, Z. The Regulation of Ferroptosis by Tumor Suppressor P53 and Its Pathway. Int. J. Mol. Sci. 2020, 21, 8387. [Google Scholar] [CrossRef] [PubMed]
- Gai, C.; Liu, C.; Wu, X.; Yu, M.; Zheng, J.; Zhang, W.; Lv, S.; Li, W. MT1DP Loaded by Folate-Modified Liposomes Sensitizes Erastin-Induced Ferroptosis via Regulating MiR-365a-3p/NRF2 Axis in Non-Small Cell Lung Cancer Cells. Cell Death Dis. 2020, 11, 751. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Luo, Y.; Chen, S.; Wang, G.; Jin, W.; Jiang, W.; Li, M.; Wang, Y.; Yu, J.; Wei, H.; et al. Deubiquitylase USP52 Promotes Bladder Cancer Progression by Modulating Ferroptosis through Stabilizing SLC7A11/XCT. Adv. Sci. 2024, 11, 2403995. [Google Scholar] [CrossRef]
- Yang, M.; Lai, C.L. SARS-CoV-2 Infection: Can Ferroptosis Be a Potential Treatment Target for Multiple Organ Involvement? Cell Death Discov. 2020, 6, 130. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Xu, Y.; Zhang, K.; Shen, L.; Deng, M. Ferroptosis in COVID-19-Related Liver Injury: A Potential Mechanism and Therapeutic Target. Front. Cell. Infect. Microbiol. 2022, 12, 922511. [Google Scholar] [CrossRef]
- Sousa, R.A.L.; Yehia, A.; Abulseoud, O.A. Attenuation of Ferroptosis as a Potential Therapeutic Target for Neuropsychiatric Manifestations of Post-COVID Syndrome. Front. Neurosci. 2023, 17, 1237153. [Google Scholar] [CrossRef]
- Lei, X.; Zhao, G.; Guo, R.; Nui, C. Ferroptosis in Sepsis: The Mechanism, the Role and the Therapeutic Potential. Front. Immunol. 2022, 13, 956361. [Google Scholar] [CrossRef] [PubMed]
- Amaral, E.P.; Costa, D.L.; Namasivayam, S.; Riteau, N.; Kamenyeva, O.; Mittereder, L.; Mayer-Barber, K.D.; Andrade, B.B.; Sher, A. A Major Role for Ferroptosis in Mycobacterium Tuberculosis–Induced Cell Death and Tissue Necrosis. J. Exp. Med. 2019, 216, 556–570. [Google Scholar] [CrossRef]
- Ma, R.; Fang, L.; Chen, L.; Wang, X.; Jiang, J.; Gao, L. Ferroptotic Stress Promotes Macrophages against Intracellular Bacteria. Theranostics 2022, 12, 2266–2289. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Wang, L.; Wu, Y.; Yu, Y.; Yao, Y.; Yang, H.; Hao, C. Lipid Metabolism in Ferroptosis: Mechanistic Insights and Therapeutic Potential. Front. Immunol. 2025, 16, 1545339. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef]
- Mortensen, M.S.; Ruiz, J.; Watts, J.L. Polyunsaturated Fatty Acids Drive Lipid Peroxidation during Ferroptosis. Cells 2023, 12, 804. [Google Scholar] [CrossRef]
- Vani Raju, M.; Kaniyur Chandrasekaran, M.; Muthaiyan Ahalliya, R.; Velliyur Kanniappan, G. Reconnoitering the Role of Lipid Metabolites in Ferroptosis. Adv. Redox Res. 2025, 14, 100117. [Google Scholar] [CrossRef]
- Collin, F. Chemical Basis of Reactive Oxygen Species Reactivity and Involvement in Neurodegenerative Diseases. Int. J. Mol. Sci. 2019, 20, 2407. [Google Scholar] [CrossRef]
- Su, L.-J.; Zhang, J.-H.; Gomez, H.; Murugan, R.; Hong, X.; Xu, D.; Jiang, F.; Peng, Z.-Y. Reactive Oxygen Species-Induced Lipid Peroxidation in Apoptosis, Autophagy, and Ferroptosis. Oxid. Med. Cell. Longev. 2019, 2019, 5080843. [Google Scholar] [CrossRef]
- Ma, X.-H.; Liu, J.-H.-Z.; Liu, C.-Y.; Sun, W.-Y.; Duan, W.-J.; Wang, G.; Kurihara, H.; He, R.-R.; Li, Y.-F.; Chen, Y.; et al. ALOX15-Launched PUFA-Phospholipids Peroxidation Increases the Susceptibility of Ferroptosis in Ischemia-Induced Myocardial Damage. Signal Transduct. Target. Ther. 2022, 7, 288. [Google Scholar] [CrossRef]
- Kim, J.W.; Lee, J.-Y.; Oh, M.; Lee, E.-W. An Integrated View of Lipid Metabolism in Ferroptosis Revisited via Lipidomic Analysis. Exp. Mol. Med. 2023, 55, 1620–1631. [Google Scholar] [CrossRef]
- Chandimali, N.; Bak, S.G.; Park, E.H.; Lim, H.-J.; Won, Y.-S.; Kim, E.-K.; Park, S.-I.; Lee, S.J. Free Radicals and Their Impact on Health and Antioxidant Defenses: A Review. Cell Death Discov. 2025, 11, 19. [Google Scholar] [CrossRef]
- Li Pomi, F.; Gammeri, L.; Borgia, F.; Di Gioacchino, M.; Gangemi, S. Oxidative Stress and Skin Diseases: The Role of Lipid Peroxidation. Antioxidants 2025, 14, 555. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Luo, Q.; Zhao, Y.; Ren, P.; Jin, Y.; Zhou, J. The Ferroptosis–Mitochondrial Axis in Depression: Unraveling the Feedforward Loop of Oxidative Stress, Metabolic Homeostasis Dysregulation, and Neuroinflammation. Antioxidants 2025, 14, 613. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Tang, D.; Wang, Y.; Li, X.X.; Bao, H.; Tang, C.; Dong, X.; Li, X.X.; Yang, Q.; Yan, Y.; et al. The Mechanism of Ferroptosis and Its Related Diseases. Mol. Biomed. 2023, 4, 33. [Google Scholar] [CrossRef] [PubMed]
- Pope, L.E.; Dixon, S.J. Regulation of Ferroptosis by Lipid Metabolism. Trends Cell Biol. 2023, 33, 1077–1087. [Google Scholar] [CrossRef]
- Lyamzaev, K.G.; Panteleeva, A.A.; Simonyan, R.A.; Avetisyan, A.V.; Chernyak, B.V. The Critical Role of Mitochondrial Lipid Peroxidation in Ferroptosis: Insights from Recent Studies. Biophys. Rev. 2023, 15, 875–885. [Google Scholar] [CrossRef]
- Panov, A.V.; Dikalov, S.I. Cardiolipin, Perhydroxyl Radicals, and Lipid Peroxidation in Mitochondrial Dysfunctions and Aging. Oxid. Med. Cell. Longev. 2020, 2020, 1323028. [Google Scholar] [CrossRef]
- dos Santos, A.F.; Fazeli, G.; Xavier da Silva, T.N.; Friedmann Angeli, J.P. Ferroptosis: Mechanisms and Implications for Cancer Development and Therapy Response. Trends Cell Biol. 2023, 33, 1062–1076. [Google Scholar] [CrossRef]
- Weaver, K.; Skouta, R. The Selenoprotein Glutathione Peroxidase 4: From Molecular Mechanisms to Novel Therapeutic Opportunities. Biomedicines 2022, 10, 891. [Google Scholar] [CrossRef]
- Wei, C. The Role of Glutathione Peroxidase 4 in Neuronal Ferroptosis and Its Therapeutic Potential in Ischemic and Hemorrhagic Stroke. Brain Res. Bull. 2024, 217, 111065. [Google Scholar] [CrossRef] [PubMed]
- Costa, I.; Barbosa, D.J.; Benfeito, S.; Silva, V.; Chavarria, D.; Borges, F.; Remião, F.; Silva, R. Molecular Mechanisms of Ferroptosis and Their Involvement in Brain Diseases. Pharmacol. Ther. 2023, 244, 108373. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Pratt, D.A. Ferroptosis: A Flexible Constellation of Related Biochemical Mechanisms. Mol. Cell 2023, 83, 1030–1042. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Meng, Y.; Li, D.; Yao, L.; Le, J.; Liu, Y.; Sun, Y.; Zeng, F.; Chen, X.; Deng, G. Ferroptosis in Cancer: From Molecular Mechanisms to Therapeutic Strategies. Signal Transduct. Target. Ther. 2024, 9, 55. [Google Scholar] [CrossRef]
- Piccolo, M.; Ferraro, M.G.; Iazzetti, F.; Santamaria, R.; Irace, C. Insight into Iron, Oxidative Stress and Ferroptosis: Therapy Targets for Approaching Anticancer Strategies. Cancers 2024, 16, 1220. [Google Scholar] [CrossRef]
- Cheng, Y.; Song, Y.; Chen, H.; Li, Q.; Gao, Y.; Lu, G.; Luo, C. Ferroptosis Mediated by Lipid Reactive Oxygen Species: A Possible Causal Link of Neuroinflammation to Neurological Disorders. Oxid. Med. Cell. Longev. 2021, 2021, 5005136. [Google Scholar] [CrossRef]
- Lv, H.; Shang, P. The Significance, Trafficking and Determination of Labile Iron in Cytosol, Mitochondria and Lysosomes. Metallomics 2018, 10, 899–916. [Google Scholar] [CrossRef]
- Zhang, Y.; Lu, Y.; Jin, L. Iron Metabolism and Ferroptosis in Physiological and Pathological Pregnancy. Int. J. Mol. Sci. 2022, 23, 9395. [Google Scholar] [CrossRef]
- Abdukarimov, N.; Kokabi, K.; Kunz, J. Ferroptosis and Iron Homeostasis: Molecular Mechanisms and Neurodegenerative Disease Implications. Antioxidants 2025, 14, 527. [Google Scholar] [CrossRef]
- Antonelli, A.; Battaglia, A.M.; Sacco, A.; Petriaggi, L.; Giorgio, E.; Barone, S.; Biamonte, F.; Giudice, A. Ferroptosis and Oral Squamous Cell Carcinoma: Connecting the Dots to Move Forward. Front. Oral Health 2024, 5, 1461022. [Google Scholar] [CrossRef]
- Recalcati, S.; Cairo, G. Macrophages and Iron: A Special Relationship. Biomedicines 2021, 9, 1585. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Feng, Y.; Li, Y.; He, P.; Zhou, Q.; Tian, Y.; Yao, R.; Yao, Y. Ferritinophagy: A Novel Insight into the Double-edged Sword in Ferritinophagy—Ferroptosis Axis and Human Diseases. Cell Prolif. 2024, 57, e13621. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Zhou, Y.-L.; Mao, J.-A.; Tang, L.-F.; Xu, J.; Wang, Z.-X.; He, Y.; Li, M. NCOA4-Mediated Ferritinophagy Is Involved in Ionizing Radiation-Induced Ferroptosis of Intestinal Epithelial Cells. Redox Biol. 2022, 55, 102413. [Google Scholar] [CrossRef] [PubMed]
- Nadimpalli, H.P.; Katsioudi, G.; Arpa, E.S.; Chikhaoui, L.; Arpat, A.B.; Liechti, A.; Palais, G.; Tessmer, C.; Hofmann, I.; Galy, B.; et al. Diurnal Control of Iron Responsive Element Containing MRNAs through Iron Regulatory Proteins IRP1 and IRP2 Is Mediated by Feeding Rhythms. Genome Biol. 2024, 25, 128. [Google Scholar] [CrossRef]
- Catapano, A.; Cimmino, F.; Petrella, L.; Pizzella, A.; D’Angelo, M.; Ambrosio, K.; Marino, F.; Sabbatini, A.; Petrelli, M.; Paolini, B.; et al. Iron Metabolism and Ferroptosis in Health and Diseases: The Crucial Role of Mitochondria in Metabolically Active Tissues. J. Nutr. Biochem. 2025, 140, 109888. [Google Scholar] [CrossRef]
- Ren, Y.; Mao, X.; Xu, H.; Dang, Q.; Weng, S.; Zhang, Y.; Chen, S.; Liu, S.; Ba, Y.; Zhou, Z.; et al. Ferroptosis and EMT: Key Targets for Combating Cancer Progression and Therapy Resistance. Cell. Mol. Life Sci. 2023, 80, 263. [Google Scholar] [CrossRef]
- Ma, T.; Du, J.; Zhang, Y.; Wang, Y.; Wang, B.; Zhang, T. GPX4-Independent Ferroptosis—A New Strategy in Disease’s Therapy. Cell Death Discov. 2022, 8, 434. [Google Scholar] [CrossRef]
- Lee, J.; Roh, J.-L. Unleashing Ferroptosis in Human Cancers: Targeting Ferroptosis Suppressor Protein 1 for Overcoming Therapy Resistance. Antioxidants 2023, 12, 1218. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, Y.; Liao, Y.; Zhu, C.; Zou, Z. GPX4, Ferroptosis, and Diseases. Biomed. Pharmacother. 2024, 174, 116512. [Google Scholar] [CrossRef]
- Ufer, C.; Wang, C.C.; Fähling, M.; Schiebel, H.; Thiele, B.J.; Billett, E.E.; Kuhn, H.; Borchert, A. Translational Regulation of Glutathione Peroxidase 4 Expression through Guanine-Rich Sequence-Binding Factor 1 Is Essential for Embryonic Brain Development. Genes Dev. 2008, 22, 1838–1850. [Google Scholar] [CrossRef]
- Berndt, C.; Alborzinia, H.; Amen, V.S.; Ayton, S.; Barayeu, U.; Bartelt, A.; Bayir, H.; Bebber, C.M.; Birsoy, K.; Böttcher, J.P.; et al. Ferroptosis in Health and Disease. Redox Biol. 2024, 75, 103211. [Google Scholar] [CrossRef] [PubMed]
- Jomova, K.; Alomar, S.Y.; Alwasel, S.H.; Nepovimova, E.; Kuca, K.; Valko, M. Several Lines of Antioxidant Defense against Oxidative Stress: Antioxidant Enzymes, Nanomaterials with Multiple Enzyme-Mimicking Activities, and Low-Molecular-Weight Antioxidants. Arch. Toxicol. 2024, 98, 1323–1367. [Google Scholar] [CrossRef] [PubMed]
- Avolio, R.; Matassa, D.S.; Criscuolo, D.; Landriscina, M.; Esposito, F. Modulation of Mitochondrial Metabolic Reprogramming and Oxidative Stress to Overcome Chemoresistance in Cancer. Biomolecules 2020, 10, 135. [Google Scholar] [CrossRef] [PubMed]
- Li, F.-J.; Long, H.-Z.; Zhou, Z.-W.; Luo, H.-Y.; Xu, S.-G.; Gao, L.-C. System Xc−/GSH/GPX4 Axis: An Important Antioxidant System for the Ferroptosis in Drug-Resistant Solid Tumor Therapy. Front. Pharmacol. 2022, 13, 910292. [Google Scholar] [CrossRef]
- Bersuker, K.; Hendricks, J.M.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R.; et al. The CoQ Oxidoreductase FSP1 Acts Parallel to GPX4 to Inhibit Ferroptosis. Nature 2019, 575, 688–692. [Google Scholar] [CrossRef]
- Zhang, C.; Liu, X.; Jin, S.; Chen, Y.; Guo, R. Ferroptosis in Cancer Therapy: A Novel Approach to Reversing Drug Resistance. Mol. Cancer 2022, 21, 47. [Google Scholar] [CrossRef]
- Xu, H.; Yang, L.; Wu, Y.; Lei, H. Double-Edged Sword Effect of GPX4 in Skin Homeostasis and Diseases. Arch. Dermatol. Res. 2025, 317, 404. [Google Scholar] [CrossRef]
- Yang, Y.; Zuo, S.; Li, L.; Kuang, X.; Li, J.; Sun, B.; Wang, S.; He, Z.; Sun, J. Iron-Doxorubicin Prodrug Loaded Liposome Nanogenerator Programs Multimodal Ferroptosis for Efficient Cancer Therapy. Asian J. Pharm. Sci. 2021, 16, 784–793. [Google Scholar] [CrossRef]
- Wang, F.; Min, J. DHODH Tangoing with GPX4 on the Ferroptotic Stage. Signal Transduct. Target. Ther. 2021, 6, 244. [Google Scholar] [CrossRef]
- Lyamzaev, K.G.; Panteleeva, A.A.; Simonyan, R.A.; Avetisyan, A.V.; Chernyak, B.V. Mitochondrial Lipid Peroxidation Is Responsible for Ferroptosis. Cells 2023, 12, 611. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Kroemer, G. Peroxisome: The New Player in Ferroptosis. Signal Transduct. Target. Ther. 2020, 5, 273. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Guo, Z. Recent Progress in Ferroptosis: Inducers and Inhibitors. Cell Death Discov. 2022, 8, 501. [Google Scholar] [CrossRef] [PubMed]
- Salnikow, K. Role of Iron in Cancer. Semin. Cancer Biol. 2021, 76, 189–194. [Google Scholar] [CrossRef]
- Tu, H.; Tang, L.J.; Luo, X.J.; Ai, K.L.; Peng, J. Insights into the Novel Function of System Xc– in Regulated Cell Death. Eur. Rev. Med. Pharmacol. Sci. 2021, 25, 1650–1662. [Google Scholar] [CrossRef]
- Pasini, A.M.F.; Stranieri, C.; Girelli, D.; Busti, F.; Cominacini, L. Is Ferroptosis a Key Component of the Process Leading to Multiorgan Damage in COVID-19? Antioxidants 2021, 10, 1677. [Google Scholar] [CrossRef]
- Brown, S.M.; Sinha, B.K.; Cannon, R.E. A Role for INOS in Erastin Mediated Reduction of P-Glycoprotein Transport Activity. Cancers 2024, 16, 1733. [Google Scholar] [CrossRef]
- Li, S.; He, Y.; Chen, K.; Sun, J.; Zhang, L.; He, Y.; Yu, H.; Li, Q. RSL3 Drives Ferroptosis through NF- κ B Pathway Activation and GPX4 Depletion in Glioblastoma. Oxid. Med. Cell. Longev. 2021, 2021, 2915019. [Google Scholar] [CrossRef]
- Cheff, D.M.; Huang, C.; Scholzen, K.C.; Gencheva, R.; Ronzetti, M.H.; Cheng, Q.; Hall, M.D.; Arnér, E.S.J. The Ferroptosis Inducing Compounds RSL3 and ML162 Are Not Direct Inhibitors of GPX4 but of TXNRD1. Redox Biol. 2023, 62, 102703. [Google Scholar] [CrossRef]
- Zhang, X.; Guo, Y.; Li, H.; Han, L. FIN56, a Novel Ferroptosis Inducer, Triggers Lysosomal Membrane Permeabilization in a TFEB-Dependent Manner in Glioblastoma. J. Cancer 2021, 12, 6610–6619. [Google Scholar] [CrossRef]
- Chen, X.; Li, J.; Kang, R.; Klionsky, D.J.; Tang, D. Ferroptosis: Machinery and Regulation. Autophagy 2021, 17, 2054–2081. [Google Scholar] [CrossRef]
- Gaschler, M.M.; Andia, A.A.; Liu, H.; Csuka, J.M.; Hurlocker, B.; Vaiana, C.A.; Heindel, D.W.; Zuckerman, D.S.; Bos, P.H.; Reznik, E.; et al. FINO2 Initiates Ferroptosis through GPX4 Inactivation and Iron Oxidation. Nat. Chem. Biol. 2018, 14, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.L.; Chen, J.X.; Zhu, P.; Zhang, C.B.; Zhou, Y.; Duan, J.X. Focus on Ferroptosis Regulation: Exploring Novel Mechanisms and Applications of Ferroptosis Regulator. Life Sci. 2022, 307, 120868. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A.; Satapathy, T.; Sen, K.; Sahu, S. Pharmacological Targeting of Ferroptosis in Cancer Treatment. J. Drug Deliv. Ther. 2024, 14, 205–221. [Google Scholar] [CrossRef]
- Zhao, C.; Yu, Y.; Yin, G.; Xu, C.; Wang, J.; Wang, L.; Zhao, G.; Ni, S.; Zhang, H.; Zhou, B.; et al. Sulfasalazine Promotes Ferroptosis through AKT-ERK1/2 and P53-SLC7A11 in Rheumatoid Arthritis. Inflammopharmacology 2024, 32, 1277–1294. [Google Scholar] [CrossRef]
- Zheng, J.; Sato, M.; Mishima, E.; Sato, H.; Proneth, B.; Conrad, M. Sorafenib Fails to Trigger Ferroptosis across a Wide Range of Cancer Cell Lines. Cell Death Dis. 2021, 12, 698. [Google Scholar] [CrossRef]
- Gu, Y.; Li, Y.; Wang, J.; Zhang, L.; Zhang, J.; Wang, Y. Targeting Ferroptosis: Paving New Roads for Drug Design and Discovery. Eur. J. Med. Chem. 2023, 247, 115015. [Google Scholar] [CrossRef]
- Zilka, O.; Shah, R.; Li, B.; Friedmann Angeli, J.P.; Griesser, M.; Conrad, M.; Pratt, D.A. On the Mechanism of Cytoprotection by Ferrostatin-1 and Liproxstatin-1 and the Role of Lipid Peroxidation in Ferroptotic Cell Death. ACS Cent. Sci. 2017, 3, 232–243. [Google Scholar] [CrossRef]
- Scarpellini, C.; Klejborowska, G.; Lanthier, C.; Hassannia, B.; Vanden Berghe, T.; Augustyns, K. Beyond Ferrostatin-1: A Comprehensive Review of Ferroptosis Inhibitors. Trends Pharmacol. Sci. 2023, 44, 902–916. [Google Scholar] [CrossRef]
- Bao, C.; Liu, C.; Liu, Q.; Hua, L.; Hu, J.; Li, Z.; Xu, S. Liproxstatin-1 Alleviates LPS/IL-13-Induced Bronchial Epithelial Cell Injury and Neutrophilic Asthma in Mice by Inhibiting Ferroptosis. Int. Immunopharmacol. 2022, 109, 108770. [Google Scholar] [CrossRef]
- Cheon, Y., II; Kim, J.M.; Shin, S.C.; Kim, H.S.; Lee, J.C.; Park, G.C.; Sung, E.S.; Lee, M.; Lee, B.J. Effect of Deferoxamine and Ferrostatin-1 on Salivary Gland Dysfunction in Ovariectomized Rats. Aging 2023, 15, 2418–2432. [Google Scholar] [CrossRef]
- Zhang, M.; Chen, X.; Zhang, Y. Mechanisms of Vitamins Inhibiting Ferroptosis. Antioxidants 2024, 13, 1571. [Google Scholar] [CrossRef] [PubMed]
- Griesser, M.; Shah, R.; Van Kessel, A.T.; Zilka, O.; Haidasz, E.A.; Pratt, D.A. The Catalytic Reaction of Nitroxides with Peroxyl Radicals and Its Relevance to Their Cytoprotective Properties. J. Am. Chem. Soc. 2018, 140, 3798–3808. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Gou, S.; Zhang, Q.; Yong, X.; Gan, B.; Jia, D. FSP1 Oxidizes NADPH to Suppress Ferroptosis. Cell Res. 2023, 33, 967–970. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Tsui, M.G.; Tsang, J.K.W.; Goit, R.K.; Yao, K.M.; So, K.F.; Lam, W.C.; Lo, A.C.Y. Involvement of FSP1-CoQ10-NADH and GSH-GPx-4 Pathways in Retinal Pigment Epithelium Ferroptosis. Cell Death Dis. 2022, 13, 468. [Google Scholar] [CrossRef]
- Entezari, S.; Haghi, S.M.; Norouzkhani, N.; Sahebnazar, B.; Vosoughian, F.; Akbarzadeh, D.; Islampanah, M.; Naghsh, N.; Abbasalizadeh, M.; Deravi, N. Iron Chelators in Treatment of Iron Overload. J. Toxicol. 2022, 2022, 4911205. [Google Scholar] [CrossRef]
- Horinouchi, T.; Mazaki, Y.; Miwa, S. Mechanism of Cytotoxicity Induced by the Cigarette Smoke Extract (CSE) of Heated Tobacco Products in Vascular Smooth Muscle Cells: A Comparative Study of the Cytotoxic Effects of CSE and the Ferroptosis Inducer, Erastin. J. Pharmacol. Sci. 2024, 154, 86–96. [Google Scholar] [CrossRef]
- Agrawal, S.; Fox, J.; Baskaran, T.; Fox, J. Brain Mitochondrial Iron Accumulates in Huntington’s Disease, Mediates Mitochondrial Dysfunction, and Can Be Removed Pharmacologically. Free Radic. Biol. Med. 2018, 120, 317–329. [Google Scholar] [CrossRef]
- Liu, X.; Du, Y.; Liu, J.; Cheng, L.; He, W.; Zhang, W. Ferrostatin-1 Alleviates Cerebral Ischemia/Reperfusion Injury through Activation of the AKT/GSK3β Signaling Pathway. Brain Res. Bull. 2023, 193, 146–157. [Google Scholar] [CrossRef]
- Lin, L.; Ling, X.; Chen, T.; Zhou, Q.; Huang, J.; Huang, L.; Lin, X.; Lin, L. Inhibition of Hippocampal Neuronal Ferroptosis by Liproxstatin-1 Improves Learning and Memory Function in Aged Mice with Perioperative Neurocognitive Dysfunction. J. Inflamm. Res. 2025, 18, 2991–3007. [Google Scholar] [CrossRef]
- Sampilvanjil, A.; Karasawa, T.; Yamada, N.; Komada, T.; Higashi, T.; Baatarjav, C.; Watanabe, S.; Kamata, R.; Ohno, N.; Takahashi, M. Cigarette Smoke Extract Induces Ferroptosis in Vascular Smooth Muscle Cells. Am. J. Physiol.—Heart Circ. Physiol. 2020, 318, H508–H518. [Google Scholar] [CrossRef] [PubMed]
- Stepanić, V.; Kučerová-Chlupáčová, M. Review and Chemoinformatic Analysis of Ferroptosis Modulators with a Focus on Natural Plant Products. Molecules 2023, 28, 475. [Google Scholar] [CrossRef] [PubMed]
- Lillo-Moya, J.; Rojas-Solé, C.; Muñoz-Salamanca, D.; Panieri, E.; Saso, L.; Rodrigo, R. Targeting Ferroptosis against Ischemia/Reperfusion Cardiac Injury. Antioxidants 2021, 10, 667. [Google Scholar] [CrossRef]
- Cruz-Gregorio, A.; Aranda-Rivera, A.K. Quercetin and Ferroptosis. Life 2023, 13, 1730. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.W.; Cai, S.; Zhao, T.S.; Li, M.; Tian, Y. Green Tea Derivative (−)-Epigallocatechin-3-Gallate (EGCG) Confers Protection against Ionizing Radiation-Induced Intestinal Epithelial Cell Death Both in Vitro and in Vivo. Free Radic. Biol. Med. 2020, 161, 175–186. [Google Scholar] [CrossRef]
- Chen, Y.; Huang, L.; Zhang, H.; Diao, X.; Zhao, S.; Zhou, W. Reduction in Autophagy by (-)-Epigallocatechin-3-Gallate (EGCG): A Potential Mechanism of Prevention of Mitochondrial Dysfunction After Subarachnoid Hemorrhage. Mol. Neurobiol. 2017, 54, 392–405. [Google Scholar] [CrossRef]
- Jiang, Y.; Hui, D.; Pan, Z.; Yu, Y.; Liu, L.; Yu, X.; Wu, C.; Sun, M. Curcumin Promotes Ferroptosis in Hepatocellular Carcinoma via Upregulation of ACSL4. J. Cancer Res. Clin. Oncol. 2024, 150, 429. [Google Scholar] [CrossRef]
- Hu, Y.; Guo, N.; Yang, T.; Yan, J.; Wang, W.; Li, X. The Potential Mechanisms by Which Artemisinin and Its Derivatives Induce Ferroptosis in the Treatment of Cancer. Oxid. Med. Cell. Longev. 2022, 2022, 1458143. [Google Scholar] [CrossRef]
- Di Giacomo, C.; Malfa, G.A.; Tomasello, B.; Bianchi, S.; Acquaviva, R. Natural Compounds and Glutathione: Beyond Mere Antioxidants. Antioxidants 2023, 12, 1445. [Google Scholar] [CrossRef]
- Wang, W.; Zhang, N. Oridonin Inhibits Hela Cell Proliferation via Downregulation of Glutathione Metabolism: A New Insight from Metabolomics. J. Pharm. Pharmacol. 2023, 75, 837–845. [Google Scholar] [CrossRef]
- Wang, X.; Li, Y.; Han, L.; Li, J.; Liu, C.; Sun, C. Role of Flavonoids in the Treatment of Iron Overload. Front. Cell Dev. Biol. 2021, 9, 685364. [Google Scholar] [CrossRef]
- Wang, I.C.; Lin, J.H.; Lee, W.S.; Liu, C.H.; Lin, T.Y.; Yang, K.T. Baicalein and Luteolin Inhibit Ischemia/Reperfusion-Induced Ferroptosis in Rat Cardiomyocytes. Int. J. Cardiol. 2023, 375, 74–86. [Google Scholar] [CrossRef]
- Jiang, Z.; Sun, H.; Miao, J.; Sheng, Q.; Xu, J.; Gao, Z.; Zhang, X.; Song, Y.; Chen, K. The Natural Flavone Acacetin Protects against High-Fat Diet-Induced Lipid Accumulation in the Liver via the Endoplasmic Reticulum Stress/Ferroptosis Pathway. Biochem. Biophys. Res. Commun. 2023, 640, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Farhan, M. Insights on the Role of Polyphenols in Combating Cancer Drug Resistance. Biomedicines 2023, 11, 1709. [Google Scholar] [CrossRef] [PubMed]
- Houessinon, A.; François, C.; Sauzay, C.; Louandre, C.; Mongelard, G.; Godin, C.; Bodeau, S.; Takahashi, S.; Saidak, Z.; Gutierrez, L.; et al. Metallothionein-1 as a Biomarker of Altered Redox Metabolism in Hepatocellular Carcinoma Cells Exposed to Sorafenib. Mol. Cancer 2016, 15, 38. [Google Scholar] [CrossRef]
- Wang, Q.; Bin, C.; Xue, Q.; Gao, Q.; Huang, A.; Wang, K.; Tang, N. GSTZ1 Sensitizes Hepatocellular Carcinoma Cells to Sorafenib-Induced Ferroptosis via Inhibition of NRF2/GPX4 Axis. Cell Death Dis. 2021, 12, 426. [Google Scholar] [CrossRef]
- Li, X.; Yu, Q.; Zhao, R.; Guo, X.; Liu, C.; Zhang, K.; Zhang, W.; Liu, J.; Yu, J.; Wang, S.; et al. Designer Exosomes for Targeted Delivery of a Novel Therapeutic Cargo to Enhance Sorafenib-Mediated Ferroptosis in Hepatocellular Carcinoma. Front. Oncol. 2022, 12, 898156. [Google Scholar] [CrossRef]
- Li, X.; Li, Y.; Lian, P.; Lv, Q.; Liu, F. Silencing LncRNA HCG18 Regulates GPX4-Inhibited Ferroptosis by Adsorbing MiR-450b-5p to Avert Sorafenib Resistance in Hepatocellular Carcinoma. Hum. Exp. Toxicol. 2023, 42, 1–14. [Google Scholar] [CrossRef]
- Zhao, Y.; Li, Y.; Zhang, R.; Wang, F.; Wang, T.; Jiao, Y. The Role of Erastin in Ferroptosis and Its Prospects in Cancer Therapy. Onco Targets Ther. 2020, 13, 5429–5441. [Google Scholar] [CrossRef]
- Devericks, E.N.; Brosnan, B.H.; Ho, A.N.; Glenny, E.M.; Malian, H.M.; Teegarden, D.; Wendt, M.K.; Coleman, M.F.; Hursting, S.D. Glutathione Peroxidase 4 (GPX4) and Obesity Interact to Impact Tumor Progression and Treatment Response in Triple Negative Breast Cancer. Cancer Metab. 2025, 13, 11. [Google Scholar] [CrossRef]
- Yuan, J.; Liu, C.; Jiang, C.; Liu, N.; Yang, Z.; Xing, H. RSL3 Induces Ferroptosis by Activating the NF-ΚB Signalling Pathway to Enhance the Chemosensitivity of Triple-Negative Breast Cancer Cells to Paclitaxel. Sci. Rep. 2025, 15, 1654. [Google Scholar] [CrossRef]
- Huo, L.; Liu, C.; Yuan, Y.; Liu, X.; Cao, Q. Pharmacological Inhibition of Ferroptosis as a Therapeutic Target for Sepsis-Associated Organ Damage. Eur. J. Med. Chem. 2023, 257, 115438. [Google Scholar] [CrossRef]
- Shikuma, A.; Kami, D.; Maeda, R.; Suzuki, Y.; Sano, A.; Taya, T.; Ogata, T.; Konkel, A.; Matoba, S.; Schunck, W.H.; et al. Amelioration of Endotoxemia by a Synthetic Analog of Omega-3 Epoxyeicosanoids. Front. Immunol. 2022, 13, 825171. [Google Scholar] [CrossRef]
- Chen, G.Q.; Benthani, F.A.; Wu, J.; Liang, D.; Bian, Z.X.; Jiang, X. Artemisinin Compounds Sensitize Cancer Cells to Ferroptosis by Regulating Iron Homeostasis. Cell Death Differ. 2020, 27, 242–254. [Google Scholar] [CrossRef]
- Kerkhove, L.; Geirnaert, F.; Rifi, A.L.; Law, K.L.; Gutiérrez, A.; Oudaert, I.; Corbet, C.; Gevaert, T.; Dufait, I.; De Ridder, M. Repurposing Sulfasalazine as a Radiosensitizer in Hypoxic Human Colorectal Cancer. Cancers 2023, 15, 2363. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Shen, G.; Fan, M.; Zheng, P. Lipid Metabolic Reprogramming and Associated Ferroptosis in Osteosarcoma: From Molecular Mechanisms to Potential Targets. J. Bone Oncol. 2025, 51, 100660. [Google Scholar] [CrossRef] [PubMed]
- Mallick, R.; Bhowmik, P.; Duttaroy, A.K. Targeting Fatty Acid Uptake and Metabolism in Cancer Cells: A Promising Strategy for Cancer Treatment. Biomed. Pharmacother. 2023, 167, 115591. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Shi, F.; Cao, F.; Wang, L.; She, J.; He, B.; Xu, X.; Kong, L.; Cai, B. Neutrophils in Tissue Injury and Repair: Molecular Mechanisms and Therapeutic Targets. MedComm 2025, 6, e70184. [Google Scholar] [CrossRef]
- Liang, D.; Minikes, A.M.; Jiang, X. Ferroptosis at the Intersection of Lipid Metabolism and Cellular Signaling. Mol. Cell 2022, 82, 2215–2227. [Google Scholar] [CrossRef]
- Zhao, Q.; Yu, H.; Shi, M.; Wang, X.; Fan, Z.; Wang, Z. Tumor Microenvironment Characteristics of Lipid Metabolism Reprogramming Related to Ferroptosis and EndMT Influencing Prognosis in Gastric Cancer. Int. Immunopharmacol. 2024, 137, 112433. [Google Scholar] [CrossRef]
- Bhol, N.K.; Bhanjadeo, M.M.; Singh, A.K.; Dash, U.C.; Ojha, R.R.; Majhi, S.; Duttaroy, A.K.; Jena, A.B. The Interplay between Cytokines, Inflammation, and Antioxidants: Mechanistic Insights and Therapeutic Potentials of Various Antioxidants and Anti-Cytokine Compounds. Biomed. Pharmacother. 2024, 178, 117177. [Google Scholar] [CrossRef]
- Wang, H.; Liu, Y.; Che, S.; Li, X.; Tang, D.; Lv, S.; Zhao, H. Deciphering the Link: Ferroptosis and Its Role in Glioma. Front. Immunol. 2024, 15, 1346585. [Google Scholar] [CrossRef]
- Broadfield, L.A.; Pane, A.A.; Talebi, A.; Swinnen, J.V.; Fendt, S.-M. Lipid Metabolism in Cancer: New Perspectives and Emerging Mechanisms. Dev. Cell 2021, 56, 1363–1393. [Google Scholar] [CrossRef]
- Zhu, K.; Cai, Y.; Lan, L.; Luo, N. Tumor Metabolic Reprogramming and Ferroptosis: The Impact of Glucose, Protein, and Lipid Metabolism. Int. J. Mol. Sci. 2024, 25, 13413. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Wang, X.; Song, C.; He, Z.; Wang, R.; Xu, Y.; Jiang, G.; Wan, Y.; Mei, J.; Mao, W. The Role of Lipid Metabolic Reprogramming in Tumor Microenvironment. Theranostics 2023, 13, 1774–1808. [Google Scholar] [CrossRef]
- Yan, R.; Lin, B.; Jin, W.; Tang, L.; Hu, S.; Cai, R. NRF2, a Superstar of Ferroptosis. Antioxidants 2023, 12, 1739. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Peng, J.; Cheng, P.; Yang, C.; Gong, S.; Zhang, L.; Zhang, T.; Peng, J. Mechanistic Elucidation of Ferroptosis and Ferritinophagy: Implications for Advancing Our Understanding of Arthritis. Front. Physiol. 2024, 15, 1290234. [Google Scholar] [CrossRef] [PubMed]
- Gryzik, M.; Asperti, M.; Denardo, A.; Arosio, P.; Poli, M. NCOA4-Mediated Ferritinophagy Promotes Ferroptosis Induced by Erastin, but Not by RSL3 in HeLa Cells. Biochim. Biophys. Acta—Mol. Cell Res. 2021, 1868, 118913. [Google Scholar] [CrossRef]
- Liu, Y.-C.; Gong, Y.-T.; Sun, Q.-Y.; Wang, B.; Yan, Y.; Chen, Y.-X.; Zhang, L.-J.; Zhang, W.-D.; Luan, X. Ferritinophagy Induced Ferroptosis in the Management of Cancer. Cell. Oncol. 2024, 47, 19–35. [Google Scholar] [CrossRef]
- Nairz, M.; Weiss, G. Iron in Infection and Immunity. Mol. Asp. Med. 2020, 75, 100864. [Google Scholar] [CrossRef]
- Nairz, M.; Dichtl, S.; Schroll, A.; Haschka, D.; Tymoszuk, P.; Theurl, I.; Weiss, G. Iron and Innate Antimicrobial Immunity—Depriving the Pathogen, Defending the Host. J. Trace Elem. Med. Biol. 2018, 48, 118–133. [Google Scholar] [CrossRef] [PubMed]
- Georgieva, E.; Ananiev, J.; Yovchev, Y.; Arabadzhiev, G.; Abrashev, H.; Abrasheva, D.; Atanasov, V.; Kostandieva, R.; Mitev, M.; Petkova-Parlapanska, K.; et al. COVID-19 Complications: Oxidative Stress, Inflammation, and Mitochondrial and Endothelial Dysfunction. Int. J. Mol. Sci. 2023, 24, 14876. [Google Scholar] [CrossRef] [PubMed]
- Khasheii, B.; Mahmoodi, P.; Mohammadzadeh, A. Siderophores: Importance in Bacterial Pathogenesis and Applications in Medicine and Industry. Microbiol. Res. 2021, 250, 126790. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Hyun, D.-H. The Interplay between Intracellular Iron Homeostasis and Neuroinflammation in Neurodegenerative Diseases. Antioxidants 2023, 12, 918. [Google Scholar] [CrossRef]
- Rodriguez, G.M.; Sharma, N.; Biswas, A.; Sharma, N. The Iron Response of Mycobacterium Tuberculosis and Its Implications for Tuberculosis Pathogenesis and Novel Therapeutics. Front. Cell. Infect. Microbiol. 2022, 12, 876667. [Google Scholar] [CrossRef]
- Ward, J.L.; Torres-Gonzalez, M.; Ammons, M.C.B. The Influence of Viral Infections on Iron Homeostasis and the Potential for Lactoferrin as a Therapeutic in the Age of the SARS-CoV-2 Pandemic. Nutrients 2022, 14, 3090. [Google Scholar] [CrossRef]
- Qiu, B.; Zandkarimi, F.; Saqi, A.; Castagna, C.; Tan, H.; Sekulic, M.; Miorin, L.; Hibshoosh, H.; Toyokuni, S.; Uchida, K.; et al. Fatal COVID-19 Pulmonary Disease Involves Ferroptosis. Nat. Commun. 2024, 15, 3816. [Google Scholar] [CrossRef]
- Li, S.; Zhang, G.; Hu, J.; Tian, Y.; Fu, X. Ferroptosis at the Nexus of Metabolism and Metabolic Diseases. Theranostics 2024, 14, 5826–5852. [Google Scholar] [CrossRef]
- Bhowmick, S.; Banerjee, S.; Shridhar, V.; Mondal, S. Reprogrammed Immuno-Metabolic Environment of Cancer: The Driving Force of Ferroptosis Resistance. Mol. Cancer 2025, 24, 161. [Google Scholar] [CrossRef]
- Haschka, D.; Hoffmann, A.; Weiss, G. Iron in Immune Cell Function and Host Defense. Semin. Cell Dev. Biol. 2021, 115, 27–36. [Google Scholar] [CrossRef]
- Jinson, S.; Zhang, Z.; Lancaster, G.I.; Murphy, A.J.; Morgan, P.K. Iron, Lipid Peroxidation, and Ferroptosis Play Pathogenic Roles in Atherosclerosis. Cardiovasc. Res. 2025, 121, 44–61. [Google Scholar] [CrossRef]
- Ru, Q.; Li, Y.; Chen, L.; Wu, Y.; Min, J.; Wang, F. Iron Homeostasis and Ferroptosis in Human Diseases: Mechanisms and Therapeutic Prospects. Signal Transduct. Target. Ther. 2024, 9, 271. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.J.; Xu, G.L. Overexpression of Acyl-CoA Ligase 4 (ACSL4) in Patients with Hepatocellular Carcinoma and Its Prognosis. Med. Sci. Monit. 2017, 23, 4343–4350. [Google Scholar] [CrossRef] [PubMed]
- Mielke Cabello, L.A.; Meresman, G.; Darici, D.; Carnovale, N.; Heitkötter, B.; Schulte, M.; Espinoza-Sánchez, N.A.; Le, Q.K.; Kiesel, L.; Schäfer, S.D.; et al. Assessment of the Ferroptosis Regulators: Glutathione Peroxidase 4, Acyl-Coenzyme A Synthetase Long-Chain Family Member 4, and Transferrin Receptor 1 in Patient-Derived Endometriosis Tissue. Biomolecules 2024, 14, 876. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Xu, Z.; Cheng, Y.; Huang, R.; Xie, Y.; Tsai, H.I.; Zha, H.; Xi, L.; Wang, K.; Cheng, X.; et al. Fe3+-Binding Transferrin Nanovesicles Encapsulating Sorafenib Induce Ferroptosis in Hepatocellular Carcinoma. Biomater. Res. 2023, 27, 63. [Google Scholar] [CrossRef]
- Werth, E.G.; Rajbhandari, P.; Stockwell, B.R.; Brown, L.M. Time Course of Changes in Sorafenib-Treated Hepatocellular Carcinoma (HCC) Cells Suggests Involvement of PhosphoRegulated Signaling in Ferroptosis Induction. Proteomics 2020, 20, e2000006. [Google Scholar] [CrossRef]
- Legendre, C.; Garcion, E. Iron Metabolism: A Double-Edged Sword in the Resistance of Glioblastoma to Therapies. Trends Endocrinol. Metab. 2015, 26, 322–331. [Google Scholar] [CrossRef]
- Caverzan, M.D.; Ibarra, L.E. Advancing Glioblastoma Treatment through Iron Metabolism: A Focus on TfR1 and Ferroptosis Innovations. Int. J. Biol. Macromol. 2024, 278, 134777. [Google Scholar] [CrossRef]
- Zhao, J.; Zang, F.; Huo, X.; Zheng, S. Novel Approaches Targeting Ferroptosis in Treatment of Glioma. Front. Neurol. 2023, 14, 1292160. [Google Scholar] [CrossRef]
- Zhang, P.; Zhou, C.; Ren, X.; Jing, Q.; Gao, Y.; Yang, C.; Shen, Y.; Zhou, Y.; Hu, W.; Jin, F.; et al. Inhibiting the Compensatory Elevation of XCT Collaborates with Disulfiram/Copper-Induced GSH Consumption for Cascade Ferroptosis and Cuproptosis. Redox Biol. 2024, 69, 103007. [Google Scholar] [CrossRef]
- Abdel Hadi, N.; Reyes-Castellanos, G.; Carrier, A. Targeting Redox Metabolism in Pancreatic Cancer. Int. J. Mol. Sci. 2021, 22, 1534. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Guo, Y.; Wang, T.; Wang, L.; Yan, Y.; Xia, L.; Bam, R.; Yang, Z.; Lee, H.; Iwawaki, T.; et al. IRE1α Determines Ferroptosis Sensitivity through Regulation of Glutathione Synthesis. Nat. Commun. 2024, 15, 4114. [Google Scholar] [CrossRef] [PubMed]
- Bottoni, L.; Minetti, A.; Realini, G.; Pio, E.; Giustarini, D.; Rossi, R.; Rocchio, C.; Franci, L.; Salvini, L.; Catona, O.; et al. NRF2 Activation by Cysteine as a Survival Mechanism for Triple-Negative Breast Cancer Cells. Oncogene 2024, 43, 1701–1713. [Google Scholar] [CrossRef] [PubMed]
- Desterke, C.; Xiang, Y.; Elhage, R.; Duruel, C.; Chang, Y.; Hamaï, A. Ferroptosis Inducers Upregulate PD-L1 in Recurrent Triple-Negative Breast Cancer. Cancers 2024, 16, 155. [Google Scholar] [CrossRef]
- Fang, K.; Xu, Z.; Jiang, S.; Yan, C.; Tang, D.; Huang, Y. Integrated Profiling Uncovers Prognostic, Immunological, and Pharmacogenomic Features of Ferroptosis in Triple-Negative Breast Cancer. Front. Immunol. 2022, 13, 985861. [Google Scholar] [CrossRef]
- Chu, B.; Kon, N.; Chen, D.; Li, T.; Liu, T.; Jiang, L.; Song, S.; Tavana, O.; Gu, W. ALOX12 Is Required for P53-Mediated Tumour Suppression through a Distinct Ferroptosis Pathway. Nat. Cell Biol. 2019, 21, 579–591. [Google Scholar] [CrossRef]
- Wang, X.; Chen, Y.; Yang, X.; Cheng, L.; He, Z.; Xin, Y.; Huang, S.; Meng, F.; Zhang, P.; Luo, L. Activation of ALOX12 by a Multi-Organelle-Orienting Photosensitizer Drives ACSL4-Independent Cell Ferroptosis. Cell Death Dis. 2022, 13, 1040. [Google Scholar] [CrossRef]
- Li, Y.; Xu, H.; Shi, J.; Li, C.; Li, M.; Zhang, X.; Xue, Q.; Qiu, J.; Cui, L.; Sun, Y.; et al. Regulation of the P53/SLC7A11/GPX4 Pathway by Gentamicin Induces Ferroptosis in HEI-OC1 Cells. Otol. Neurotol. 2024, 45, 947–953. [Google Scholar] [CrossRef]
- Friedmann Angeli, J.P.; Krysko, D.V.; Conrad, M. Ferroptosis at the Crossroads of Cancer-Acquired Drug Resistance and Immune Evasion. Nat. Rev. Cancer 2019, 19, 405–414. [Google Scholar] [CrossRef]
- Greenwood, H.E.; Barber, A.R.; Edwards, R.S.; Tyrrell, W.E.; George, M.E.; dos Santos, S.N.; Baark, F.; Tanc, M.; Khalil, E.; Falzone, A.; et al. Imaging NRF2 Activation in Non-Small Cell Lung Cancer with Positron Emission Tomography. Nat. Commun. 2024, 15, 10484. [Google Scholar] [CrossRef]
- El-Gohary, R.M.; Okasha, A.H.; Abd El-Azeem, A.H.; Abdel Ghafar, M.T.; Ibrahim, S.; Hegab, I.I.; Farghal, E.E.; Shalaby, S.A.F.; Elshora, O.A.; ElMehy, A.E.; et al. Uncovering the Cardioprotective Potential of Diacerein in Doxorubicin Cardiotoxicity: Mitigating Ferritinophagy-Mediated Ferroptosis via Upregulating NRF2/SLC7A11/GPX4 Axis. Antioxidants 2024, 13, 493. [Google Scholar] [CrossRef]
- Koppula, P.; Lei, G.; Zhang, Y.; Yan, Y.; Mao, C.; Kondiparthi, L.; Shi, J.; Liu, X.; Horbath, A.; Das, M.; et al. A Targetable CoQ-FSP1 Axis Drives Ferroptosis- and Radiation-Resistance in KEAP1 Inactive Lung Cancers. Nat. Commun. 2022, 13, 2206. [Google Scholar] [CrossRef]
- Mei, M.; You, Y.; Tan, N.; He, X.; Huang, J. EZH1 Deficiency Promotes Ferroptosis Resistance by Activating NRF2 in Sepsis-Associated Liver Injury. Clin. Epigenet. 2025, 17, 96. [Google Scholar] [CrossRef] [PubMed]
- Floros, K.V.; Cai, J.Y.; Jacob, S.; Kurupi, R.; Fairchild, C.K.; Shende, M.; Coon, C.M.; Powell, K.M.; Belvin, B.R.; Hu, B.; et al. MYCN-Amplified Neuroblastoma Is Addicted to Iron and Vulnerable to Inhibition of the System Xc-/Glutathione Axis. Cancer Res. 2021, 81, 1896–1908. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.S.; Choi, B.; Choi, H.; Ko, M.J.; Kim, D.H.; Kim, D.H. Enhanced Natural Killer Cell Anti-Tumor Activity with Nanoparticles Mediated Ferroptosis and Potential Therapeutic Application in Prostate Cancer. J. Nanobiotechnol 2022, 20, 428. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, S.; Liu, M.; Yang, Z.; Huang, R. Research Progress on Ferroptosis and Nanotechnology-Based Treatment in Triple-Negative Breast Cancer. Breast Cancer Targets Ther. 2024, 16, 347–358. [Google Scholar] [CrossRef]
- Li, J.; He, D.; Li, S.; Xiao, J.; Zhu, Z. Ferroptosis: The Emerging Player in Remodeling Triple-Negative Breast Cancer. Front. Immunol. 2023, 14, 1284057. [Google Scholar] [CrossRef]
- He, G.; Zhang, Y.; Feng, Y.; Chen, T.; Liu, M.; Zeng, Y.; Yin, X.; Qu, S.; Huang, L.; Ke, Y.; et al. SBFI26 Induces Triple-Negative Breast Cancer Cells Ferroptosis via Lipid Peroxidation. J. Cell. Mol. Med. 2024, 28, e18212. [Google Scholar] [CrossRef]
- Wu, Y.C.; Huang, C.S.; Hsieh, M.S.; Huang, C.M.; Setiawan, S.A.; Yeh, C.T.; Kuo, K.T.; Liu, S.C. Targeting of FSP1 Regulates Iron Homeostasis in Drug-Tolerant Persister Head and Neck Cancer Cells via Lipid-Metabolism-Driven Ferroptosis. Aging 2024, 16, 627–647. [Google Scholar] [CrossRef]
- Tian, R.L.; Wang, T.X.; Huang, Z.X.; Yang, Z.; Guan, K.L.; Xiong, Y.; Wang, P.; Ye, D. Temsirolimus Inhibits FSP1 Enzyme Activity to Induce Ferroptosis and Restrain Liver Cancer Progression. J. Mol. Cell Biol. 2025, 16, mjae036. [Google Scholar] [CrossRef]
- Zhi, D.; Yang, T.; Yang, J.; Fu, S.; Zhang, S. Targeting Strategies for Superparamagnetic Iron Oxide Nanoparticles in Cancer Therapy. Acta Biomater. 2020, 102, 13–34. [Google Scholar] [CrossRef] [PubMed]
- Pang, S.; Geng, C.; Fan, Z.; Hou, M.; Mao, H.; Tao, S.; Wang, J.; Wu, Y.; Wei, K.; Li, Y.; et al. Synergistic Effect of Layered Double Hydroxides Nanodosage Form to Induce Apoptosis and Ferroptosis in Breast Cancer. Int. J. Nanomed. 2024, 19, 4199–4215. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.W.; Kim, M.J.; Han, T.H.; Lee, J.Y.; Kim, S.; Kim, H.; Oh, K.J.; Kim, W.K.; Han, B.S.; Bae, K.H.; et al. FSP1 Confers Ferroptosis Resistance in KEAP1 Mutant Non-Small Cell Lung Carcinoma in NRF2-Dependent and -Independent Manner. Cell Death Dis. 2023, 14, 567. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Wu, L.; Chen, Z.; Yang, J.; Chen, X.; Yu, F.; Zheng, F.; Lin, X. MiR-141 Activates Nrf2-Dependent Antioxidant Pathway via down-Regulating the Expression of Keap1 Conferring the Resistance of Hepatocellular Carcinoma Cells to 5-Fluorouracil. Cell. Physiol. Biochem. 2015, 35, 2333–2348. [Google Scholar] [CrossRef]
- Gao, J.; Wang, Q.; Tang, Y.D.; Zhai, J.; Hu, W.; Zheng, C. When Ferroptosis Meets Pathogenic Infections. Trends Microbiol. 2023, 31, 468–479. [Google Scholar] [CrossRef]
- Bagayoko, S.; Meunier, E. Emerging Roles of Ferroptosis in Infectious Diseases. FEBS J. 2022, 289, 7869–7890. [Google Scholar] [CrossRef]
- Qu, G.; Liu, H.; Li, J.; Huang, S.; Zhao, N.; Zeng, L.; Deng, J. GPX4 Is a Key Ferroptosis Biomarker and Correlated with Immune Cell Populations and Immune Checkpoints in Childhood Sepsis. Sci. Rep. 2023, 13, 11358. [Google Scholar] [CrossRef]
- Tong, J.; Lan, X.T.; Zhang, Z.; Liu, Y.; Sun, D.Y.; Wang, X.J.; Ou-Yang, S.X.; Zhuang, C.L.; Shen, F.M.; Wang, P.; et al. Ferroptosis Inhibitor Liproxstatin-1 Alleviates Metabolic Dysfunction-Associated Fatty Liver Disease in Mice: Potential Involvement of PANoptosis. Acta Pharmacol. Sin. 2023, 44, 1014–1028. [Google Scholar] [CrossRef]
- Guo, R.; Fang, X.; Shang, K.; Wen, J.; Ding, K. Induction of Ferroptosis: A New Strategy for the Control of Bacterial Infections. Microbiol. Res. 2024, 284, 127728. [Google Scholar] [CrossRef]
- Wang, J.; Zhu, J.; Ren, S.; Zhang, Z.; Niu, K.; Li, H.; Wu, W.; Peng, C. The Role of Ferroptosis in Virus Infections. Front. Microbiol. 2023, 14, 1279655. [Google Scholar] [CrossRef]
- Chen, J.; Fu, J.; Zhao, S.; Zhang, X.; Chao, Y.; Pan, Q.; Sun, H.; Zhang, J.; Li, B.; Xue, T.; et al. Free Radical and Viral Infection: A Review from the Perspective of Ferroptosis. Vet. Sci. 2023, 10, 456. [Google Scholar] [CrossRef]
- Zarkovic, N.; Jakovcevic, A.; Mataic, A.; Jaganjac, M.; Vukovic, T.; Waeg, G.; Zarkovic, K. Post-Mortem Findings of Inflammatory Cells and the Association of 4-Hydroxynonenal with Systemic Vascular and Oxidative Stress in Lethal COVID-19. Cells 2022, 11, 444. [Google Scholar] [CrossRef] [PubMed]
- Kaur, S.; Bansal, R.; Kollimuttathuillam, S.; Gowda, A.M.; Singh, B.; Mehta, D.; Maroules, M. The Looming Storm: Blood and Cytokines in COVID-19. Blood Rev. 2021, 46, 100743. [Google Scholar] [CrossRef] [PubMed]
- Ali, I.; Muhammad, S.; Naqvi, S.S.Z.H.; Wei, L.; Yan, W.; Khan, M.F.; Shah, W. Hepatitis B Virus-Associated Liver Carcinoma: The Role of Iron Metabolism and Its Modulation. J. Viral Hepat. 2025, 32, e14016. [Google Scholar] [CrossRef]
- Huang, R.; Wu, J.; Ma, Y.; Kang, K. Molecular Mechanisms of Ferroptosis and Its Role in Viral Pathogenesis. Viruses 2023, 15, 2373. [Google Scholar] [CrossRef]
- You, H.; Wang, L.; Bu, F.; Meng, H.; Huang, C.; Fang, G.; Li, J. Ferroptosis: Shedding Light on Mechanisms and Therapeutic Opportunities in Liver Diseases. Cells 2022, 11, 3301. [Google Scholar] [CrossRef]
- Poonkuzhi Naseef, P.; Elayadeth-Meethal, M.; Mohammed Salim, K.T.; Anjana, A.; Muhas, C.; Abdul Vajid, K.; Saheer Kuruniyan, M. Therapeutic Potential of Induced Iron Depletion Using Iron Chelators in Covid-19. Saudi J. Biol. Sci. 2022, 29, 1947–1956. [Google Scholar] [CrossRef]
- Xiao, L.; Huang, H.; Fan, S.; Zheng, B.; Wu, J.; Zhang, J.; Pi, J.; Xu, J.F. Ferroptosis: A Mixed Blessing for Infectious Diseases. Front. Pharmacol. 2022, 13, 992734. [Google Scholar] [CrossRef]
- Fei, Y.; Ding, Y. The Role of Ferroptosis in Neurodegenerative Diseases. Front. Cell. Neurosci. 2024, 18, 1475934. [Google Scholar] [CrossRef]
- Liu, L.; Cui, Y.; Chang, Y.Z.; Yu, P. Ferroptosis-Related Factors in the Substantia Nigra Are Associated with Parkinson’s Disease. Sci. Rep. 2023, 13, 15365. [Google Scholar] [CrossRef]
- Thorwald, M.A.; Godoy-Lugo, J.A.; Garcia, G.; Silva, J.; Kim, M.; Christensen, A.; Mack, W.J.; Head, E.; O’Day, P.A.; Benayoun, B.A.; et al. Iron-Associated Lipid Peroxidation in Alzheimer’s Disease Is Increased in Lipid Rafts with Decreased Ferroptosis Suppressors, Tested by Chelation in Mice. Alzheimer’s Dement. 2025, 21, e14541. [Google Scholar] [CrossRef] [PubMed]
- Tu, L.F.; Zhang, T.Z.; Zhou, Y.F.; Zhou, Q.Q.; Gong, H.B.; Liang, L.; Hai, L.N.; You, N.X.; Su, Y.; Chen, Y.J.; et al. GPX4 Deficiency-Dependent Phospholipid Peroxidation Drives Motor Deficits of ALS. J. Adv. Res. 2023, 43, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Wen, S.; Aki, T.; Unuma, K.; Uemura, K. Chemically Induced Models of Parkinson’s Disease: History and Perspectives for the Involvement of Ferroptosis. Front. Cell. Neurosci. 2020, 14, 581191. [Google Scholar] [CrossRef] [PubMed]
- Kaftan Öcal, G.; Armagan, G. Induction of Ferroptotic Cell Death by Neuromelanin Pigments in Dopaminergic Cells. ACS Chem. Neurosci. 2025, 16, 1500–1510. [Google Scholar] [CrossRef]
- Ayton, S.; Barton, D.; Brew, B.; Brodtmann, A.; Clarnette, R.; Desmond, P.; Devos, D.; Ellis, K.A.; Fazlollahi, A.; Fradette, C.; et al. Deferiprone in Alzheimer Disease: A Randomized Clinical Trial. JAMA Neurol. 2025, 25, 11–18. [Google Scholar] [CrossRef]
- Martin-Bastida, A.; Ward, R.J.; Newbould, R.; Piccini, P.; Sharp, D.; Kabba, C.; Patel, M.C.; Spino, M.; Connelly, J.; Tricta, F.; et al. Brain Iron Chelation by Deferiprone in a Phase 2 Randomised Double-Blinded Placebo Controlled Clinical Trial in Parkinson’s Disease. Sci. Rep. 2017, 7, 1398. [Google Scholar] [CrossRef]
- Lee, J.; Roh, J.-L. Lipid Metabolism in Ferroptosis: Unraveling Key Mechanisms and Therapeutic Potential in Cancer. Biochim. Biophys. Acta—Rev. Cancer 2025, 1880, 189258. [Google Scholar] [CrossRef]
- Punziano, C.; Trombetti, S.; Cesaro, E.; Grosso, M.; Faraonio, R. Antioxidant Systems as Modulators of Ferroptosis: Focus on Transcription Factors. Antioxidants 2024, 13, 298. [Google Scholar] [CrossRef]
- Mas-Bargues, C.; Escrivá, C.; Dromant, M.; Borrás, C.; Viña, J. Lipid Peroxidation as Measured by Chromatographic Determination of Malondialdehyde. Human Plasma Reference Values in Health and Disease. Arch. Biochem. Biophys. 2021, 709, 108941. [Google Scholar] [CrossRef]
- Ioannidis, M.; Tjepkema, J.; Uitbeijerse, M.R.P.; van den Bogaart, G. Immunomodulatory Effects of 4-Hydroxynonenal. Redox Biol. 2025, 85, 103719. [Google Scholar] [CrossRef]
- Milne, G.L.; Nogueira, M.S.; Gao, B.; Sanchez, S.C.; Amin, W.; Thomas, S.; Oger, C.; Galano, J.-M.; Murff, H.J.; Yang, G.; et al. Identification of Novel F2-Isoprostane Metabolites by Specific UDP-Glucuronosyltransferases. Redox Biol. 2024, 70, 103020. [Google Scholar] [CrossRef]
- Gerhardtova, I.; Jankech, T.; Majerova, P.; Piestansky, J.; Olesova, D.; Kovac, A.; Jampilek, J. Recent Analytical Methodologies in Lipid Analysis. Int. J. Mol. Sci. 2024, 25, 2249. [Google Scholar] [CrossRef]
- Tsikas, D. GC–MS and GC–MS/MS Measurement of Malondialdehyde (MDA) in Clinical Studies: Pre-Analytical and Clinical Considerations. J. Mass Spectrom. Adv. Clin. Lab 2023, 30, 10–24. [Google Scholar] [CrossRef] [PubMed]
- Tyurina, Y.Y.; Tyurin, V.A.; Anthonymuthu, T.; Amoscato, A.A.; Sparvero, L.J.; Nesterova, A.M.; Baynard, M.L.; Sun, W.; He, R.; Khaitovich, P.; et al. Redox Lipidomics Technology: Looking for a Needle in a Haystack. Chem. Phys. Lipids 2019, 221, 93–107. [Google Scholar] [CrossRef] [PubMed]
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.F.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized Arachidonic and Adrenic PEs Navigate Cells to Ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Sokol, K.H.; Lee, C.J.; Rogers, T.J.; Waldhart, A.; Ellis, A.E.; Madireddy, S.; Daniels, S.R.; House, R.J.; Ye, X.; Olesnavich, M.; et al. Lipid Availability Influences Ferroptosis Sensitivity in Cancer Cells by Regulating Polyunsaturated Fatty Acid Trafficking. Cell Chem. Biol. 2025, 32, 408–422.e6. [Google Scholar] [CrossRef]
- Dhas, N.; Kudarha, R.; Tiwari, R.; Tiwari, G.; Garg, N.; Kumar, P.; Kulkarni, S.; Kulkarni, J.; Soman, S.; Hegde, A.R.; et al. Recent Advancements in Nanomaterial-Mediated Ferroptosis-Induced Cancer Therapy: Importance of Molecular Dynamics and Novel Strategies. Life Sci. 2024, 346, 122629. [Google Scholar] [CrossRef]
- Sturtevant, D.; Aziz, M.; Romsdahl, T.B.; Corley, C.D.; Chapman, K.D. In Situ Localization of Plant Lipid Metabolites by Matrix-Assisted Laser Desorption/Ionization Mass Spectrometry Imaging (MALDI-MSI). Methods Mol. Biol. 2021, 2295, 417–438. [Google Scholar] [CrossRef]
- Liang, Y.; Feng, Q.; Wang, Z. Mass Spectrometry Imaging as a New Method: To Reveal the Pathogenesis and the Mechanism of Traditional Medicine in Cerebral Ischemia. Front. Pharmacol. 2022, 13, 887050. [Google Scholar] [CrossRef]
- Feng, H.; Stockwell, B.R. Unsolved Mysteries: How Does Lipid Peroxidation Cause Ferroptosis? PLoS Biol. 2018, 16, e2006203. [Google Scholar] [CrossRef]
- Arbatskiy, M.; Balandin, D.; Akberdin, I.; Churov, A. A Systems Biology Approach Towards a Comprehensive Understanding of Ferroptosis. Int. J. Mol. Sci. 2024, 25, 11782. [Google Scholar] [CrossRef]
- Nguyen, C.T.N.; Kim, S.M.; Kang, Y.P. Mass Spectrometry-Based Approaches to Explore Metabolism Regulating Ferroptosis. BMB Rep. 2022, 55, 413–416. [Google Scholar] [CrossRef]
- Ryan, F.; Blex, C.; Ngo, T.D.; Kopp, M.A.; Michalke, B.; Venkataramani, V.; Curran, L.; Schwab, J.M.; Ruprecht, K.; Otto, C.; et al. Ferroptosis Inhibitor Improves Outcome after Early and Delayed Treatment in Mild Spinal Cord Injury. Acta Neuropathol. 2024, 147, 106. [Google Scholar] [CrossRef]
- Michalke, B.; Willkommen, D.; Venkataramani, V. Iron Redox Speciation Analysis Using Capillary Electrophoresis Coupled to Inductively Coupled Plasma Mass Spectrometry (CE-ICP-MS). Front. Chem. 2019, 7, 136. [Google Scholar] [CrossRef]
- Kirkwood-Donelson, K.I.; Jarmusch, A.K.; Bortner, C.D.; Merrick, B.A.; Sinha, B.K. Metabolic Consequences of Erastin-Induced Ferroptosis in Human Ovarian Cancer Cells: An Untargeted Metabolomics Study. Front. Mol. Biosci. 2025, 11, 1520876. [Google Scholar] [CrossRef]
- Wang, B.; Yao, K.; Hu, Z. Advances in Mass Spectrometry-Based Single-Cell Metabolite Analysis. TrAC Trends Anal. Chem. 2023, 163, 117075. [Google Scholar] [CrossRef]
- Yangyun, W.; Guowei, S.; Shufen, S.; Jie, Y.; Rui, Y.; Yu, R. Everolimus Accelerates Erastin and RSL3-Induced Ferroptosis in Renal Cell Carcinoma. Gene 2022, 809, 145992. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Guo, Y.; Qian, Y.; Liu, X.; Li, G.; Wang, J.; Yang, X.; Wu, M.; Fan, Y.; Luo, H.; et al. Ferroptosis-Inducing Compounds Synergize with Docetaxel to Overcome Chemoresistance in Docetaxel-Resistant Non-Small Cell Lung Cancer Cells. Eur. J. Med. Chem. 2024, 276, 116670. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, T.; Li, X.; Sheng, H.; Ma, X.; Hao, L. Ferroptosis-Inducing Nanomedicine for Cancer Therapy. Front. Pharmacol. 2021, 12, 735965. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; He, R.; Liu, Y.; Zhang, J.; Xu, H.; Zhang, T.; Chen, L.; Yang, G.; Zhang, J.; Liu, J.; et al. Exploiting Cell Death and Tumor Immunity in Cancer Therapy: Challenges and Future Directions. Front. Cell Dev. Biol. 2024, 12, 1416115. [Google Scholar] [CrossRef]
- Yu, H.; Yan, J.; Li, Z.; Yang, L.; Ju, F.; Sun, Y. Recent Trends in Emerging Strategies for Ferroptosis-Based Cancer Therapy. Nanoscale Adv. 2023, 5, 1271–1290. [Google Scholar] [CrossRef]
- Lee, J.; Roh, J.L. Targeting Nrf2 for Ferroptosis-Based Therapy: Implications for Overcoming Ferroptosis Evasion and Therapy Resistance in Cancer. Biochim. Biophys. Acta—Mol. Basis Dis. 2023, 1869, 166788. [Google Scholar] [CrossRef]
- Liu, D.; Huang, S.Y.; Sun, J.H.; Zhang, H.C.; Cai, Q.L.; Gao, C.; Li, L.; Cao, J.; Xu, F.; Zhou, Y.; et al. Sepsis-Induced Immunosuppression: Mechanisms, Diagnosis and Current Treatment Options. Mil. Med. Res. 2022, 9, 56. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Li, X.; Wang, S.; Miao, R.; Zhong, J. Targeting Iron Metabolism and Ferroptosis as Novel Therapeutic Approaches in Cardiovascular Diseases. Nutrients 2023, 15, 591. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Sun, M.; Cao, F.; Chen, Y.; Zhang, L.; Li, H.; Cao, J.; Song, J.; Ma, Y.; Mi, W.; et al. The Ferroptosis Inhibitor Liproxstatin-1 Ameliorates LPS-Induced Cognitive Impairment in Mice. Nutrients 2022, 14, 4599. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Chen, Z.; Zhou, X.; Li, G.; Zhang, C.; Yang, Y. Ferroptosis and Multi-Organ Complications in COVID-19: Mechanisms and Potential Therapies. Front. Genet. 2023, 14, 1187985. [Google Scholar] [CrossRef]
- Naeem, M.; Hoque, M.Z.; Ovais, M.; Basheer, C.; Ahmad, I. Stimulus-Responsive Smart Nanoparticles-Based CRISPR-Cas Delivery for Therapeutic Genome Editing. Int. J. Mol. Sci. 2021, 22, 11300. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Compound | Mechanisms of Action | Key Characteristics |
|---|---|---|
| Erastin | Inhibits the system Xc− transporter, reducing cystine uptake and depleting GSH levels. | Enhances the uptake of chemotherapeutic drugs in resistant tumors by modulating iNOS. |
| RSL3 | Directly binds and inhibits GPX4 via covalent modification of its active site. | Highly effective in tumors with elevated GPX4 expression (e.g., non-small cell lung cancer). |
| FIN56 | Degrades GPX4 and activates squalene synthase, depleting CoQ10 antioxidant. | Dual-action: reduces GPX4 protein levels and increases oxidative stress. |
| FINO2 | Indirectly disrupts GPX4 and oxidizes intracellular iron to generate ROS. | Contains endoperoxide/hydroxyl structure; selective for cancer cells (e.g., HT-1080, RS411). |
| Sulfasalazine | Inhibits system Xc−, depleting GSH. | FDA-approved for inflammatory diseases (e.g., rheumatoid arthritis); exhibits anti-cancer properties. |
| Sorafenib | Disrupts the system Xc− function. | Oral multitarget kinase inhibitor used for thyroid, liver, and renal cell carcinomas. |
| Compound | Mechanisms of Action | Key Characteristics |
|---|---|---|
| Liproxstatin-1 | Radical-trapping antioxidant | Stops lipid peroxidation chain reactions; more effective in lipid membranes than in solution. |
| Ferrostatin-1 | Radical-trapping antioxidant | Neutralizes lipid peroxyl radicals more efficiently than vitamin E. |
| Vitamin E | Lipophilic antioxidant | Neutralizes lipid radicals, but is weaker than synthetic RTAs. |
| Coenzyme Q10 (CoQ10) | Lipid-soluble antioxidant | FSP1-CoQ10-NADPH axis regenerates reduced CoQ10. |
| Deferoxamine (DFO) | Iron chelator | Binds free iron to block the Fenton reaction. |
| Deferiprone (DFP) | Iron chelator | Binds free iron to inhibit iron-dependent lipid peroxidation. |
| Compound Class | Compound | Mechanisms of Action | Key Effects |
|---|---|---|---|
| Polyphenols | Quercetin | ↓ GPX4 & SLC7A11 expression ↑ NCOA4-dependent ferritinophagy ↑ Iron uptake | GSH depletion ↑ Lipid peroxidation ↑ ROS production |
| EGCG | Suppresses GPX4 Blocks the NRF2 pathway ↓ Mitochondrial membrane potential | ↑ Lipid peroxide accumulation Sensitizes cancer cells | |
| Curcumin | ↓ SLC7A11 Activates ACSL4 Iron chelation | Lipid remodeling ↑ Fenton reactions ↑ OS | |
| Terpenoids | Artemisinin derivatives | Iron-activated endoperoxide bridges ↓ Mevalonate pathway Inhibits GCLC | ↑ Carbon-centered radicals ↓ CoQ10 production ↑ Lipid peroxidation |
| Parthenolide | Inhibits GCLC | ↓ GSH levels ↓ GPX4 activity | |
| Flavonoids | Baicalein/Luteolin | ↑ TFR1 expression ↓ Ferritin ↓ GPX4 Activates ACSL4 | ↑ Iron uptake ↑ PUFAs incorporation ↑ Membrane oxidation |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pawłowska, M.; Nuszkiewicz, J.; Jarek, D.J.; Woźniak, A. Ferroptosis and Metabolic Dysregulation: Emerging Chemical Targets in Cancer and Infection. Molecules 2025, 30, 3020. https://doi.org/10.3390/molecules30143020
Pawłowska M, Nuszkiewicz J, Jarek DJ, Woźniak A. Ferroptosis and Metabolic Dysregulation: Emerging Chemical Targets in Cancer and Infection. Molecules. 2025; 30(14):3020. https://doi.org/10.3390/molecules30143020
Chicago/Turabian StylePawłowska, Marta, Jarosław Nuszkiewicz, Dorian Julian Jarek, and Alina Woźniak. 2025. "Ferroptosis and Metabolic Dysregulation: Emerging Chemical Targets in Cancer and Infection" Molecules 30, no. 14: 3020. https://doi.org/10.3390/molecules30143020
APA StylePawłowska, M., Nuszkiewicz, J., Jarek, D. J., & Woźniak, A. (2025). Ferroptosis and Metabolic Dysregulation: Emerging Chemical Targets in Cancer and Infection. Molecules, 30(14), 3020. https://doi.org/10.3390/molecules30143020