Advances in α-Arylation of Carbonyl Compounds: Diaryliodonium Salts as Arylating Agents

 ,
,
Abstract
1. Introduction
2. The Racemic α-Arylation of Carbonyl Compounds
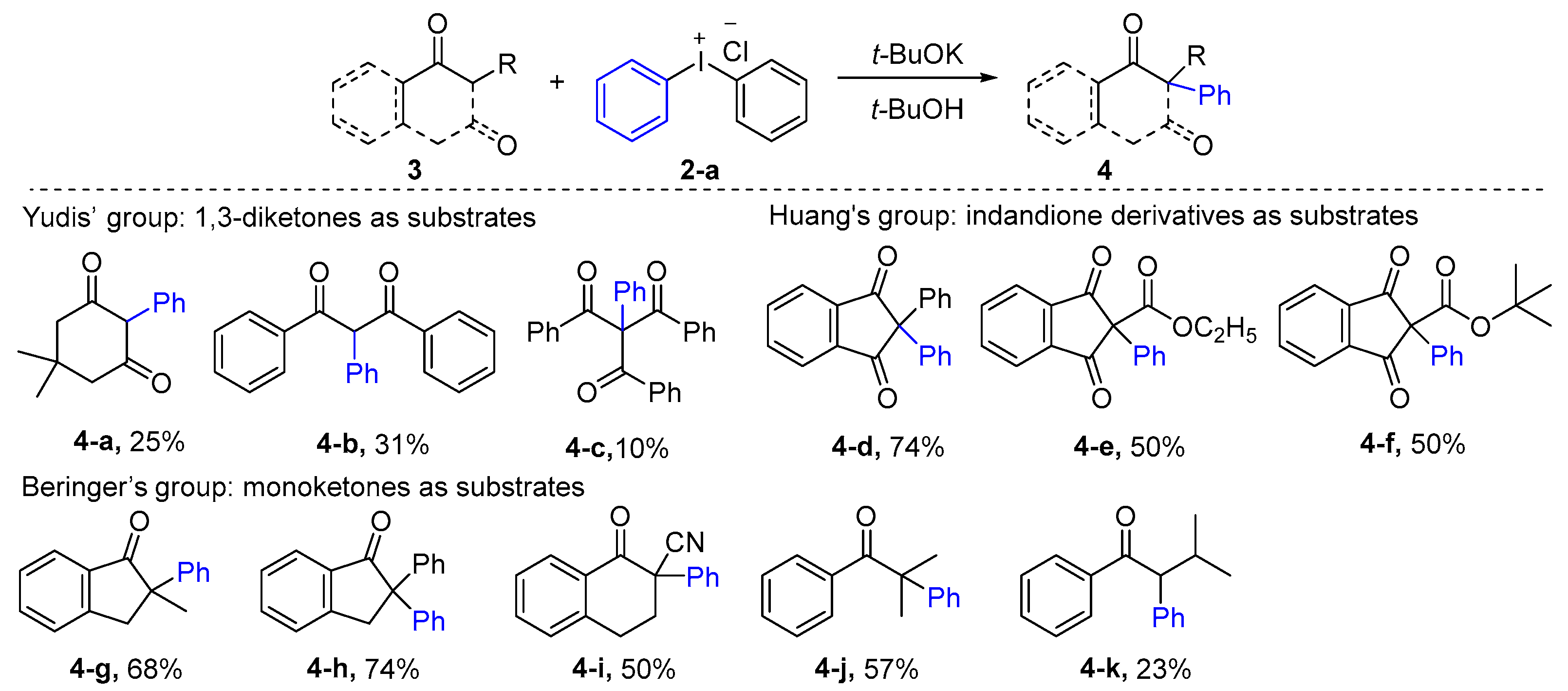
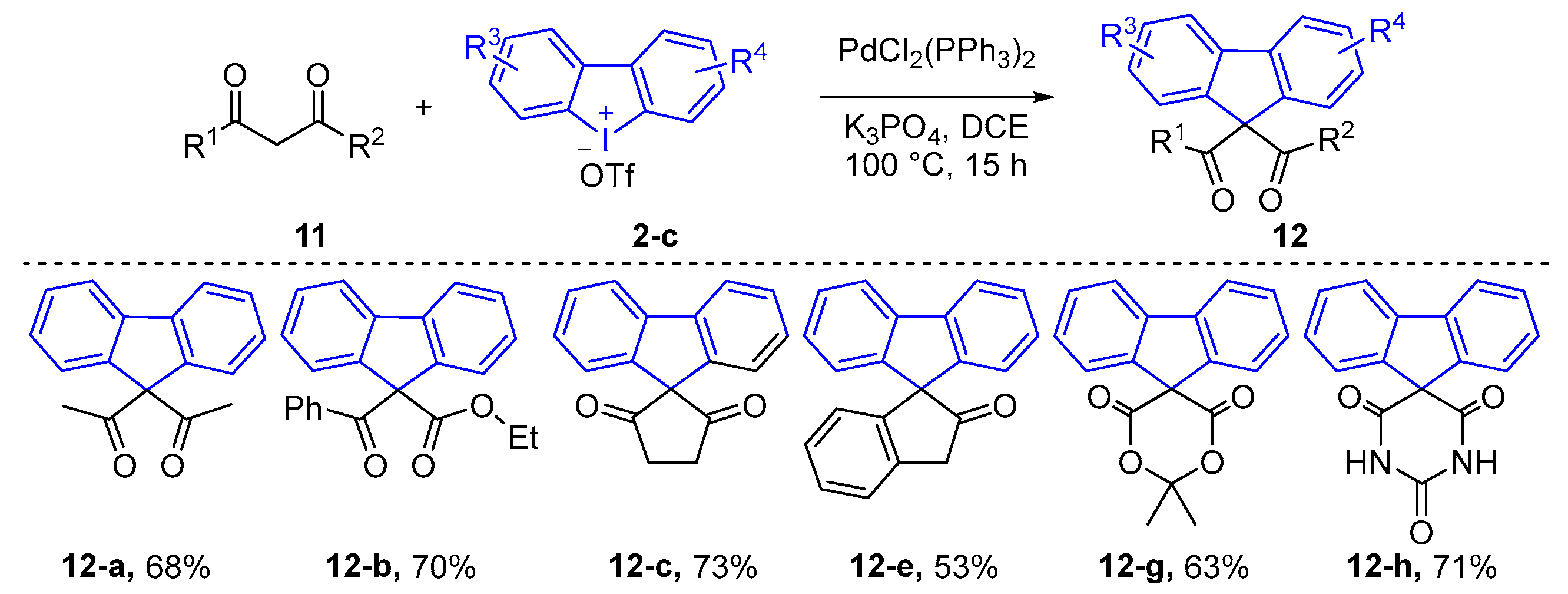
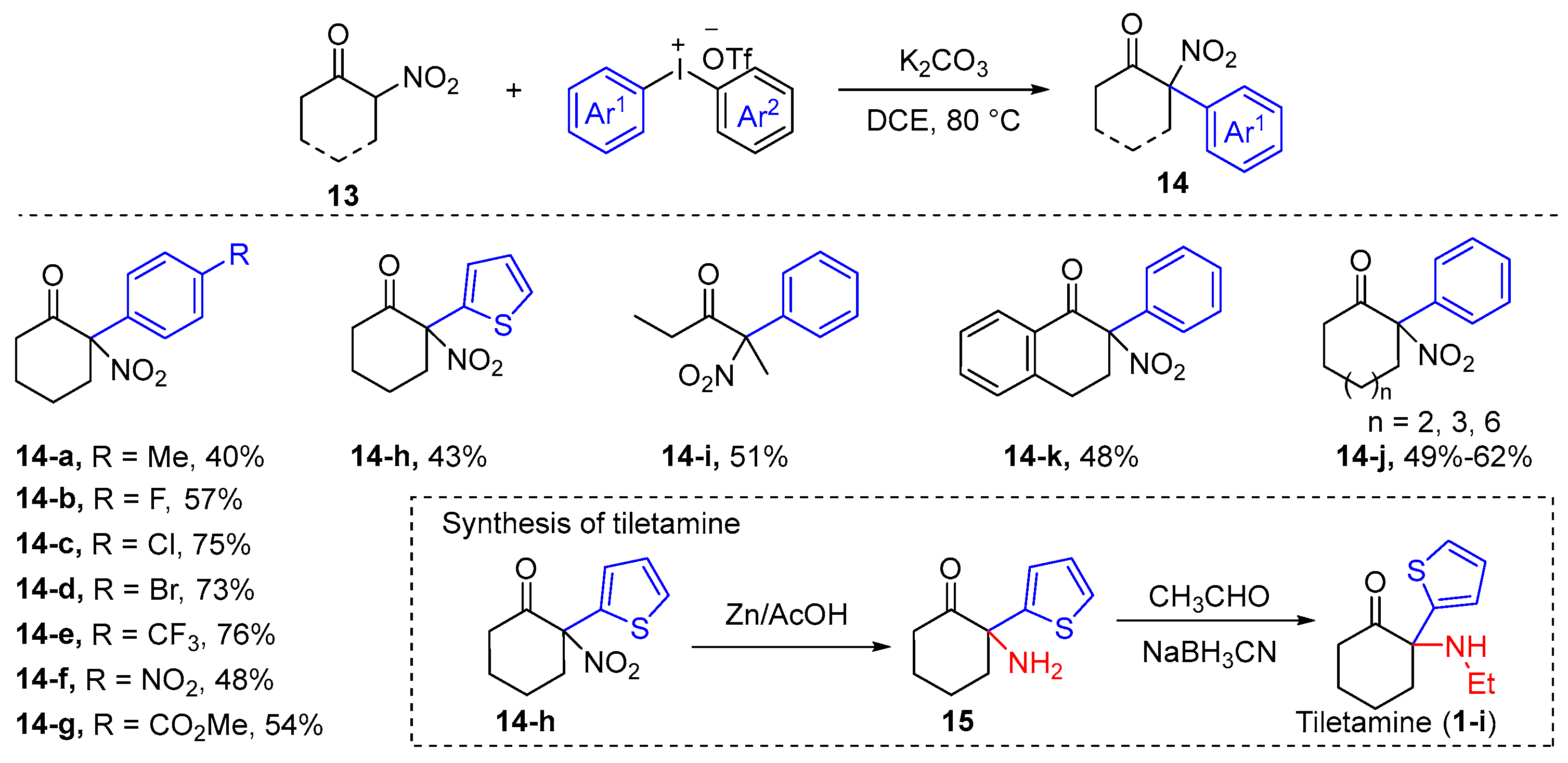
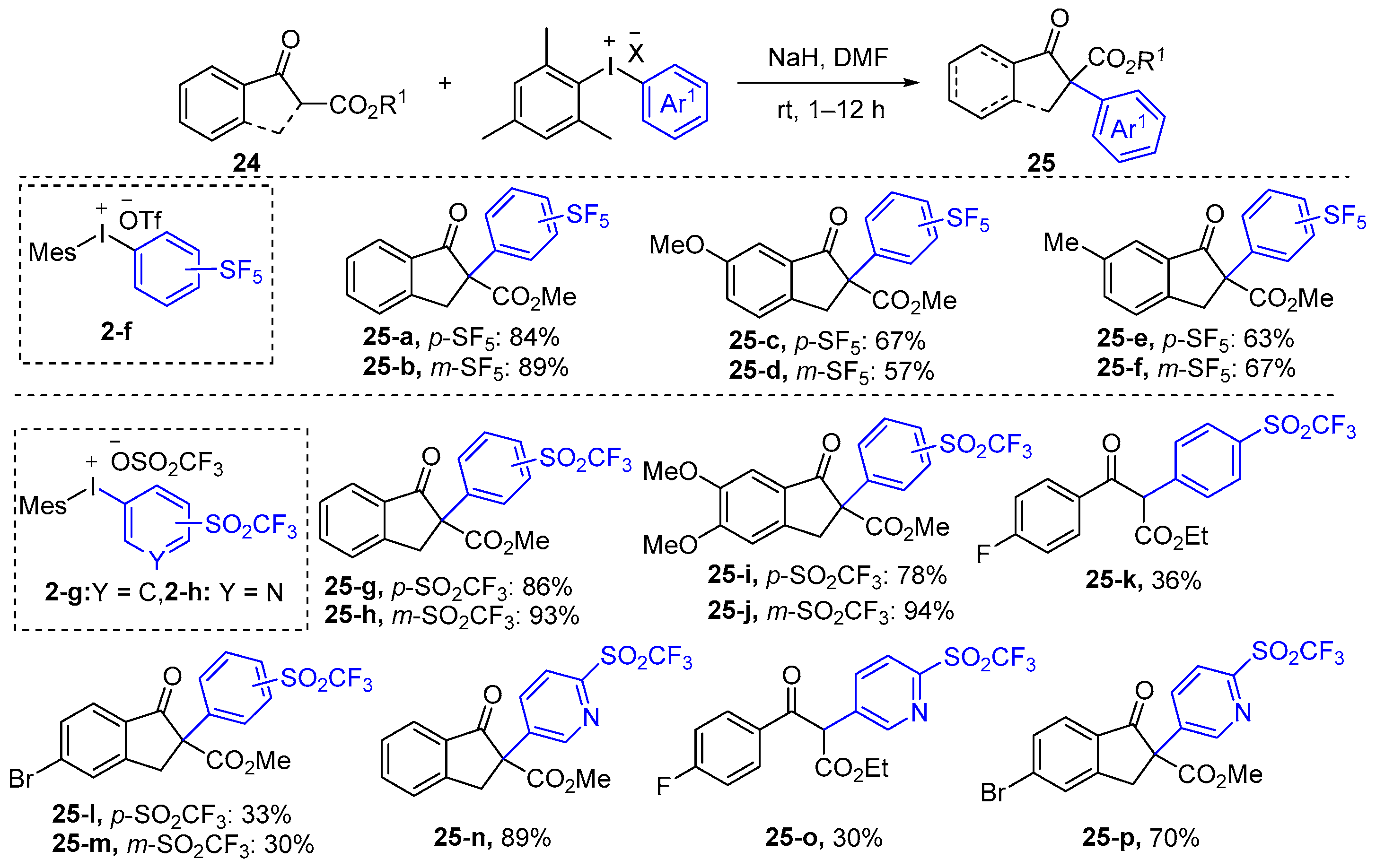
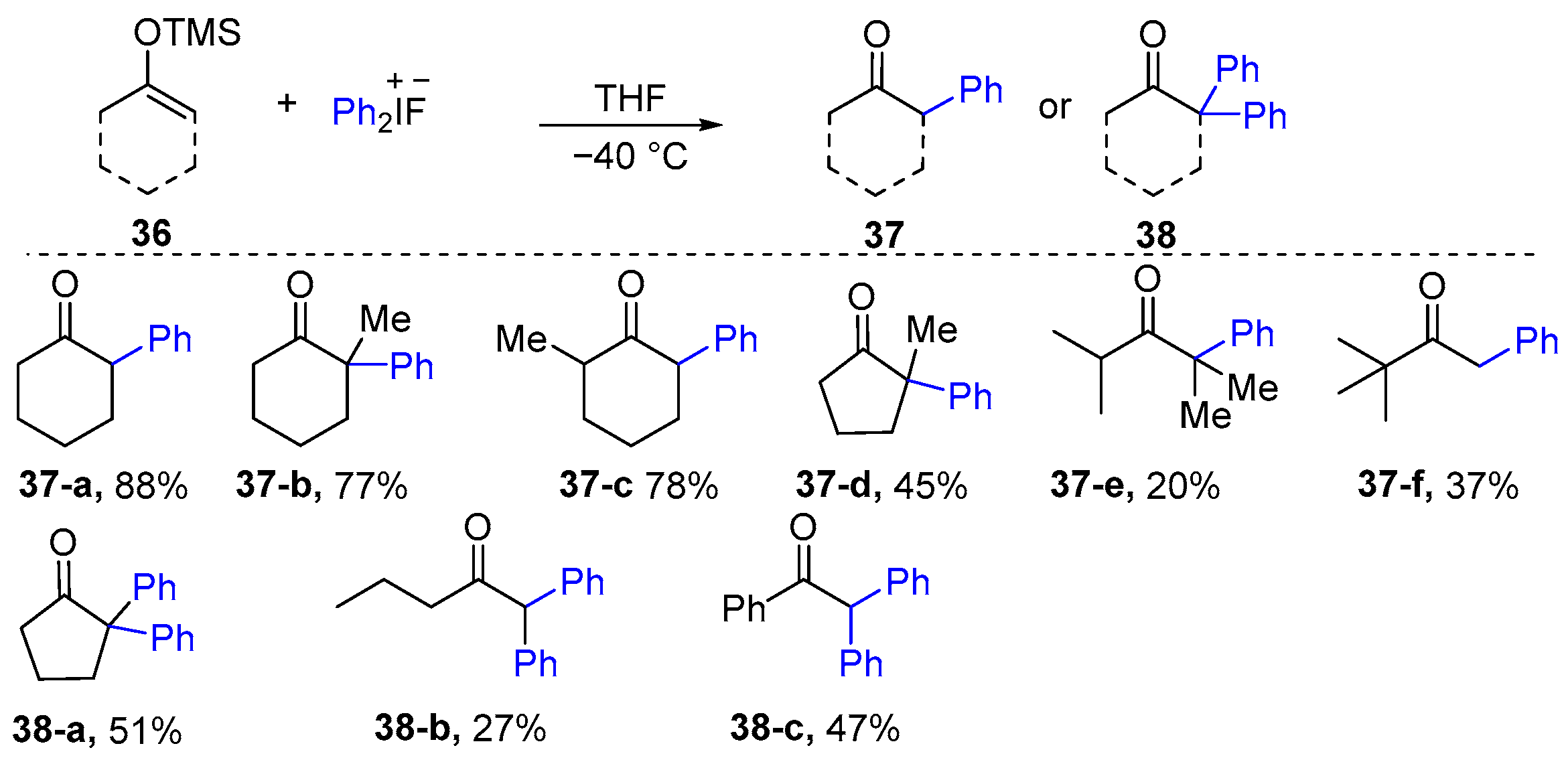
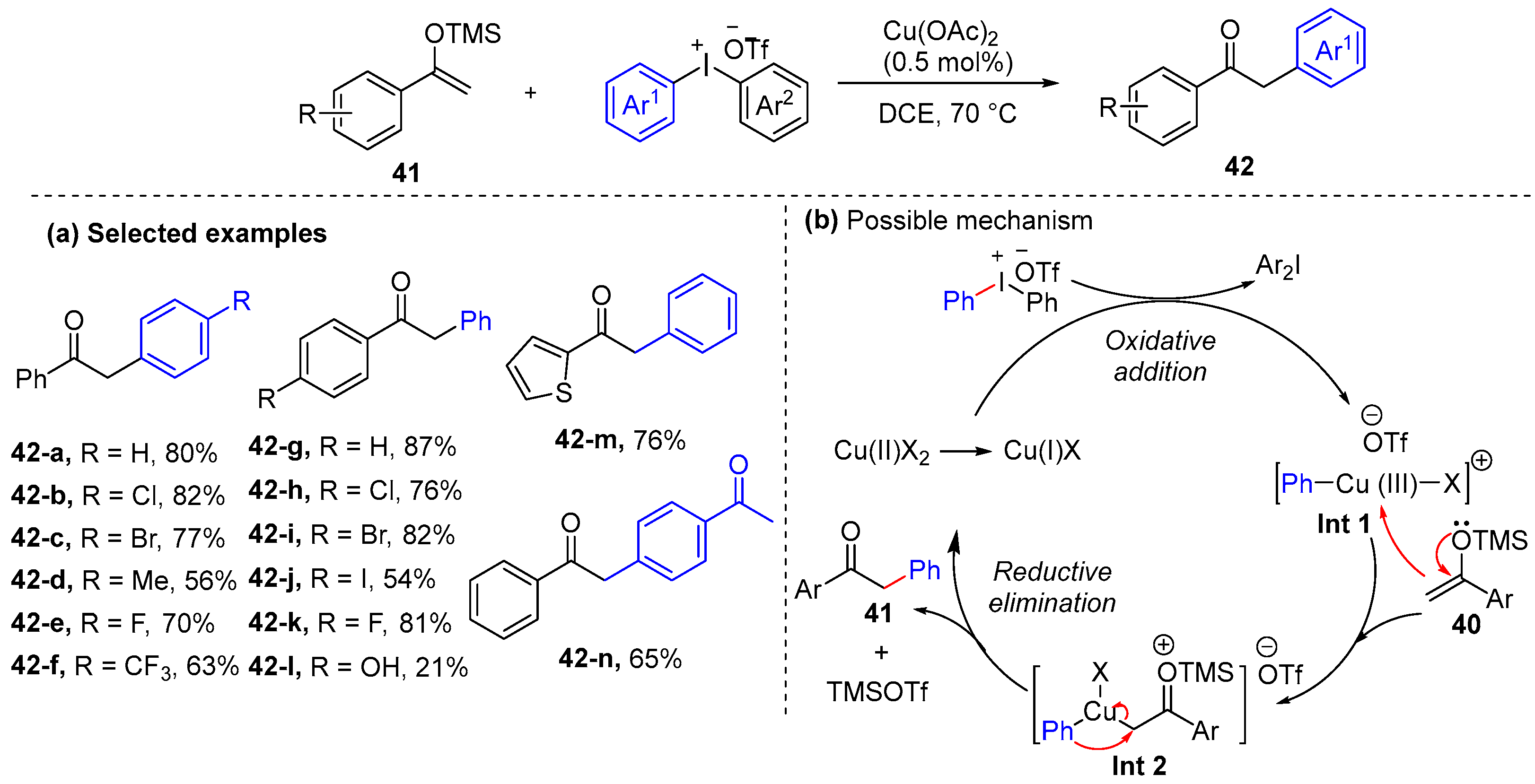
2.1. α-Arylation of Keto Carbonyl Compounds
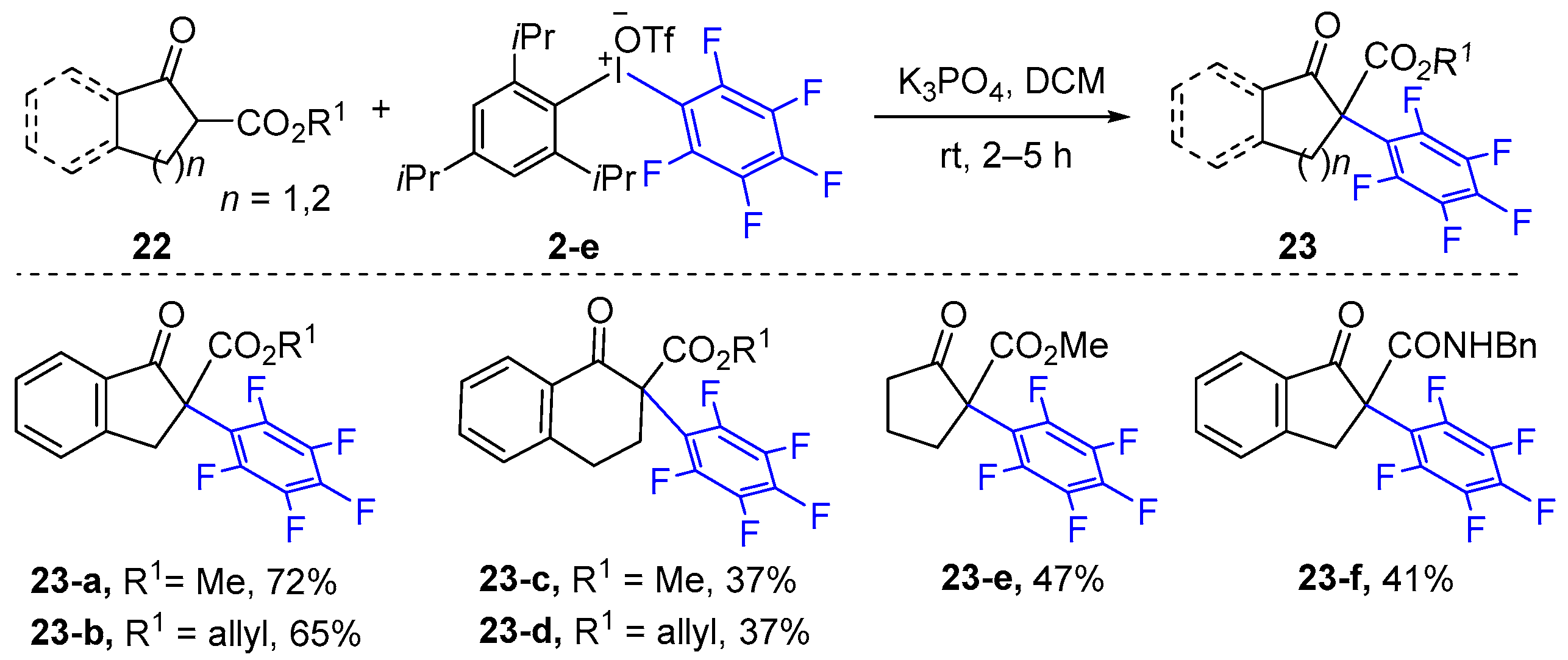
2.2. α-Arylation of Ester Carbonyl Compounds
2.3. α-Arylation of Silyl Enol Ethers
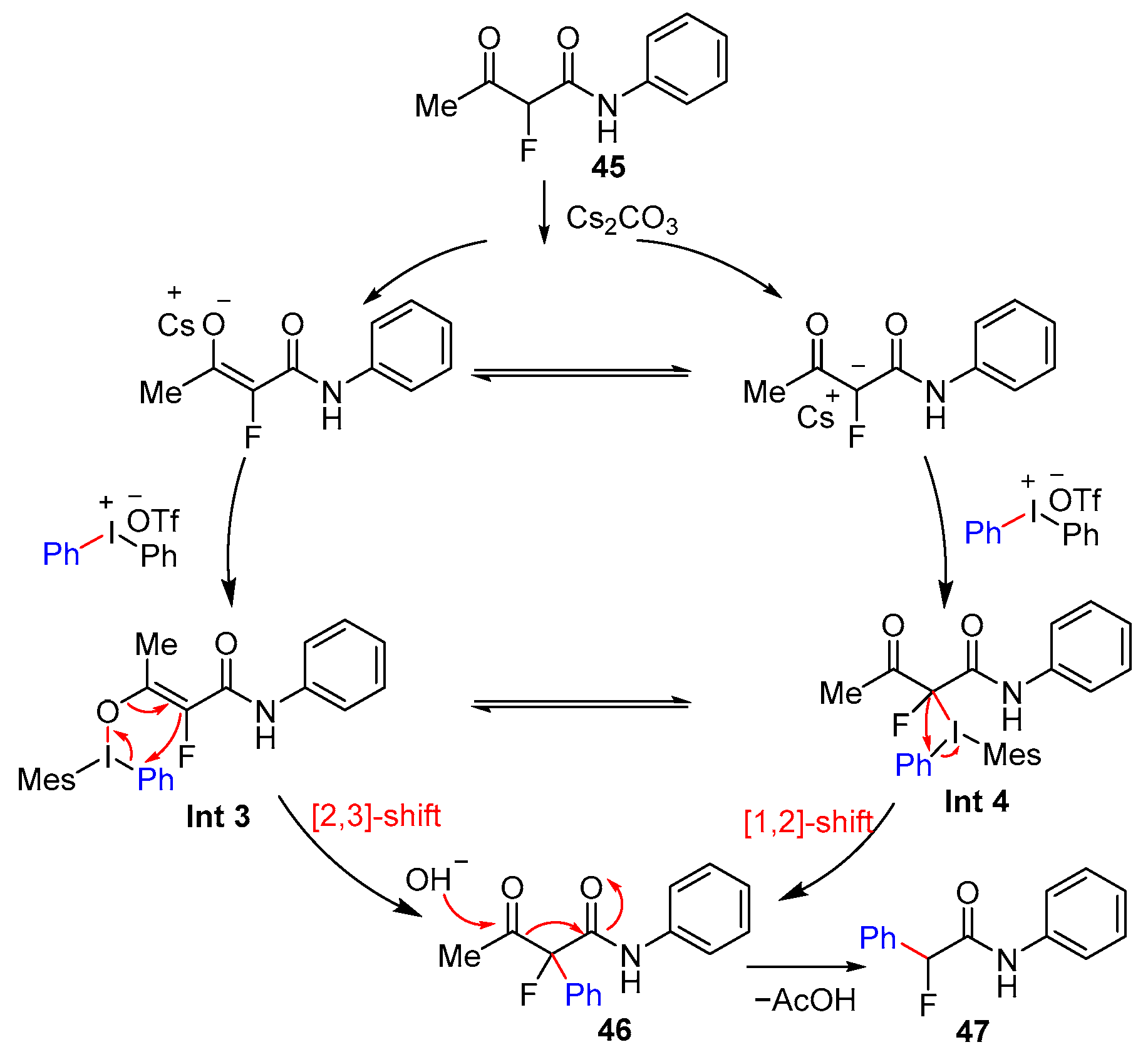
2.4. α-Arylation of Amides
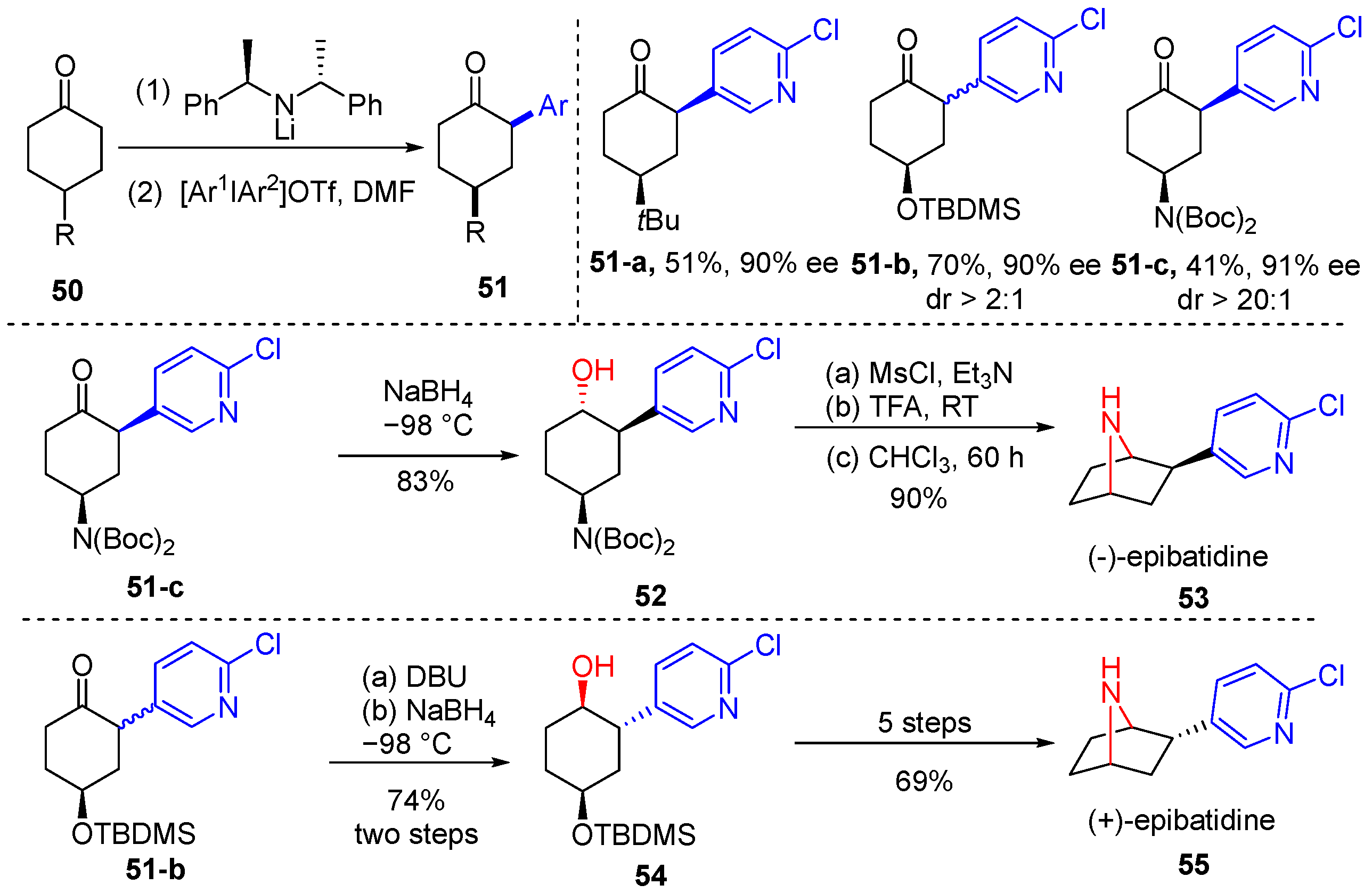
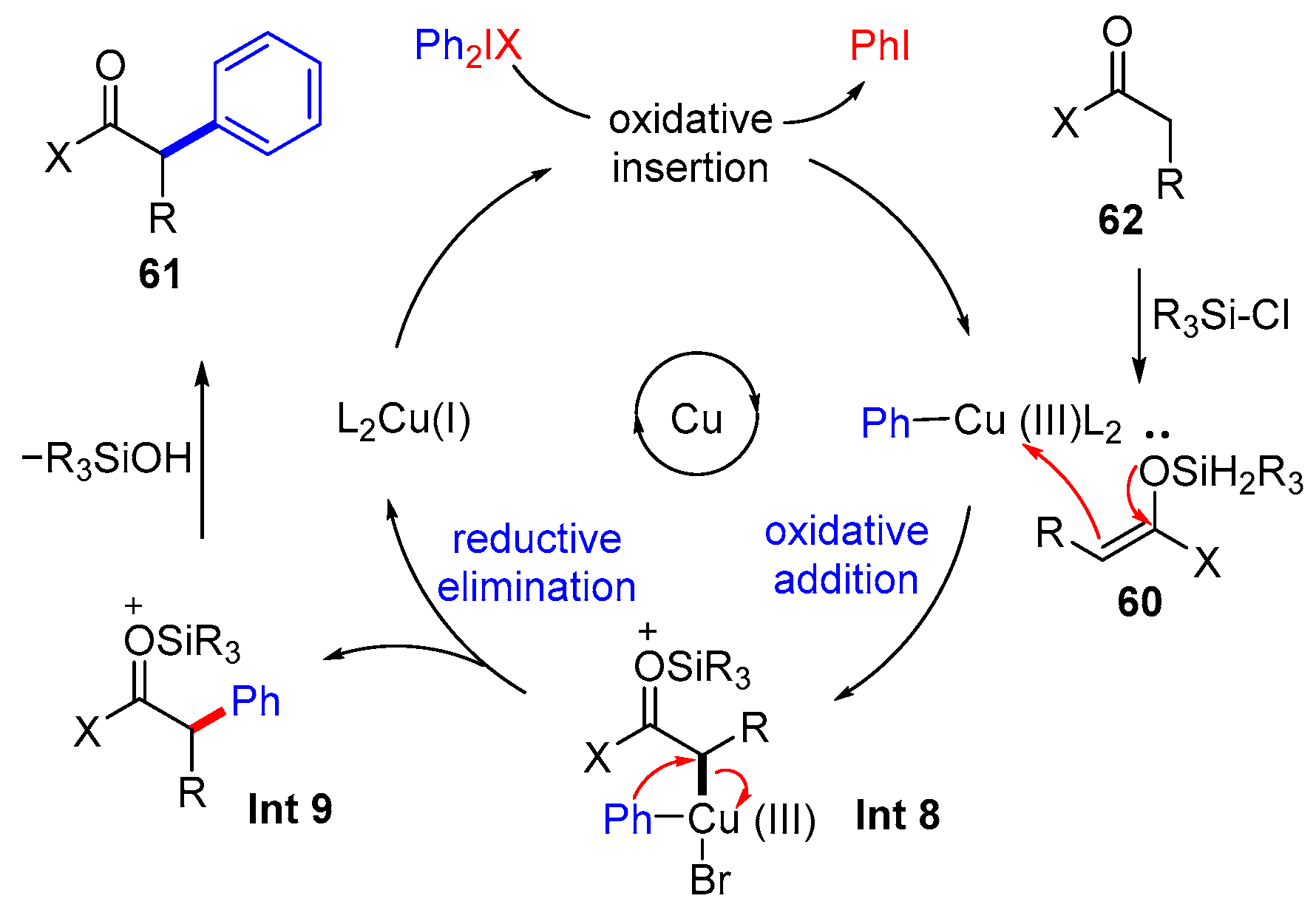
3. The Asymmetric α-Arylation of Carbonyl Compounds
4. Summary and Outlook
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bellina, F.; Rossi, R. Transition metal-catalyzed direct arylation of substrates with activated sp3-hybridized C-H bonds and some of their synthetic equivalents with aryl halides and pseudohalides. Chem. Rev. 2010, 110, 1082–1146. [Google Scholar] [CrossRef] [PubMed]
- Aoki, S.; Watanabe, Y.; Sanagawa, M.; Setiawan, A.; Kotoku, N.; Kobayashi, M. Cortistatins A, B, C, and D, Anti-angiogenic steroidal alkaloids, from the marine sponge Corticium simplex. J. Am. Chem. Soc. 2006, 128, 3148–3149. [Google Scholar] [CrossRef] [PubMed]
- Doan, T.H.; Ho, V.D.; Le, T.B.H.; Le, T.A.; Pham, T.K.; Nguyen, T.H.; Raal, A. Two new abietane diterpenes huperphlegmarins A and B from Huperzia Phlegmaria. Nat. Prod. Res. 2019, 33, 2051–2059. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Tanaka, T.; Iinuma, M.; Nakaya, K.; Takahashi, Y.; Sawa, R.; Murata, J.; Darnaedi, D. Three new resveratrol oligomers from the stem bark of Vatica pauciflora. J. Nat. Prod. 2004, 67, 932–937. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, J.A.; Akarasereenont, P.; Thiemermann, C.; Flower, R.J.; Vane, J.R. Selectivity of nonsteroidal anti-inflammatory drugs as inhibitors of constitutive and inducible cyclooxygenase. Proc. Natl. Acad. Sci. USA 1993, 90, 11693–11697. [Google Scholar] [CrossRef] [PubMed]
- Katayev, D.; Kündig, E.P. Catalytic enantioselective synthesis of a 3-aryl-3-benzyloxindole (3-aryl-3-benzyl-1,3-dihydro-2H-indol-2-one) exhibiting antitumor activity. Helv. Chim. Acta 2012, 95, 2287–2295. [Google Scholar] [CrossRef]
- Khwaja, F.; Allen, J.; Lynch, J.; Andrews, P.; Djakiew, D. Ibuprofen inhibits survival of bladder cancer cells by induced expression of the p75NTR tumor suppressor protein. Cancer Res. 2004, 64, 6207–6213. [Google Scholar] [CrossRef] [PubMed]
- Palomer, A.; Pérez, J.J.; Navea, S.; Llorens, O.; Pascual, J.; García, L.; Mauleón, D. Modeling cyclooxygenase inhibition. Implication of active site hydration on the selectivity of ketoprofen analogues. J. Med. Chem. 2000, 43, 2280–2284. [Google Scholar] [CrossRef] [PubMed]
- Zanos, P.; Moaddel, R.; Morris, P.J.; Riggs, L.M.; Highland, J.N.; Georgiou, P.; Pereira, E.F.R.; Albuquerque, E.X.; Thomas, C.J.; Zarate, C.A.; et al. Ketamine and ketamine metabolite pharmacology: Insights into therapeutic mechanisms. Pharmacol. Rev. 2018, 70, 621–660. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Yang, S.; Zhou, X.; Chen, Q.; Tu, E.; Zhang, B.; Shi, L.; Zhou, X. Severe tremors induced by tiletamine e-cigarette and alcohol use: A case report. Front. Psychiatry 2025, 16, 1537822. [Google Scholar] [CrossRef] [PubMed]
- Han, B.; He, X.H.; Liu, Y.Q.; He, G.; Peng, C.; Li, J.L. Asymmetric organocatalysis: An enabling technology for medicinal chemistry. Chem. Soc. Rev. 2021, 50, 1522–1586. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Rueping, M. Metallaphotoredox catalysis for sp3C-H functionalizations through hydrogen atom transfer (HAT). Chem. Soc. Rev. 2023, 52, 4099–4120. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, P.; Mandal, R.; Garg, N.; Sundararaju, B. Recent advances in transition metal-catalyzed asymmetric electrocatalysis. Coord. Chem. Rev. 2021, 444, 214065–241088. [Google Scholar] [CrossRef]
- Biffis, A.; Centomo, P.; Del Zotto, A.; Zecca, M. Pd metal catalysts for cross-couplings and related reactions in the 21st century: A critical review. Chem. Rev. 2018, 118, 2249–2295. [Google Scholar] [CrossRef] [PubMed]
- Swathy, K.J.; Saranya, P.V.; Anilkumar, G. Recent advances in palladium-catalyzed α-arylation reactions. Appl. Organomet. Chem. 2024, 38, 7508. [Google Scholar] [CrossRef]
- Mamgain, R.; Sakthivel, K.; Singh, F.V. Recent advances in transition-metal-free arylation reactions involving hypervalent iodine salts. Beilstein J. Org. Chem. 2024, 20, 2891–2920. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.Q.; MacMillan, D.W.C. A metallaphotoredox strategy for the cross-electrophile coupling of α-chloro carbonyls with aryl halides. Angew. Chem. Int. Ed. 2019, 58, 14584–14588. [Google Scholar] [CrossRef] [PubMed]
- Pandey, G.; Tiwari, S.K.; Budakoti, A.; Sahani, P.K. Transition-metal-free photoredox intermolecular α-arylation of ketones. Org. Chem. Front. 2018, 5, 2610–2614. [Google Scholar] [CrossRef]
- Hossain, M.M.; Shaikh, A.C.; Moutet, J.; Gianetti, T.L. Photocatalytic α-arylation of cyclic ketones. Nat. Synth. 2022, 1, 147–157. [Google Scholar] [CrossRef]
- Hartmann, C.; Meyer, V. Ueber eine neue klasse jodhaltiger, stickstofffreier organischer basen. Ber. Dtsch. Chem. Ges. 1894, 27, 426–432. [Google Scholar] [CrossRef]
- Takenaga, N.; Kumar, R.; Dohi, T. Heteroaryliodonium(III) salts as highly reactive electrophiles. Front. Chem. 2020, 8, 599026. [Google Scholar] [CrossRef] [PubMed]
- Bielawski, M.; Olofsson, B. High-yielding one-pot synthesis of diaryliodonium triflates from arenes and iodine or aryl iodides. Chem. Commun. 2007, 24, 2521–2523. [Google Scholar] [CrossRef] [PubMed]
- Bielawski, M.; Zhu, M.; Olofsson, B. Efficient and general one-pot synthesis of diaryliodonium triflates: Optimization, scope and limitations. Adv. Synth. Catal. 2007, 349, 2610–2618. [Google Scholar] [CrossRef]
- Bielawski, M.; Krämer, K.; Olofsson, B. Efficent.one-pot synthesis of bis (4-tert butylphenyl) iodonium triflate. Org. Synth. 2009, 86, 308–314. [Google Scholar]
- Koser, G.F.; Wettach, R.H.; Smith, C.S. New methodology in iodonium salt synthesis. Reactions of [hydroxy(tosyloxy)iodo] arenes with aryltrimethylsilanes. J. Org. Chem. 1980, 45, 1543–1544. [Google Scholar] [CrossRef]
- Bielawski, M.; Aili, D.; Olofsson, B. Regiospecific one-pot synthesis of diaryliodonium tetrafluoroborates from arylboronic acids and aryl iodides. J. Org. Chem. 2008, 73, 4602–4607. [Google Scholar] [CrossRef] [PubMed]
- Cardinale, J.; Ermert, J.; Coenen, H.H. Convenient preparation of (4-iodophenyl) aryliodonium salts. Tetrahedron 2012, 68, 4112–4116. [Google Scholar] [CrossRef]
- Chun, J.-H.; Pike, V.W. Regiospecific syntheses of functionalized diaryliodonium tosylates via [hydroxy(tosyloxy)iodo] arenes generated in situ from (Diacetoxyiodo) arenes. J. Org. Chem. 2012, 77, 1931–1938. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Itani, I.; Toyoda, Y.; Morimoto, K.; Dohi, T.; Kita, Y. Synthesis of boron-substituted diaryliodonium salts and selective transformation into functionalized aryl boronates. Angew. Chem. Int. Ed. 2012, 51, 12555–12558. [Google Scholar] [CrossRef] [PubMed]
- Jalalian, N.; Olofsson, B. Design and asymmetric synthesis of chiral diaryliodonium salts. Tetrahedron 2010, 66, 5793–5800. [Google Scholar] [CrossRef]
- Yoshimura, A.; Zhdankin, V.V. Advances in synthetic applications of hypervalent iodine compounds. Chem. Rev. 2016, 116, 3328–3435. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Chen, C.; Xi, C.J. β-Arylation of oxime ethers using diaryliodonium salts through activation of inert C(sp3)–H bonds using a palladium catalyst. Chem. Sci. 2016, 7, 1383–1387. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Li, M.; Xi, C.J.; Chen, C. Direct vicinal disubstitution of diaryliodonium salts by pyridine N-oxides and N-amidates by a 1,3-radical rearrangement. Angew. Chem. Int. Ed. 2013, 52, 7574–7578. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, M.; Chen, C. Copper (II)-catalyzed three-component cascade annulation of diaryliodoniums, nitriles, and alkynes: A regioselective synthesis of multiply substituted quinolines. Angew. Chem. Int. Ed. 2013, 52, 5323–5327. [Google Scholar] [CrossRef] [PubMed]
- Cao, C.K.; Sheng, J.; Chen, C. Cu-catalyzed cascade annulation of diaryliodonium salts and nitriles: Synthesis of nitrogen-containing heterocycles. Synlett 2017, 49, 5081–5092. [Google Scholar] [CrossRef]
- Merritt, E.A.; Olofsson, B. Diaryliodonium salts: A journey from obscurity to fame. Angew. Chem. Int. Ed. 2009, 48, 9052–9070. [Google Scholar] [CrossRef] [PubMed]
- Yusubov, M.S.; Maskaev, A.V.; Zhdankin, V.V. Iodonium salts in organic synthesis. Arkivoc 2011, 2011, 370–409. [Google Scholar] [CrossRef]
- Aradi, K.; Tóth, B.; Tolnai, G.; Novák, Z. Diaryliodonium salts in organic syntheses: A useful compound class for novel arylation strategies. Synlett 2016, 27, 1456–1485. [Google Scholar] [CrossRef]
- Fañanás-Mastral, M. Copper-catalyzed arylation with diaryliodonium salts. Synthesis 2017, 49, 1905–1930. [Google Scholar] [CrossRef]
- Stuart, D.R. Aryl transfer selectivity in metal-free reactions of unsymmetrical diaryliodonium salts. Chem. Eur. J. 2017, 23, 15852–15863. [Google Scholar] [CrossRef] [PubMed]
- Singhal, R.; Choudhary, S.P.; Malik, B.; Pilania, M. Cyclic diaryliodonium salts: Applications and overview. Org. Biomol. Chem. 2023, 21, 4358–4378. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Z.; Xia, C. Progresses of diaryliodonium salts in organic reactions. Chin. J. Org. Chem. 2013, 33, 2119–2130. [Google Scholar] [CrossRef]
- Wang, D.; Li, Q.; Li, M.; Du, Z.; Fu, Y. Recent progress in arylation reactions with diaryliodonium salts. Curr. Org. Chem. 2021, 25, 1298–1320. [Google Scholar] [CrossRef]
- Silva, L.F. Hypervalent iodine–mediated ring contraction reactions. Molecules 2006, 21, 421–434. [Google Scholar] [CrossRef] [PubMed]
- Karandikar, S.S.; Bhattacharjee, A.; Metze, B.E.; Javaly, N.; Valente, E.J.; McCormick, T.M.; Stuart, D.R. Orbital analysis of bonding in diarylhalonium salts and relevance to periodic trends in structure and reactivity. Chem. Sci. 2022, 13, 653–6540. [Google Scholar] [CrossRef] [PubMed]
- Le Du, E.; Waser, J. Recent progress in alkynylation with hypervalent iodine reagents. Chem. Commun. 2023, 59, 1589–1604. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, A.; Zhdankin, V.V. Recent progress in synthetic applications of hypervalent iodine (III) reagents. Chem. Rev. 2024, 124, 11108–11186. [Google Scholar] [CrossRef] [PubMed]
- Dong, D.-Q.; Hao, S.-H.; Wang, Z.-L.; Chen, C. Hypervalent iodine: A powerful electrophile for asymmetric α-functionalization of carbonyl compounds. Org. Biomol. Chem. 2014, 12, 4278–4289. [Google Scholar] [CrossRef] [PubMed]
- Olofsson, B. Arylation with diaryliodonium salts. In Hypervalent Iodine Chemistry; Wirth, T., Ed.; Topics in Current Chemistry; Springer: Cham, Switzerland, 2015; Volume 373, pp. 135–166. [Google Scholar]
- Prier, C.K.; Rankic, D.A.; MacMillan, D.W.C. Visible light photoredox catalysis with transition metal complexes: Applications in organic synthesis. Chem. Rev. 2013, 113, 5322–5363. [Google Scholar] [CrossRef] [PubMed]
- Romero, N.A.; Nicewicz, D.A. Organic photoredox catalysis. Chem. Rev. 2016, 116, 10075–10166. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Zhao, C.; Zhou, J.; Hu, H.-S.; Li, J.; Wu, P.; Chen, C. Wet carbonate-promoted radical arylation of vinyl pinacolboronates with diaryliodonium salts yields substituted olefins. Commun. Chem. 2020, 3, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.-J.; Hu, X.-S.; Zhou, Y.; Zhou, J.; Yu, J.-S. Catalytic enantioselective α-arylation of carbonyl enolates and related compounds. ACS Catal. 2020, 10, 955–993. [Google Scholar] [CrossRef]
- Singh, P.R.; Banerjee, A.; Simlandy, A.K. Advances in catalytic enantioselective transformations using diaryliodonium reagents. ACS Catal. 2025, 15, 3096–3115. [Google Scholar] [CrossRef]
- Marshall Beringer, F.; Forgione, P.S.; Yudis, M.D. Diaryliodonium salts—XII: The phenylation of dimedone, dibenzoylmethane and tribenzoylmethane. Tetrahedron 1960, 8, 49–63. [Google Scholar] [CrossRef]
- Beringer, F.M.; Galton, S.A.; Huang, S.J. Diaryliodonium salts. XVII. The phenylation of 1,3-indandiones. J. Am. Chem. Soc. 1962, 84, 2819–2823. [Google Scholar] [CrossRef]
- Beringer, F.M.; Daniel, W.J.; Galton, S.A.; Rubin, G. Phenylation of monoketones with diphenyliodonium chloride. J. Org. Chem. 1966, 31, 4315–4318. [Google Scholar] [CrossRef]
- Ryan, J.H.; Stang, P.J. Direct α-arylation of ketones: The reaction of cyclic ketone enolates with diphenyliodonium triflate. Tetrahedron Lett. 1997, 38, 5061–5064. [Google Scholar] [CrossRef]
- Liu, Z.-J.; Han, J.; Zhang, Y. tert-Butoxide-mediated arylation of 1-acetylindolin-3-ones with diaryliodonium salts. Synlett 2015, 26, 2593–2597. [Google Scholar]
- Pan, J.-L.; Chen, T.; Zhang, Z.-Q.; Li, Y.-F.; Zhang, X.-M.; Zhang, F.-M. A Cu-mediated one-pot Michael addition/α-arylation strategy using diaryliodonium salt: Direct and efficient approach to α-aryl-β substituted cyclic ketone scaffolds. Chem. Commun. 2016, 52, 2382–2385. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Luo, H.; Wu, F.; Zhu, D.; Ganesan, A.; Huang, P.; Wen, S. Synthesis of fluorenes with an all-carbon quaternary center via palladium-catalyzed dual arylation using cyclic diaryliodonium triflates. Adv. Syn. Catal. 2017, 359, 1152–1156. [Google Scholar] [CrossRef]
- An, Y.; Zhang, X.M.; Li, Z.Y.; Xiong, W.H.; Yu, R.D.; Zhang, F.M. Transition-metal-free α-arylation of nitroketones with diaryliodonium salts for the synthesis of tertiary α-aryl, α-nitro ketones. Chem. Commun. 2019, 55, 119–122. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.-W.; Hou, Z.-C.; Chen, C.; Zhang, L.-H.; Chen, M.-E.; Zhang, F.-M. Enantioselective total syntheses of six natural and two proposed meroterpenoids from Psoralea Corylifolia. Chem. Sci. 2023, 14, 5699–5704. [Google Scholar] [CrossRef] [PubMed]
- Beringer, F.M.; Forgione, P.S. Diaryliodonium salts. XVIII. The phenylation of esters in t-butyl alcohol1-3. J. Org. Chem. 1963, 28, 714–717. [Google Scholar] [CrossRef]
- Chen, Z.; Jin, Y.; Stang, P.J. Polyvalent iodine in synthesis. 1. An efficient route to isopropylidene arylmalonates (5-aryl-substituted Meldrum’s acid). J. Org. Chem. 1987, 52, 4115–4117. [Google Scholar] [CrossRef]
- Oh, C.H.; Kim, J.S.; Jung, H.H. Highly efficient arylation of malonates with diaryliodonium salts. J. Org. Chem. 1999, 64, 1338–1340. [Google Scholar] [CrossRef]
- Matsuzaki, K.; Okuyama, K.; Tokunaga, E.; Shiro, M.; Shibata, N. Sterically demanding unsymmetrical diaryl-λ3-iodanes for electrophilic pentafluorophenylation and an approach to α-pentafluorophenyl carbonyl compounds with an all-carbon stereocenter. ChemistryOpen 2014, 3, 233–237. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, K.; Okuyama, K.; Tokunaga, E.; Saito, N.; Shiro, M.; Shibata, N. Synthesis of diaryliodonium salts having pentafluorosulfanylarenes and their application to electrophilic pentafluorosulfanylarylation of C-, O-, N-, and S-nucleophiles. Org. Lett. 2015, 17, 3038–3041. [Google Scholar] [CrossRef] [PubMed]
- Das, P.; Shibata, N. Electrophilic Triflyl-arylation and triflyl-pyridylation by unsymmetrical aryl/pyridyl-λ3-iodonium salts: Synthesis of aryl and pyridyl triflones. J. Org. Chem. 2017, 82, 11915–11924. [Google Scholar] [CrossRef] [PubMed]
- Monastyrskyi, A.; Namelikonda, N.K.; Manetsch, R. Metal-free arylation of ethyl acetoacetate with hypervalent diaryliodonium salts: An immediate access to diverse 3-aryl-4(1H)-quinolones. J. Org. Chem. 2015, 80, 2513–2520. [Google Scholar] [CrossRef] [PubMed]
- Dey, C.; Lindstedt, E.; Olofsson, B. Metal-free C-arylation of nitro compounds with diaryliodonium salts. Org. Lett. 2015, 17, 4554–4557. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; Han, J.; Wang, L. tert-Butoxide-mediated arylation of 2-substituted cyanoacetates with diaryliodonium salts. Adv. Syn. Catal. 2016, 358, 940–946. [Google Scholar] [CrossRef]
- Kikushima, K.; Yamada, K.; Umekawa, N.; Yoshio, N.; Kita, Y.; Dohi, T. Decarboxylative arylation with diaryliodonium(iii) salts: Alternative approach for catalyst-free difluoroenolate coupling to aryldifluoromethyl ketones. Green Chem. 2023, 25, 1790–1796. [Google Scholar] [CrossRef]
- Chen, K.; Koser, G.F. Direct and regiocontrolled synthesis of α-phenyl ketones from silyl enol ethers and diphenyliodonium fluoride. J. Org. Chem. 1991, 56, 5764–5767. [Google Scholar] [CrossRef]
- Iwama, T.; Birman, V.B.; Kozmin, S.A.; Rawal, V.H. Regiocontrolled synthesis of carbocycle-fused indoles via arylation of silyl enol ethers with o-nitrophenylphenyliodonium fluoride. Org. Lett. 1999, 1, 673–676. [Google Scholar] [CrossRef] [PubMed]
- Bouquin, M.; Jaroschik, F.; Taillefer, M. Versatile and base-free copper-catalyzed α-arylations of aromatic ketones using diaryliodonium salts. Tetrahedron Lett. 2021, 75, 153208–153212. [Google Scholar] [CrossRef]
- Zaheer, M.K.; Gupta, E.; Kant, R.; Mohanan, K. Metal-free α-arylation of α-fluoro-α-nitroacetamides employing diaryliodonium salts. Chem. Commun. 2020, 56, 153–156. [Google Scholar] [CrossRef] [PubMed]
- Zaheer, M.K.; Vaishanv, N.K.; Kant, R.; Mohanan, K. Utilization of unsymmetric diaryliodonium salts in α-Arylation of α-fluoroacetoacetamides. Chem. Asian J. 2020, 15, 4297–4301. [Google Scholar] [CrossRef] [PubMed]
- Ochiai, M.; Kitagawa, Y.; Takayama, N.; Takaoka, Y.; Shiro, M. Synthesis of chiral diaryliodonium salts, 1,1′-binaphthyl-2-yl(phenyl)iodonium tetrafluoroborates: Asymmetric α-phenylation of β-keto ester enolates. J. Am. Chem. Soc. 1999, 121, 9233–9234. [Google Scholar] [CrossRef]
- Aggarwal, V.K.; Olofsson, B. Enantioselective α-arylation of cyclohexanones with diaryliodonium salts: Application to the synthesis of (-)-epibatidine. Angew. Chem. Int. Ed. 2005, 44, 5516–5519. [Google Scholar] [CrossRef] [PubMed]
- Allen, A.E.; MacMillan, D.W.C. Enantioselective α-Arylation of aldehydes via the productive merger of iodonium salts and organocatalysis. J. Am. Chem. Soc. 2011, 133, 4260–4263. [Google Scholar] [CrossRef] [PubMed]
- Harvey, J.S.; Simonovich, S.P.; Jamison, C.R.; MacMillan, D.W. Enantioselective α-arylation of carbonyls via Cu(I)-bisoxazoline catalysis. J. Am. Chem. Soc. 2011, 133, 13782–13785. [Google Scholar] [CrossRef] [PubMed]
- Bigot, A.; Williamson, A.E.; Gaunt, M.J. Enantioselective α-arylation of N-acyloxazolidinones with copper(II)-bisoxazoline catalysts and diaryliodonium salts. J. Am. Chem. Soc. 2011, 133, 13778–13781. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Dong, S.; Zhang, Y.; Kuang, Y.; Liu, X.; Lin, L.; Feng, X. Chiral scandium(III)-catalyzed enantioselective α-arylation of N-unprotected 3-substituted oxindoles with diaryliodonium salts. Angew. Chem. Int. Ed. 2013, 52, 10245–10249. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Lin, L.; Liu, Y.; Li, X.; Liu, X.; Feng, X. Nickel(II)-catalyzed enantioselective α-vinylation of β-keto amides/esters with hypervalent iodine salts. Org. Lett. 2016, 18, 5540–5543. [Google Scholar] [CrossRef] [PubMed]
- Escudero-Casao, M.; Licini, G.; Orlandi, M. Enantioselective α-arylation of ketones via a novel Cu(I)-bis(phosphine) dioxide catalytic system. J. Am. Chem. Soc. 2021, 143, 3289–3294. [Google Scholar] [CrossRef] [PubMed]
- Norrby, P.-O.; Petersen, T.B.; Bielawski, M.; Olofsson, B. α-Arylation by rearrangement: On the reaction of enolates with diaryliodonium salts. Chem. Eur. J. 2010, 16, 8251–8254. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Graskemper, J.W.; Qin, L.; DiMagno, S.G. Regiospecific reductive elimination from diaryliodonium salts. Angew. Chem. Int. Ed. 2010, 49, 4079–4083. [Google Scholar] [CrossRef] [PubMed]
- Malmgren, J.; Santoro, S.; Jalalian, N.; Himo, F.; Olofsson, B. Arylation with Unsymmetrical Diaryliodonium Salts: A Chemoselectivity Study. Chem.–Eur. J. 2013, 19, 10334–10342. [Google Scholar] [CrossRef] [PubMed]
- Trost, B.M. The atom economy-A search for synthetic efficiency. Science 1991, 254, 1471–1477. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Chen, S.; Jiang, X. Atom-economical applications of diaryliodonium salts. Chem. Asian J. 2018, 13, 2195–2207. [Google Scholar] [CrossRef] [PubMed]
- Modha, S.G.; Greaney, M.F. Atom-Economical Transformation of Diaryliodonium Salts: Tandem C−H and N−H Arylation of Indoles. J. Am. Chem. Soc. 2015, 137, 1416–1419. [Google Scholar] [CrossRef] [PubMed]
- Linde, E.; Bulfield, D.; Kervefors, G.; Purkait, N.; Olofsson, B. Diarylation of N- and O-nucleophiles through a metal-free cascade reaction. Chem 2022, 8, 850–865. [Google Scholar] [CrossRef]
- Wu, T.; Greaney, M.F. Chemoselective cascade arylation. Chem 2022, 8, 609–611. [Google Scholar] [CrossRef]
- Silva, L.F., Jr.; Olofsson, B. Hypervalent iodine reagents in the total synthesis of natural products. Nat. Prod. Rep. 2011, 28, 1722–1754. [Google Scholar] [CrossRef] [PubMed]


























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Substrate | Catalyst Systems | Enantioselectivity | Efficiency | Functional Group Tolerance | Substrate Scope | Ref. | |
| Substrate | Diaryliodonium Salts | |||||||
| 1 | 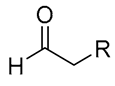 | Combination of copper and organic catalysts (Cat. 3·TCA and CuBr) | 90–94% ee | Cat. 3·TCA (10–40 mol%) CuBr (5–10 mol%) NaHCO3, 23 °C 67–95% yield | CO2Et, NBoc, NHCbz, Alkene, etc. | F, CF3, OMe, NO2, etc. | R = Alkyl, iPr, Bn; Ar1 = Aryl, Thiophene, Pyridine, Naphthalene group, etc. | [81] |
| 2 |  | Cu(II)-bis(oxazoline) | 17–94% ee | Cat. 3 (5–10 mol%), 0–10 °C, 6–20 h, 40–99% yield | NMeCbz, Alkene, etc. | CO2Et, F, CF3, OMe, etc. | R = Alkyl, Aryl, Indole, iPr, Bn; Ar1 = Aryl, Thiophene, Naphthalene group, etc. | [82] |
| 3 | 90–94% ee | Cat. 4 (10–20 mol%), −20–0 °C, 24 h 65–96% yield | NMeCbz, Alkene, etc. | CO2Et, F, CF3, OMe, etc. | R = Alkyl, Indole, iPr, Bn; Ar1 = Aryl, Thiophene, Naphthalene group, etc. | [83] | ||
| 4 |  | Cu(I)− Bis(phosphine) Dioxide | 45–95% ee | L3 (12 mol%) Cu(OTf)2·Tol (4 mol%) rt, 16 h 20–90% yield | NCbz, F, CF3, OMe, NO2, etc. | CO2Et, F, Cl, Br, CF3, OMe, NO2, etc. | R = Alkyl, iPr, Bn, Tetralone; Ar1 = Aryl, Pyridine group, etc. | [86] |
| 5 |  | N,N′-dioxide-Sc(OTf)3 complex | 17–99% ee | Sc(OTf)3/L1 =1:1 10 mol%, 3 Å M.S., NaHCO3, 35 °C 35 °C, 24–120 h 47–99% yield | F, Cl, Br, CN, OMe, NO2, etc. | F, Cl, Br, Ph, Me, etc. | R = Alkyl, 2-naphthylmethyl; Ar1 = Aryl, Pyridine group, etc. | [84] |
| 6 | 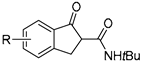 | N,N′-dioxide (L2)/Ni(OTf)2 complex | 91–99% ee | L-PisEPh/Ni(OTf)2 (1:1.2; 10 mol %) Na2CO3, 35 °C, 24–120 h 48–99% yield | F, Cl, Br, Ph, etc. | F, Cl, Br, Ph, Me, etc. | Ar1 = Aryl group | [85] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, X.-W.; Chen, J.-L.; Zhang, L.-H.; Zhang, H.; Chen, X.; Fan, X. Advances in α-Arylation of Carbonyl Compounds: Diaryliodonium Salts as Arylating Agents. Molecules 2025, 30, 3019. https://doi.org/10.3390/molecules30143019
Chen X-W, Chen J-L, Zhang L-H, Zhang H, Chen X, Fan X. Advances in α-Arylation of Carbonyl Compounds: Diaryliodonium Salts as Arylating Agents. Molecules. 2025; 30(14):3019. https://doi.org/10.3390/molecules30143019
Chicago/Turabian StyleChen, Xiao-Wei, Jia-Le Chen, Ling-Hui Zhang, Huhu Zhang, Xiaojun Chen, and Xiaohui Fan. 2025. "Advances in α-Arylation of Carbonyl Compounds: Diaryliodonium Salts as Arylating Agents" Molecules 30, no. 14: 3019. https://doi.org/10.3390/molecules30143019
APA StyleChen, X.-W., Chen, J.-L., Zhang, L.-H., Zhang, H., Chen, X., & Fan, X. (2025). Advances in α-Arylation of Carbonyl Compounds: Diaryliodonium Salts as Arylating Agents. Molecules, 30(14), 3019. https://doi.org/10.3390/molecules30143019