A Brand-New Metal Complex Catalyst-Free Approach to the Synthesis of 2,8-Dimethylimidazo[1,2-b]pyridazine-6-Carboxylic Acid—A Key Intermediate in Risdiplam Manufacturing Process

 , , ,
, , ,  , , and
, , and
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
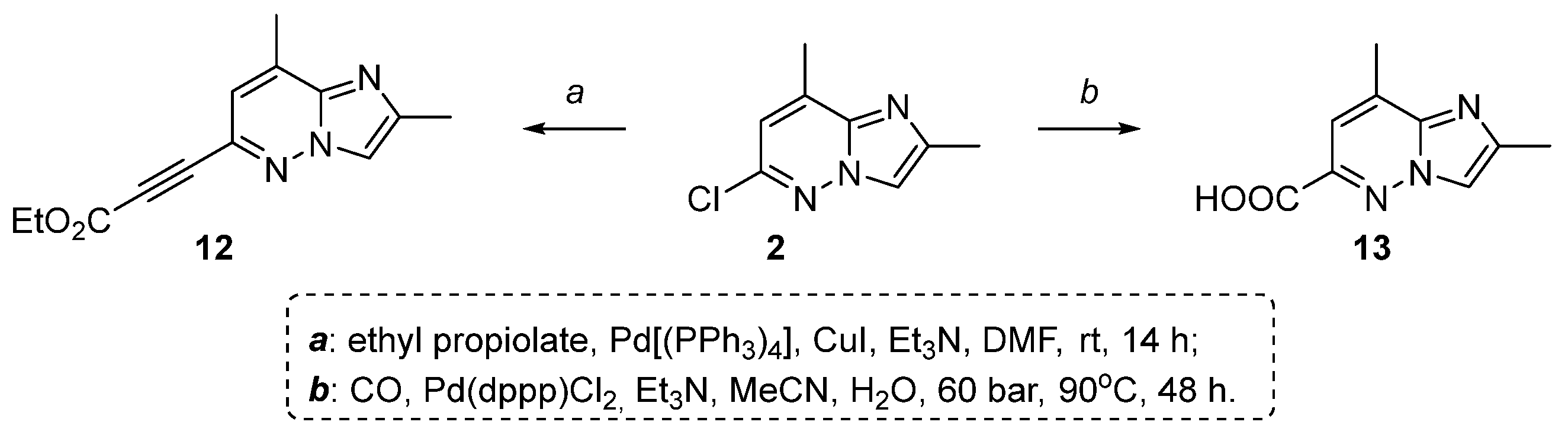
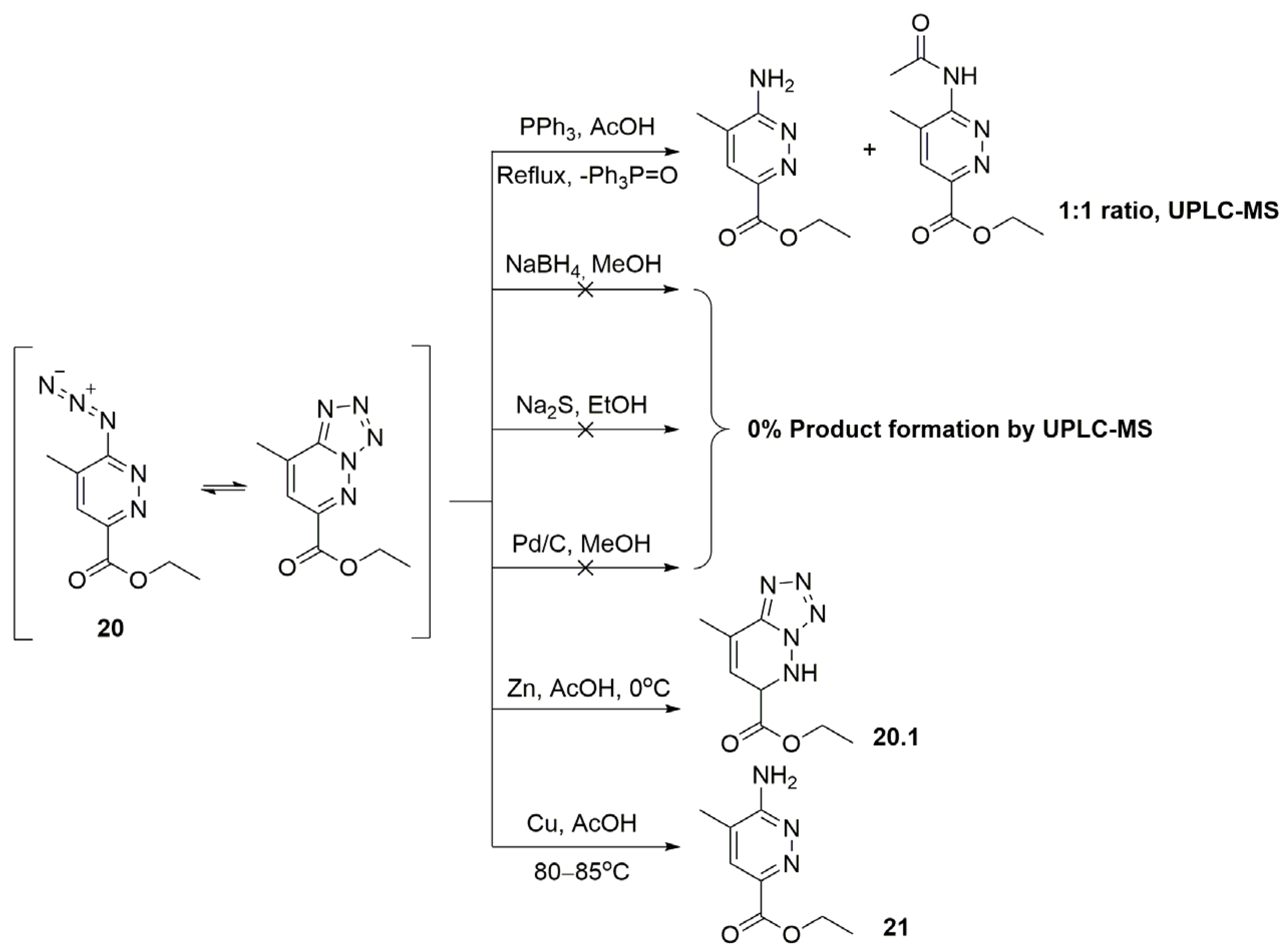
2.1. 2,8-Dimethylimidazo[1,2-b]pyridazine-6-carboxylic Acid Synthesis
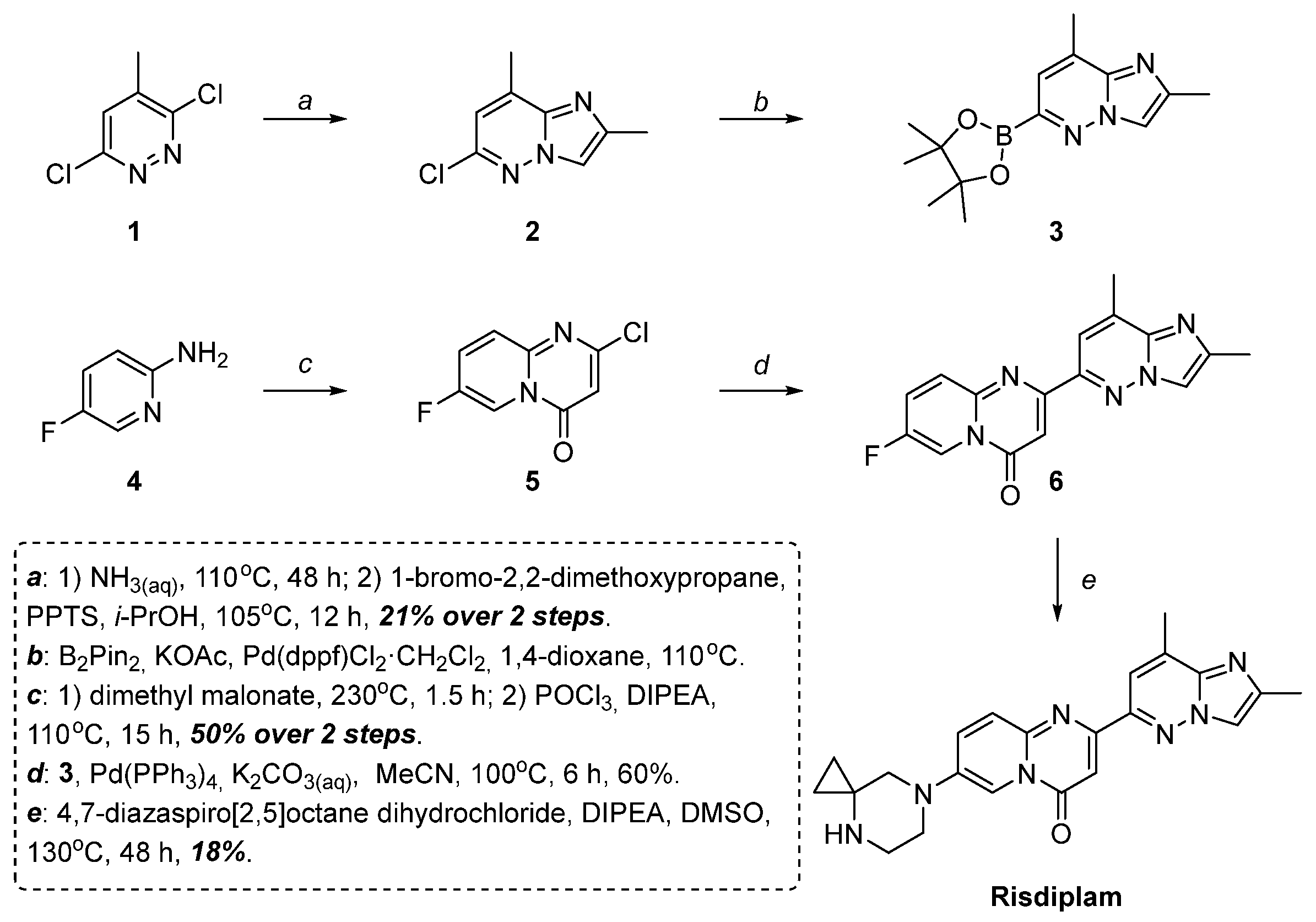
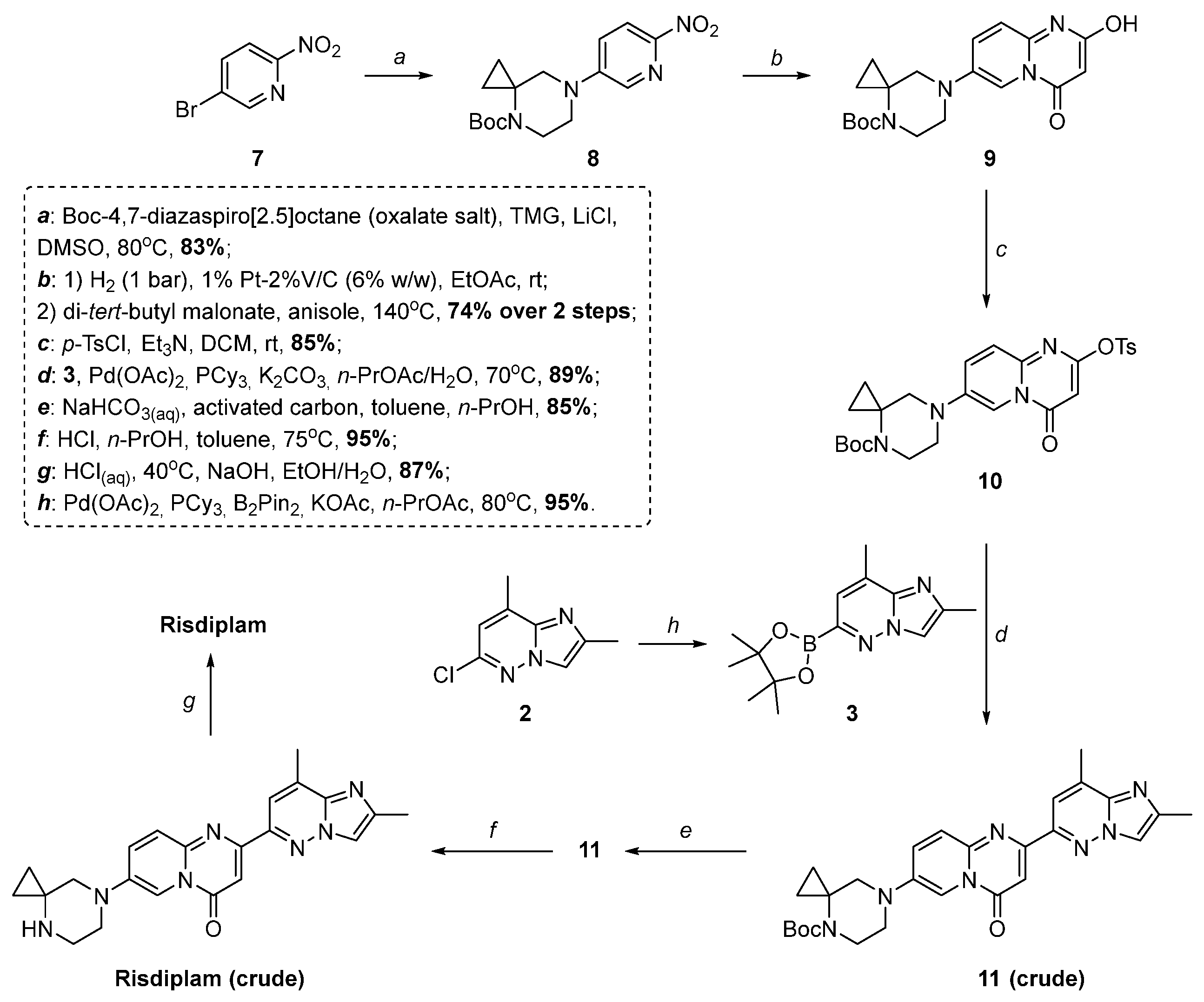
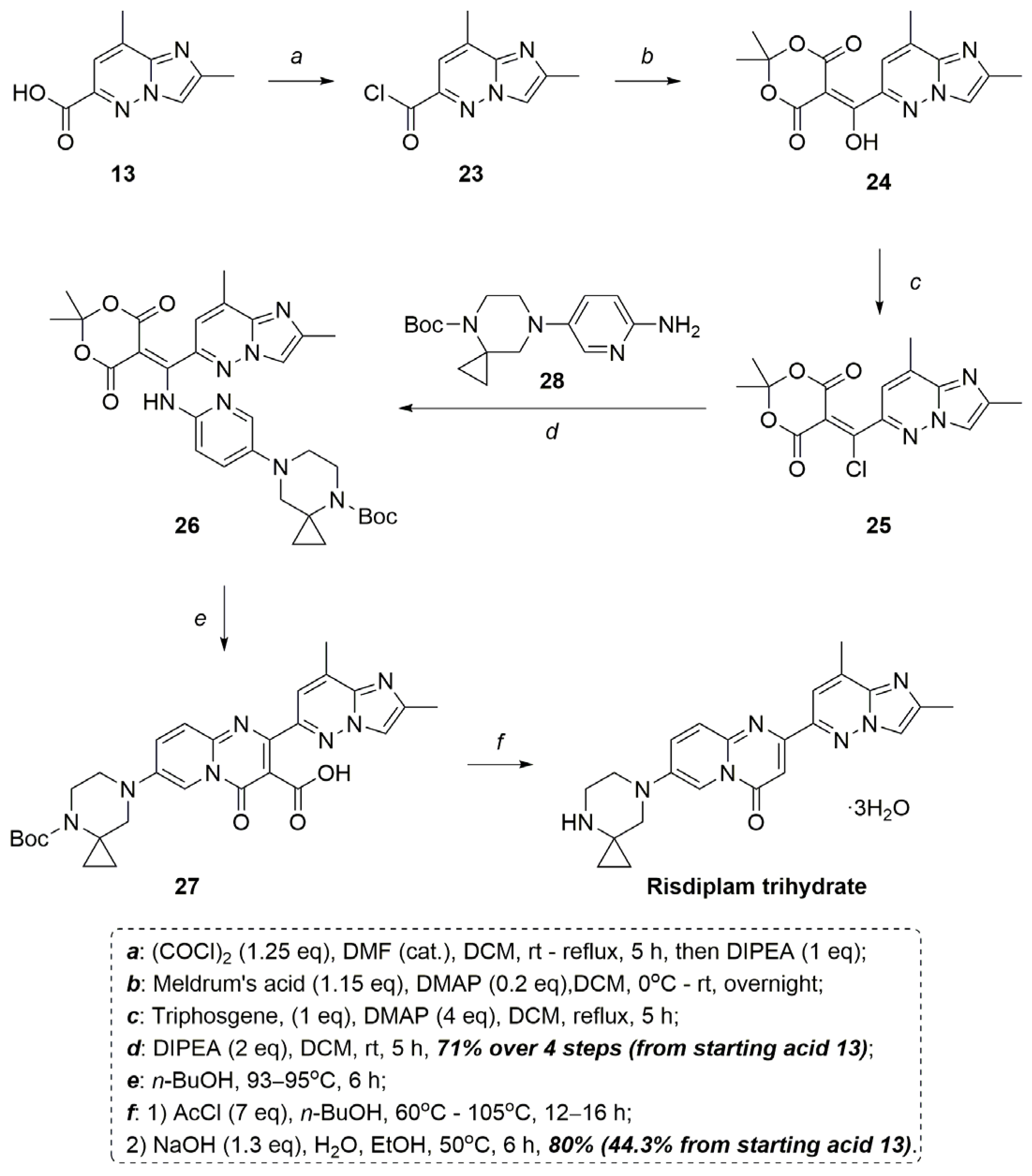
2.2. Modified Scheme for Risdiplam Synthesis
3. Materials and Methods
3.1. General
3.2. Synthetic Procedures and Compounds Characterization
3.2.1. Synthesis of 5-Methoxy-4-methyl-2,5-dioxopent-3-enoic Acid 15 [18]
3.2.2. Synthesis of 5-Methyl-6-oxo-1,6-dihydropyridazine-3-carboxylic Acid 17
- 1H NMR (400 MHz, DMSO-d6, δ): 13.36 (1H, s, 1-NH), 7.74 (1H, q, J = 1.2 Hz, 4-CH), 2.09 (3H, d, J = 1.3 Hz, 5-CH3). 13C NMR (101 MHz, DMSO-d6, δ): 164.4 (3-COOH), 162.3 (C-6), 139.9 (C-5), 137.4 (C-3), 129.4 (C-4), 16.2 (5-CH3). HRMS (ESI–): found m/z 153.0304 [M − H]−; calculated for C6H5N2O3 153.0378, mp = 256–257 °C (dec).
3.2.3. Synthesis of Ethyl 5-Methyl-6-oxo-1,6-dihydropyridazine-3-carboxylate 18
- 1H NMR (400 MHz, DMSO-d6, δ): 13.43 (1H, s, 1-NH), 7.75 (1H, q, J = 1.4 Hz, 4-CH), 4.30 (2H, q, J = 7.1 Hz, CH2CH3), 2.10 (3H, d, J = 1.3 Hz, 5-CH3), 1.30 (3H, t, J = 7.1 Hz, CH2CH3). 13C NMR (101 MHz, DMSO-d6, δ): 162.8 (3-COOCH2CH3), 162.2 (C-6), 140.2 (C-5), 136.7 (C-3), 129.1 (C-4), 61.9 (3-COOCH2CH3), 16.2 (3-COOCH2CH3), 14.5 (5-CH3). HRMS (ESI+): found m/z 183.0793 [M + H]+; calculated for C8H11N2O3 183.0691, mp = 169–170 °C.
3.2.4. Synthesis of Ethyl 6-Chloro-5-methylpyridazine-3-carboxylate 19
- 1H NMR (400 MHz, DMSO-d6, δ): 8.28 (1H, q, J = 0.9 Hz, 4-CH), 4.43 (2H, q, J = 7.1 Hz, CH2CH3), 2.47 (3H, d, J = 0.9 Hz, 5-CH3), 1.37 (3H, t, J = 7.1 Hz, CH2CH3). 13C NMR (101 MHz, DMSO-d6, δ): 163.6 (3-COOCH2CH3), 160.2 (C-6), 151.2 (C-3), 140.1 (C-5), 131.0 (C-4), 62.6 (3-COOCH2CH3), 19.0 (5-CH3), 14.5 (3-COOCH2CH3). HRMS (ESI+): found m/z 201.0452 [M + H]+; calculated for C8H1035ClN2O2 201.0353.
3.2.5. Synthesis of Ethyl 8-Methyltetrazolo[1,5-b]pyridazine-6-carboxylate (or Ethyl 6-Azido-5-methylpyridazine-3-carboxylate as Its Tautomeric Form 20)
- 1H NMR (400 MHz, DMSO-d6, δ): 8.16 (1H, q, J = 1.2 Hz, 4-CH), 4.49 (2H, q, J = 7.1 Hz, CH2CH3), 2.82 (3H, d, J = 1.2 Hz, 5-CH3), 1.41 (3H, t, J = 7.1 Hz, CH2CH3). 13C NMR (101 MHz, DMSO-d6, δ): 162.0 (3-COOCH2CH3), 147.2 (C-3), 145.3 (C-6), 139.7 (C-5), 124.2 (C-4), 63.4 (3-COOCH2CH3), 17.0 (5-CH3), 14.4 (3-COOCH2CH3). HRMS (ESI+): found m/z 208.0849 [M + H]+; calculated for C8H10N5O2 208.0756, mp = 107–108 °C.
3.2.6. Synthesis of Ethyl 6-Amino-5-methylpyridazine-3-carboxylate 21
- 1H NMR (400 MHz, DMSO-d6, δ): 7.66 (1H, d, J = 1.2 Hz, 4-CH), 6.99 (2H, br.s., 6-NH2), 4.31 (2H, q, J = 7.1 Hz, CH2CH3), 2.11 (3H, d, J = 1.0 Hz, 5-CH3), 1.32 (3H, t, J = 7.1 Hz, CH2CH3). 13C NMR (101 MHz, DMSO-d6, δ): 164.8 (3-COOCH2CH3), 162.1 (C-6), 143.2 (C-3), 128.3 (C-4), 122.4 (C-5), 61.1 (3-COOCH2CH3), 16.7 (5-CH3), 14.7 (3-COOCH2CH3). HRMS (ESI+): found m/z 182.0956 [M + H]+; calculated for C8H12N3O2 182.0851, mp = 155–156 °C.
3.2.7. Synthesis of Ethyl 8-Methyl-5,6-dihydrotetrazolo[1,5-b]pyridazine-6-carboxylate 20.1
- 1H NMR (400 MHz, DMSO-d6, δ): 7.93 (1H, d, J = 6.8 Hz, 5-NH), 6.36–6.33 (1H, m, 7-CH), 5.03-5.00 (1H, m, 6-CH), 4.09 (2H, qd, J = 7.1, 4.7 Hz, CH2CH3), 2.16 (3H, s, 8-CH3), 1.15 (3H, t, J = 7.1 Hz, CH2CH3). 13C NMR (101 MHz, DMSO-d6, δ): 168.8 (6-COOCH2CH3), 146.8 (C-4), 127.1 (C-7), 123.0 (C-8), 61.8 (3-COOCH2CH3), 57.1 (C-6), 16.2 (5-CH3), 14.3 (3-COOCH2CH3). UPLC-MS (ESI+): found m/z 210.0 [M + H]+; calculated for C8H12N5O2 210.1.
3.2.8. Synthesis of Ethyl 2,8-Dimethylimidazo[1,2-b]pyridazine-6-carboxylate 22
- 1H NMR (400 MHz, DMSO-d6, δ): 8.20 (1H, d, J = 1.0 Hz, 3-CH), 7.55 (1H, q, J = 1.1 Hz, 7-CH), 4.40 (2H, q, J = 7.1 Hz, CH2CH3), 2.60 (3H, d, J = 1.1 Hz, 8-CH3), 2.43 (3H, d, J = 0.8 Hz, 2-CH3), 1.36 (3H, t, J = 7.1 Hz, CH2CH3). 13C NMR (101 MHz, DMSO-d6, δ): 163.4 (6-COOCH2CH3), 145.3 (C-2), 142.4 (C-6), 139.6 (C-9), 136.3 (C-8), 116.0 (C-7), 115.4 (C-3), 62.3 (6-COOCH2CH3), 16.5 (8-CH3), 15.1 (2-CH3), 14.5 (3-COOCH2CH3). HRMS (ESI+): found m/z 220.1113 [M + H]+; calculated for C11H14N3O2 220.1008, mp = 109–110 °C.
3.2.9. Synthesis of 2,8-Dimethylimidazo[1,2-b]pyridazine-6-carboxylic Acid 13
- 1H NMR (400 MHz, DMSO-d6, δ): 13.62 (1H, br.s., 6-COOH), 8.14 (1H, d, J = 1.0 Hz, 3-CH), 7.53 (1H, d, J = 1.3 Hz, 7-CH), 2.58 (3H, d, J = 1.1 Hz, 8-CH3), 2.42 (3H, d, J = 0.8 Hz, 2-CH3). 13C NMR (101 MHz, DMSO-d6, δ): 164.9 (6-COOH), 145.0 (C-2), 143.3 (C-6), 139.6 (C-9), 136.0 (C-8), 116.2 (C-7), 115.3 (C-3), 16.5 (8-CH3), 15.1 (2-CH3). HRMS (ESI–): found m/z 190.0609 [M − H]+; calculated for C9H8N3O2 190.0695, mp = 271–272 °C (dec).
3.2.10. Synthesis of 2,8-Dimethylimidazo[1,2-b]pyridazine-6-carbonyl Chloride 23
3.2.11. Synthesis of 5-((2,8-Dimethylimidazo[1,2-b]pyridazin-6-yl)(hydroxy)methylene)-2,2-dimethyl-1,3-dioxane-4,6-dione 24
3.2.12. Synthesis of 5-(Chloro(2,8-dimethylimidazo[1,2-b]pyridazin-6-yl)methylene)-2,2-dimethyl-1,3-dioxane-4,6-dione 25
3.2.13. Synthesis of tert-Butyl 7-(6-(((2,2-dimethyl-4,6-dioxo-1,3-dioxan-5-ylidene)(2,8-dimethylimidazo[1,2-b]pyridazin-6-yl)methyl)amino)pyridin-3-yl)-4,7-diazaspiro[2.5]octane-4-carboxylate 26
- 1H NMR (400 MHz, DMSO-d6, δ): 12.49 (1H, s, NH), 7.93 (1H, s, 3″-CH), 7.75 (1H, d, J = 2.9 Hz, 2′-CH), 7.22 (1H, dd, J = 9.1, 3.0 Hz, 5′-CH), 7.14 (1H, d, J = 1.4 Hz, 7″-CH), 6.87 (1H, d, J = 9.0 Hz, 4′-CH), 3.50–3.47 (2H, m, 6-CH2), 3.08-3.06 (2H, m, 5-CH2), 2.95 (2H, s, 8-CH2), 2.50 (3H, s, 8″-CH3), 2.37 (3H, s, 2″-CH3), 1.77 (3H, s, (CH3)C(O)(O)(CH3)), 1.73 (3H, s, (CH3)C(O)(O)(CH3)), 1.38 (9H, s, -C(O)C(CH3)3), 0.88-0.85 (2H, m, 1-CH2, 2-CH2), 0.77-0.74 (2H, m, 1-CH2, 2-CH2). 13C NMR (101 MHz, DMSO-d6, δ): 165.5 (C=C(=NH)-), 161.2 (2C, -C(=O)-), 160.7 (C-6″), 154.6 (-C(O)C(CH3)3), 146.7 (C-6′), 142.8 (C-2″), 140.4 (C-2′), 138.4 (C-9″), 134.7 (C-2′), 134.6 (C-8′), 123.5 (C-5′), 117.1 (C-4′), 116.9 (C-7″), 114.6 (C-3″), 103.5 (C=C(=NH)-), 87.9 ((CH3)C(O)(O)(CH3)), 79.1 (-C(O)C(CH3)3), 53.3 (C-8), 45.9 (C-5), 44.9 (C-6), 37.2 (C-3), 27.9 (-C(O)C(CH3)3), 26.2 (-C(=O)-), 26.2 (-C(=O)-), 16.0 (8″-CH3), 14.4 (2″-CH3), 13.5 (C-1, C-2). UPLC-MS (ESI+): found m/z 604.2 [M + H]+; calculated for C31H38N7O6 604.3.
3.2.14. Synthesis of 7-(4-(tert-Butoxycarbonyl)-4,7-diazaspiro[2.5]octan-7-yl)-2-(2,8-dimethylimidazo[1,2-b]pyridazin-6-yl)-4-oxo-4H-pyrido[1,2-a]pyrimidine-3-carboxylic Acid 27
- 1H NMR (400 MHz, CDCl3, δ): 8.41 (1H, d, J = 2.5 Hz, 6-CH), 7.85-7.79 (2H, m, 8-CH, 9-CH), 7.64 (1H, d, J = 0.9 Hz, 3′-CH), 6.92 (1H, d, J = 1.2 Hz, 7′-CH), 3.73 (2H, dd, J = 6.1, 4.3 Hz, 6″-CH2), 3.32 (2H, t, J = 5.2 Hz, 5″-CH2), 3.10 (2H, s, 8″-CH2), 2.61 (3H, d, J = 1.1 Hz, 8′-CH3), 2.45 (3H, d, J = 0.9 Hz, 2′-CH3), 1.43 (9H, s, -C(O)C(CH3)3), 1.10–1.07 (2H, m, 1″-CH2, 2″-CH2), 0.86–0.83 (2H, m, 1″-CH2, 2″-CH2). 13C NMR (101 MHz, CDCl3, δ): 164.0 (3-COOH), 161.5 (C-4), 160.4 (C-10), 155.2 (-C(O)C(CH3)3), 151.2 (C-6′), 145.3 (C-2), 144.0 (C-7), 143.2 (C-2′), 139.3 (C-9′), 135.4 (C-8′), 132.5 (C-8), 127.3 (C-9), 116.5 (C-7′), 114.6 (C-3′), 109.7 (C-6), 102.5 (C-3), 80.8 (-C(O)C(CH3)3), 54.4 (C-8″), 46.9 (C-5″), 44.9 (C-6″), 37.1 (C-3″), 28.4 (-C(O)C(CH3)3), 16.8 (8′-CH3), 14.7 (2′-CH3), 14.3 (C-1″, C-2″). HRMS (ESI+): found m/z 546.2475 [M + H]+; calculated for C28H32N7O5 546.2387, mp = 185–186 °C (dec).
3.2.15. Synthesis of 2-(2,8-Dimethylimidazo[1,2-b]pyridazin-6-yl)-7-(4,7-diazaspiro[2.5]octan-7-yl)-4H-pyrido[1,2-a]pyrimidin-4-one (Risdiplam)
- 1H NMR (400 MHz, CDCl3, δ): 8.36 (1H, d, J = 2.4 Hz, 6-CH), 7.84 (1H, q, J = 1.1 Hz, 7′-CH), 7.72 (1H, d, J = 1.0 Hz, 3′-CH), 7.66–7.63 (1H, m, 9-CH), 7.63–7.59 (1H, m, 8-CH), 7.29 (1H, s, 3-CH), 3.19–3.16 (2H, m, 6″-CH2), 3.12–3.09 (2H, m, 5″-CH2), 3.00 (2H, s, 8″-CH2), 2.65 (3H, d, J = 1.1 Hz, 8′-CH3), 2.47 (3H, d, J = 0.8 Hz, 2′-CH3), 0.69–0.66 (2H, m, 1″-CH2, 2″-CH2), 0.59–0.56 (2H, m, 1″-CH2, 2″-CH2). 13C NMR (101 MHz, CDCl3, δ): 158.1 (C-4), 156.2 (C-6′), 148.5 (C-2), 147.2 (C-10), 144.1 (C-2′), 142.2 (C-7), 140.0 (C-9′), 135.6 (C-8′), 131.2 (C-8), 126.7 (C-9), 114.9 (C-7′), 114.7 (C-3′), 110.0 (C-6), 99.2 (C-3), 56.6 (C-8″), 49.9 (C-5″), 44.5 (C-6″), 36.5 (C-3″), 16.9 (8′-CH3), 14.9 (2′-CH3), 13.0 (C-1″, C-2″). HRMS (ESI+): found m/z 402.2051 [M + H]+; calculated for C22H24N7O 402.1964, mp = 269–270 °C (dec).
3.2.16. Synthesis of tert-Butyl 4,7-diazaspiro[2.5]octane-4-carboxylate (Oxalate Salt) 30
- 1H NMR (400 MHz, CDCl3, δ): 3.52–3.50 (2H, m, 5-CH2), 2.87–2.85 (2H, m, 6-CH2), 2.67 (2H, s, 8-CH2), 1.48 (9H, s, -C(O)C(CH3)3), 0.99–0.96 (2H, m, 1-CH2, 2-CH2), 0.75–0.71 (2H, m, 1-CH2, 2-CH2). 13C NMR (101 MHz, CDCl3, δ): 155.7 (-C(O)C(CH3)3), 79.7 (-C(O)C(CH3)3), 52.4 (C-8), 48.3 (C-5), 45.6 (C-6), 39.8 (C-3), 28.4 (-C(O)C(CH3)3), 13.6 (C-1, C-2). HRMS (ESI+): found m/z 213.1622 [M + H]+; calculated for C11H21N2O2 213.1525, mp = 55–56 °C.
3.2.17. Synthesis of tert-Butyl 7-(6-nitropyridin-3-yl)-4,7-diazaspiro[2.5]octane-4-carboxylate 31
- 1H NMR (400 MHz, DMSO-d6, δ): 8.23 (1H, d, J = 3.0 Hz, 2′-CH), 8.15 (1H, d, J = 9.2 Hz, 5′-CH), 7.46 (1H, dd, J = 9.3, 3.1 Hz, 4′-CH), 3.59 (2H, dd, J = 6.5, 3.9 Hz, 5-CH2), 3.50 (2H, dd, J = 6.6, 3.9 Hz, 6-CH2), 3.36 (2H, s, 8-CH2), 1.42 (9H, s, -C(O)C(CH3)3), 0.98–0.95 (2H, m, 1-CH2, 2-CH2), 0.88–0.85 (2H, m, 1-CH2, 2-CH2). 13C NMR (101 MHz, DMSO-d6, δ): 155.1 (-C(O)C(CH3)3), 150.6 (C-3′), 147.3 (C-6′), 133.9 (C-2′), 121.0 (C-4′), 120.2 (C-5′), 79.9 (-C(O)C(CH3)3), 52.7 (C-8), 48.5 (C-5), 45.3 (C-6), 37.8 (C-3), 28.5 (-C(O)C(CH3)3), 14.0 (C-1, C-2). HRMS (ESI+): found m/z 335.1739 [M + H]+; calculated for C16H23N4O4 335.1641, mp = 162–163 °C.
3.2.18. Synthesis of tert-Butyl 7-(6-aminopyridin-3-yl)-4,7-diazaspiro[2.5]octane-4-carboxylate 28
- 1H NMR (400 MHz, DMSO-d6, δ): 7.57 (1H, dd, J = 3.0, 0.7 Hz, 2′-CH), 7.14 (1H, dd, J = 8.9, 3.0 Hz, 4′-CH), 6.39 (1H, dd, J = 8.9, 0.7 Hz, 5′-CH), 5.40 (2H, br.s., 6′-NH2), 3.54 (2H, dd, J = 6.1, 3.9 Hz, 5-CH2), 2.89 (2H, dd, J = 6.3, 3.6 Hz, 6-CH2), 2.74 (2H, s, 8-CH2), 1.41 (9H, s, -C(O)C(CH3)3), 0.93–0.90 (2H, m, 1-CH2, 2-CH2), 0.83–0.80 (2H, m, 1-CH2, 2-CH2). 13C NMR (101 MHz, DMSO-d6, δ): 154.9 (C-6′), 155.2 (-C(O)C(CH3)3), 139.2 (C-3′), 136.5 (C-2′), 128.6 (C-4′), 108.7 (C-5′), 79.4 (-C(O)C(CH3)3), 57.2 (C-8), 49.8 (C-6), 46.0 (C-5), 38.2 (C-3), 28.5 (-C(O)C(CH3)3), 14.1 (C-1, C-2). HRMS (ESI+): found m/z 305.1974 [M + H]+; calculated for C16H25N4O2 305.1899.
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nishio, H.; Niba, E.T.E.; Saito, T.; Okamoto, K.; Takeshima, Y.; Awano, H. Spinal Muscular Atrophy: The Past, Present, and Future of Diagnosis and Treatment. Int. J. Mol. Sci. 2023, 24, 11939. [Google Scholar] [CrossRef] [PubMed]
- Macleod, M.J.; Taylor, J.E.; Lunt, P.W.; Mathew, C.G.; Robb, S.A. Prenatal Onset Spinal Muscular Atrophy. Eur. J. Paediatr. Neurol. 1999, 3, 65–72. [Google Scholar] [CrossRef]
- Dubowitz, V. Very Severe Spinal Muscular Atrophy (SMA Type 0): An Expanding Clinical Phenotype. Eur. J. Paediatr. Neurol. 1999, 3, 49–51. [Google Scholar] [CrossRef] [PubMed]
- Felderhoff-Mueser, U.; Grohmann, K.; Harder, A.; Stadelmann, C.; Zerres, K.; Bührer, C.; Obladen, M. Severe Spinal Muscular Atrophy Variant Associated With Congenital Bone Fractures. J. Child Neurol. 2002, 17, 718–721. [Google Scholar] [CrossRef]
- Kelly, T.E.; Amoroso, K.; Ferre, M.; Blanco, J.; Allinson, P.; Prior, T.W. Spinal Muscular Atrophy Variant with Congenital Fractures. Am. J. Med. Genet. 1999, 87, 65–68. [Google Scholar] [CrossRef]
- D’Amico, A.; Mercuri, E.; Tiziano, F.D.; Bertini, E. Spinal Muscular Atrophy. Orphanet J. Rare Dis. 2011, 6, 71. [Google Scholar] [CrossRef]
- Chaytow, H.; Huang, Y.-T.; Gillingwater, T.H.; Faller, K.M.E. The Role of Survival Motor Neuron Protein (SMN) in Protein Homeostasis. Cell. Mol. Life Sci. 2018, 75, 3877–3894. [Google Scholar] [CrossRef] [PubMed]
- Butchbach, M.E.R. Genomic Variability in the Survival Motor Neuron Genes (SMN1 and SMN2): Implications for Spinal Muscular Atrophy Phenotype and Therapeutics Development. Int. J. Mol. Sci. 2021, 22, 7896. [Google Scholar] [CrossRef]
- Verhaart, I.E.C.; Robertson, A.; Leary, R.; McMacken, G.; König, K.; Kirschner, J.; Jones, C.C.; Cook, S.F.; Lochmüller, H. A Multi-Source Approach to Determine SMA Incidence and Research Ready Population. J. Neurol. 2017, 264, 1465–1473. [Google Scholar] [CrossRef]
- Wirth, B.; Karakaya, M.; Kye, M.J.; Mendoza-Ferreira, N. Twenty-Five Years of Spinal Muscular Atrophy Research: From Phenotype to Genotype to Therapy, and What Comes Next. Annu. Rev. Genom. Hum. Genet. 2020, 21, 231–261. [Google Scholar] [CrossRef]
- Sarv, S.; Kahre, T.; Vaidla, E.; Pajusalu, S.; Muru, K.; Põder, H.; Gross-Paju, K.; Ütt, S.; Žordania, R.; Talvik, I.; et al. The Birth Prevalence of Spinal Muscular Atrophy: A Population Specific Approach in Estonia. Front. Genet. 2021, 12, 796862. [Google Scholar] [CrossRef] [PubMed]
- Ratni, H.; Scalco, R.S.; Stephan, A.H. Risdiplam, the First Approved Small Molecule Splicing Modifier Drug as a Blueprint for Future Transformative Medicines. ACS Med. Chem. Lett. 2021, 12, 874–877. [Google Scholar] [CrossRef] [PubMed]
- Ratni, H.; Ebeling, M.; Baird, J.; Bendels, S.; Bylund, J.; Chen, K.S.; Denk, N.; Feng, Z.; Green, L.; Guerard, M.; et al. Discovery of Risdiplam, a Selective Survival of Motor Neuron-2 (SMN2) Gene Splicing Modifier for the Treatment of Spinal Muscular Atrophy (SMA). J. Med. Chem. 2018, 61, 6501–6517. [Google Scholar] [CrossRef]
- Moessner, C.; Hoffmann-Emery, F.; Adam, J.-M.; Fantasia, S.; Fishlock, D.; Meier, R.; Wuitschik, G.; Ratni, H. Development and Optimization of the Manufacturing Process for RNA-Splicing Modifier Risdiplam RG7916. In ACS Symposium Series; American Chemical Society: Washington, DC, USA, 2022; Volume 1423, pp. 301–332. [Google Scholar]
- Wang, Z. Conrad-Limpach Quinoline Synthesis. In Comprehensive Organic Name Reactions and Reagents; Wiley: Hoboken, NJ, USA, 2010; pp. 692–696. [Google Scholar]
- Kothakonda, K.K.; Pitta, B.R.; Namana, S.B.; Rangisetty, J.B.; Pullagurla, M.R. A Novel Process for the Preparation of 7-(4,7-Diazaspiro[2.5]Octan-7-Yl)-2-(2,8 Dimethylimidazo[1,2-b]Pyridazin-6-Yl)Pyrido-4H-[1,2-a]Pyrimidin-4-One with Novel Intermediates. WO 2024/003798 A1, 29 June 2023. [Google Scholar]
- Adam, J.-M.; Pfleger, C.; Wuitschik, G. Novel Process. WO 2022/194909 A2, 16 March 2022. [Google Scholar]
- Rios, A.C.; Bera, P.P.; Moreno, J.A.; Cooper, G. Pyruvate Aldol Condensation Product: A Metabolite That Escaped Synthetic Preparation for over a Century. ACS Omega 2020, 5, 15063–15068. [Google Scholar] [CrossRef]
- McMillan, F.H.; Kun, K.A.; McMillan, C.B.; Schwartz, B.S.; King, J.A. Hydrazides of Some Pyridazonyl Substituted Acids. J. Am. Chem. Soc. 1956, 78, 407–410. [Google Scholar] [CrossRef]
- Arnott, E.A.; Chan, L.C.; Cox, B.G.; Meyrick, B.; Phillips, A. POCl 3 Chlorination of 4-Quinazolones. J. Org. Chem. 2011, 76, 1653–1661. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Gong, X.; Wang, G. 1H/2H and Azide/Tetrazole Isomerizations and Their Effects on the Aromaticity and Stability of Azido Triazoles. RSC Adv. 2015, 5, 9503–9509. [Google Scholar] [CrossRef]
- Alkorta, I.; Blanco, F.; Elguero, J. The Azido-Tetrazole Tautomerism in Azoles and Its Relationships with Aromaticity and NMR Properties. Tetrahedron 2010, 66, 5071–5081. [Google Scholar] [CrossRef]
- Deev, S.L.; Shestakova, T.S.; Charushin, V.N.; Chupakhin, O.N. Synthesis and Azido-Tetrazole Tautomerism of 3-Azido-1,2,4-Triazines. Chem. Heterocycl. Compd. 2017, 53, 963–975. [Google Scholar] [CrossRef]
- Ulomskii, E.N.; Shestakova, T.S.; Deev, S.L.; Rusinov, V.L.; Chupakhin, O.N. A New Approach to the Synthesis of Lamotrigine and Other 3,5-Diamino-1,2,4-Triazine Derivatives. Russ. Chem. Bull. 2005, 54, 726–732. [Google Scholar] [CrossRef]
- Staudinger, H.; Hauser, E. Über Neue Organische Phosphorverbindungen IV Phosphinimine. Helv. Chim. Acta 1921, 4, 861–886. [Google Scholar] [CrossRef]
- Richoll, S.M.; Colón, I. Determination of Triphenylphosphine Oxide in Active Pharmaceutical Ingredients by Hollow-Fiber Liquid-Phase Microextraction Followed by Reversed-Phase Liquid Chromatography. J. Chromatogr. A 2006, 1127, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Zhukovets, A.A.; Chernyshov, V.V.; Al’mukhametov, A.Z.; Seregina, T.A.; Revtovich, S.V.; Kasatkina, M.A.; Isakova, Y.E.; Kulikova, V.V.; Morozova, E.A.; Cherkasova, A.I.; et al. Novel Hydroxamic Acids Containing Aryl-Substituted 1,2,4- or 1,3,4-Oxadiazole Backbones and an Investigation of Their Antibiotic Potentiation Activity. Int. J. Mol. Sci. 2023, 25, 96. [Google Scholar] [CrossRef]
- Saputra, M.A.; Ngo, L.; Kartika, R. Synthesis of Vinyl Chlorides via Triphosgene–Pyridine Activation of Ketones. J. Org. Chem. 2015, 80, 8815–8820. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Korenev, G.; Gutenev, A.A.; Antipin, F.V.; Chernyshov, V.V.; Korobkina, M.P.; Nawrozkij, M.B.; Ivanov, R.A. A Brand-New Metal Complex Catalyst-Free Approach to the Synthesis of 2,8-Dimethylimidazo[1,2-b]pyridazine-6-Carboxylic Acid—A Key Intermediate in Risdiplam Manufacturing Process. Molecules 2025, 30, 3011. https://doi.org/10.3390/molecules30143011
Korenev G, Gutenev AA, Antipin FV, Chernyshov VV, Korobkina MP, Nawrozkij MB, Ivanov RA. A Brand-New Metal Complex Catalyst-Free Approach to the Synthesis of 2,8-Dimethylimidazo[1,2-b]pyridazine-6-Carboxylic Acid—A Key Intermediate in Risdiplam Manufacturing Process. Molecules. 2025; 30(14):3011. https://doi.org/10.3390/molecules30143011
Chicago/Turabian StyleKorenev, Georgiy, Alexey A. Gutenev, Fyodor V. Antipin, Vladimir V. Chernyshov, Maria P. Korobkina, Maxim B. Nawrozkij, and Roman A. Ivanov. 2025. "A Brand-New Metal Complex Catalyst-Free Approach to the Synthesis of 2,8-Dimethylimidazo[1,2-b]pyridazine-6-Carboxylic Acid—A Key Intermediate in Risdiplam Manufacturing Process" Molecules 30, no. 14: 3011. https://doi.org/10.3390/molecules30143011
APA StyleKorenev, G., Gutenev, A. A., Antipin, F. V., Chernyshov, V. V., Korobkina, M. P., Nawrozkij, M. B., & Ivanov, R. A. (2025). A Brand-New Metal Complex Catalyst-Free Approach to the Synthesis of 2,8-Dimethylimidazo[1,2-b]pyridazine-6-Carboxylic Acid—A Key Intermediate in Risdiplam Manufacturing Process. Molecules, 30(14), 3011. https://doi.org/10.3390/molecules30143011