
Development and Validation of a Highly Sensitive LC–MS/MS Method for the Precise Quantification of Sitagliptin in Human Plasma and Its Application to Pharmacokinetic Study

 , , , , and
, , , , and
Abstract
1. Introduction
2. Results and Discussion
2.1. Method Development
2.1.1. Mass Spectrometry
2.1.2. Chromatographic Conditions
2.1.3. Plasma Sample Preparation
2.2. Method Validation
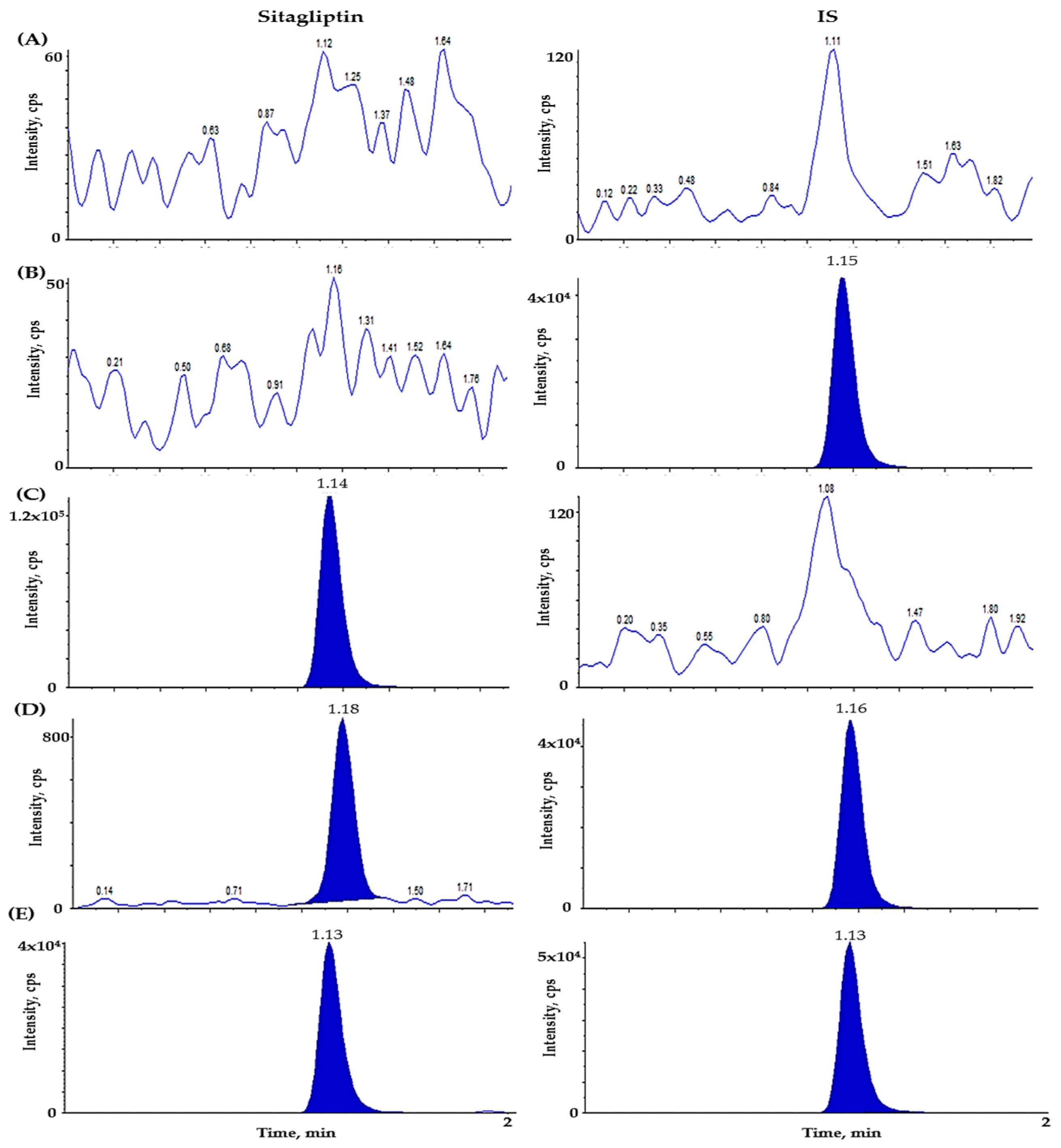
2.2.1. Selectivity and Lower Limit of Quantitation (LLOQ)
2.2.2. Linearity and Carryover
2.2.3. Precision and Accuracy
2.2.4. Recovery and Matrix Effect
2.2.5. Stability
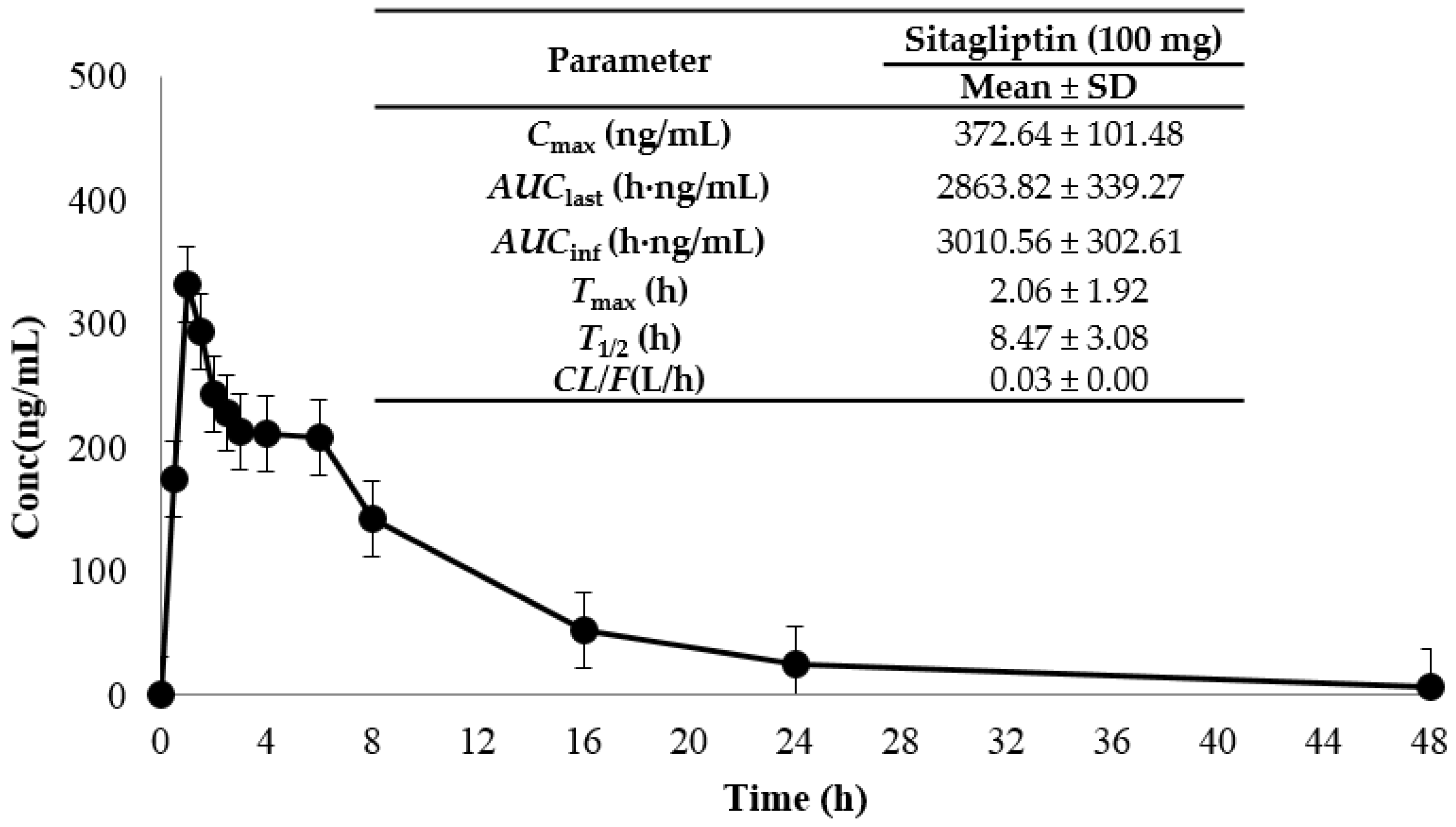
2.3. Application to a Bioequivalence Study
2.4. Incurred Sample Reanalysis (ISR)
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. Liquid Chromatographic Conditions
3.3. Mass Spectrometric Conditions
3.4. Preparation of Calibration Standard Solutions and Quality Control Samples
3.5. Plasma Sample Preparation
3.6. Method Validation
3.6.1. Selectivity and LLOQ
3.6.2. Linearity and Carryover
3.6.3. Precision and Accuracy
3.6.4. Recovery and Matrix Effect
3.6.5. Stability
3.7. Application to a Pharmacokinetic Study
3.8. Incurred Sample Reanalysis
3.9. Statistical Analysis of Bioequivalence
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- García-Aguilar, A.; Guillén, C. Targeting pancreatic beta cell death in type 2 diabetes by polyphenols. Front. Endocrinol. 2022, 13, 1052317. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, M.; Pratley, R. GLP-1 analogs and DPP-4 inhibitors in type 2 diabetes therapy: Review of head-to-head clinical trials. Front. Endocrinol. 2020, 11, 178. [Google Scholar] [CrossRef] [PubMed]
- Das, D.; Halder, D.; Bose, A.; Shaw, T.K.; Saha, C.; De, P.K.; Maji, H.S.; Pal, T.K. Determination of Metformin and Sitagliptin in Healthy Human Volunteers’ Blood Plasma and Its Bioequivalence Study Under Fasting Condition. Int. J. Appl. Pharm. 2022, 14, 42–50. [Google Scholar] [CrossRef]
- Drugs.com. Sitagliptin Monograph. Available online: https://www.drugs.com/monograph/sitagliptin.html (accessed on 8 July 2024).
- Kalra, S.; Singh, A.K.; Das, S.; Pendurthi, B.; Dharmadhikari, S.; Ahire, P.; Khandhedia, C.; Markandeywar, N.; Mane, A.; Mehta, S. Sitagliptin as an Add-on Therapy to Other Glucose-lowering Agents in Patients with Type 2 Diabetes Mellitus: A Narrative Review. J. Assoc. Physicians India 2025, 73, e13–e18. [Google Scholar] [PubMed]
- Saini, K.; Sharma, S.; Khan, Y. DPP-4 inhibitors for treating T2DM-hype or hope? an analysis based on the current literature. Front. Mol. Biosci. 2023, 10, 1130625. [Google Scholar] [CrossRef] [PubMed]
- DrugBank. Sitagliptin (DB01261). Available online: https://go.drugbank.com/drugs/DB01261 (accessed on 7 July 2024).
- Yin, R.; Xu, Y.; Wang, X.; Yang, L.; Zhao, D. Role of dipeptidyl peptidase 4 inhibitors in antidiabetic treatment. Molecules 2022, 27, 3055. [Google Scholar] [CrossRef] [PubMed]
- Texas Health and Human Service. Sitagliptin (Januvia) Monograph. Available online: https://www.hhs.texas.gov/sites/default/files/documents/doing-business-with-hhs/provider-portal/facilities-regulation/psychiatric/monograph/sitagliptin-januvia-monograph.pdf (accessed on 7 July 2024).
- Philippine Food and Drug Administration. Sitagliptin (JANUVIA) Product Information. Available online: https://verification.fda.gov.ph/files/DRP-11159_PI_01.pdf (accessed on 7 July 2024).
- Xie, Y.; Zhou, Q.; He, Q.; Wang, X.; Wang, J. Opportunities and challenges of incretin-based hypoglycemic agents treating type 2 diabetes mellitus from the perspective of physiological disposition. Acta Pharm. Sin. B 2023, 13, 2383–2402. [Google Scholar] [CrossRef] [PubMed]
- Med Central, Sitagliptin—Oral. Available online: https://www.medcentral.com/drugs/monograph/187692-307001/sitagliptin-oral (accessed on 7 July 2024).
- Ashraf, M.; Shahzad, M.N.; Hayat, M.M.; Rahman, J.; Ejaz, S.; Altaf, H.; Nasim, F.H. Development and validation of an HPLC method for the quantification of sitagliptin in plasma and tablet dosage form. Lat. Am. J. Pharm. 2015, 34, 456–461. [Google Scholar]
- Al Bratty, M.; Alhazmi, H.A.; Javed, S.A.; Lalitha, K.G.; Asmari, M.; Wölker, J.; El Deeb, S. Development and validation of LC–MS/MS method for simultaneous determination of metformin and four gliptins in human plasma. Chromatographia 2017, 80, 891–899. [Google Scholar] [CrossRef]
- Khoja, S.S.; Patel, L.J. Development and validation of new analytical LC-MS/MS method for the estimation of antidiabetic drugs ertugliflozin and sitagliptin in combined pharmaceutical dosage form. J. Pharm. Res. Int. 2021, 33, 194–204. [Google Scholar] [CrossRef]
- Nirogi, R.; Kandikere, V.; Mudigonda, K.; Komarneni, P.; Aleti, R.; Boggavarapu, R. Sensitive liquid chromatography tandem mass spectrometry method for the quantification of sitagliptin, a DPP-4 inhibitor, in human plasma using liquid–liquid extraction. Biomed. Chromatogr. 2008, 22, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Hess, C.; Musshoff, F.; Madea, B. Simultaneous identification and validated quantification of 11 oral hypoglycaemic drugs in plasma by electrospray ionisation liquid chromatography–mass spectrometry. Anal. Bioanal. Chem. 2011, 400, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Pamu, S.; Patyar, S.; Thakkalapally, L. Development and Validation of a Novel RP-HPLC Analytical Method for Sitagliptin Determination in Human Plasma. J. Pharm. Res. Int. 2021, 33, 92–101. [Google Scholar] [CrossRef]
- Han, X.; Wang, J.; Huang, J.; Peng, L. A Rapid and Sensitive Method for the Pharmacokinetic Study of Janumet (Sitagliptin and Metformin) Tablets by LC-MS/MS Coupled with Ion-Pair Solid Phase Extraction. Curr. Pharm. Anal. 2019, 15, 776–784. [Google Scholar] [CrossRef]
- Loh, G.O.K.; Wong, E.Y.L.; Tan, Y.T.F.; Lee, Y.L.; Pang, L.H.; Chin, M.C.; Damenthi, N.; Peh, K.K. Simple and rapid LC-MS/MS method for determination of sitagliptin in human plasma and application to bioequivalence study. J. Chromatogr. B 2020, 1159, 122337. [Google Scholar] [CrossRef] [PubMed]
- Lukka, P.B.; Tang, W.; Hammarstedt, A.; Conrad, T.; Heijer, M.; Karlsson, C.; Boulton, D.W. Racial Comparison of the Pharmacokinetics and Safety of Fixed-dose Combination of Dapagliflozin/Sitagliptin in Western and Korean Healthy Adults. Clin. Ther. 2024, 46, 717–725. [Google Scholar] [CrossRef] [PubMed]
- Moon, S.J.; Yu, K.-S.; Kim, M.-G. An assessment of pharmacokinetic interaction between lobeglitazone and sitagliptin after multiple oral administrations in healthy men. Clin. Ther. 2020, 42, 1047–1057. [Google Scholar] [CrossRef] [PubMed]
- Reddy, S.; Ahmed, I.; Ahmad, I.; Mukhopadhyay, A.; Thangam, S. Development and validation of a method for simultaneous estimation of metformin and sitagliptin in human plasma by LC–MS-MS and its application in a bioequivalence study. J. Chromatogr. Sci. 2015, 53, 1549–1556. [Google Scholar] [CrossRef] [PubMed]
- Scherf-Clavel, O.; Kinzig, M.; Stoffel, M.S.; Fuhr, U.; Sörgel, F. A HILIC-MS/MS assay for the quantification of metformin and sitagliptin in human plasma and urine: A tool for studying drug transporter perturbation. J. Pharm. Biomed. Anal. 2019, 175, 112754. [Google Scholar] [CrossRef] [PubMed]
- Thao, N.N.N.; Hieu, N.N.; Loan, T.T.T.; Tuan, N.D. Development, Validation, and Application for Simultaneous Assay of Metformin and Sitagliptin in Human Plasma by liquid Chromatography-Tandem Mass spectrometry. Syst. Rev. Pharm. 2020, 11, 6–13. [Google Scholar]
- Ministry of Food and Drug Safety. Guideline on Bioanalytical Method Validation. 2013. Available online: https://www.mfds.go.kr/brd/m210/down.do?brd_id=data0010&seq=13054data_tp=A&file_seq=1 (accessed on 6 December 2021).
- Food and Drug Administration. Bioanalytical Method Validation Guidance for Industry. US Department of Health and Human Services. 2018. Available online: https://www.fda.gov/media/70858/download (accessed on 6 December 2021).


{kind=link}
{kind=link}
{kind=link}
| Compound | Number | Linearity | |||
|---|---|---|---|---|---|
| Slope | Intercept | r | r2 | ||
| Sitagliptin | 1 | 0.00220 | −0.000382 | 0.9983 | 0.9966 |
| 2 | 0.00215 | −0.000367 | 0.9998 | 0.9996 | |
| 3 | 0.00214 | −0.000399 | 0.9997 | 0.9994 | |
| 4 | 0.00218 | −0.001030 | 0.9995 | 0.9990 | |
| 5 | 0.00215 | 0.000336 | 0.9998 | 0.9996 | |
| 6 | 0.00208 | 0.000278 | 0.9973 | 0.9946 | |
| 7 | 0.00211 | 0.000968 | 0.9997 | 0.9994 | |
| 8 | 0.00209 | 0.001930 | 0.9993 | 0.9986 | |
| Compound | Nominal Concentration (ng/mL) | Intra-Day (n = 5) | Inter-Day (n = 15) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Mean ± SD (ng/mL) | Precision (CV, %) a | Accuracy (%) b | Mean ± SD (ng/mL) | Precision (CV, %) a | Accuracy (%) b | ||||||
| Sitagliptin | 5 | 5.06 | ± | 0.19 | 3.72 | 101.18 | 4.91 | ± | 0.49 | 9.87 | 98.17 |
| 15 | 15.89 | ± | 0.42 | 2.67 | 105.94 | 15.04 | ± | 0.88 | 5.86 | 100.23 | |
| 300 | 289.35 | ± | 4.41 | 1.52 | 96.45 | 293.57 | ± | 6.67 | 2.27 | 97.86 | |
| 800 | 765.56 | ± | 13.85 | 1.81 | 95.70 | 777.60 | ± | 14.08 | 1.81 | 97.20 | |
| Compounds | Nominal Concentration (ng/mL) | Recovery (%) | Matrix Effect (%) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Mean ± SD | Precision (CV) | Mean ± SD | Precision (CV) | ||||||
| Sitagliptin | 15 | 83.20 | ± | 4.34 | 5.22 | 104.64 | ± | 6.72 | 6.42 |
| 300 | 81.99 | ± | 2.19 | 2.67 | 105.43 | ± | 2.65 | 2.52 | |
| 800 | 79.51 | ± | 1.34 | 1.69 | 107.30 | ± | 1.48 | 1.38 | |
| Sitagliptin-d4 | 1000 | 83.42 | ± | 2.43 | 2.91 | 103.04 | ± | 2.67 | 2.59 |
| Stability Storage Condition | Sitagliptin Concentration | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 15 ng/mL (Mean ± SD, %) | 300 ng/mL (Mean ± SD, %) | 800 ng/mL (Mean ± SD, %) | |||||||
| Solution stability (%) | |||||||||
| Stock Room temperature (3 h) | 100.89 | ± | 6.68 | 99.25 | ± | 3.59 | |||
| Working Room temperature (7 h) | 100.19 | ± | 7.76 | 95.93 | ± | 1.51 | |||
| Plasma Sample stability (%) | |||||||||
| Room temperature (7 h) | 102.57 | ± | 0.66 | 97.43 | ± | 2.55 | 96.99 | ± | 14.14 |
| Refrigeration (7 h, 4 °C) | 100.61 | ± | 0.77 | 100.02 | ± | 3.08 | 96.29 | ± | 13.25 |
| Freeze–thaw stability (3 Cycles) | 103.73 | ± | 1.08 | 98.57 | ± | 2.12 | 97.71 | ± | 7.24 |
| Autosampler (30 h, 10 °C) | 105.20 | ± | 0.49 | 97.37 | ± | 5.39 | 96.83 | ± | 1.71 |
| Deep freeze (7 h, −70 °C) | 99.61 | ± | 1.27 | 96.77 | ± | 6.79 | 97.96 | ± | 20.25 |
| Compounds | Ion Transition (m/z) | DP (V) | EP (V) | CE (V) | CXP (V) |
|---|---|---|---|---|---|
| Sitagliptin | 408.2 → 193.0 | 91.0 | 9.0 | 30.0 | 40.0 |
| Sitagliptin-d4 | 412.2 → 239.1 | 89.0 | 10.0 | 27.0 | 42.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, Y.; Shim, W.-S.; Song, E.; Park, Y.; Kim, B.-H.; Lee, S.; Chung, E.K.; Lee, K.-T. Development and Validation of a Highly Sensitive LC–MS/MS Method for the Precise Quantification of Sitagliptin in Human Plasma and Its Application to Pharmacokinetic Study. Molecules 2025, 30, 2995. https://doi.org/10.3390/molecules30142995
Song Y, Shim W-S, Song E, Park Y, Kim B-H, Lee S, Chung EK, Lee K-T. Development and Validation of a Highly Sensitive LC–MS/MS Method for the Precise Quantification of Sitagliptin in Human Plasma and Its Application to Pharmacokinetic Study. Molecules. 2025; 30(14):2995. https://doi.org/10.3390/molecules30142995
Chicago/Turabian StyleSong, Yuna, Wang-Seob Shim, Eunseo Song, Yebeen Park, Bo-Hyung Kim, Sangmin Lee, Eun Kyoung Chung, and Kyung-Tae Lee. 2025. "Development and Validation of a Highly Sensitive LC–MS/MS Method for the Precise Quantification of Sitagliptin in Human Plasma and Its Application to Pharmacokinetic Study" Molecules 30, no. 14: 2995. https://doi.org/10.3390/molecules30142995
APA StyleSong, Y., Shim, W.-S., Song, E., Park, Y., Kim, B.-H., Lee, S., Chung, E. K., & Lee, K.-T. (2025). Development and Validation of a Highly Sensitive LC–MS/MS Method for the Precise Quantification of Sitagliptin in Human Plasma and Its Application to Pharmacokinetic Study. Molecules, 30(14), 2995. https://doi.org/10.3390/molecules30142995