

Tricyclic Isatin Derivatives as Anti-Inflammatory Compounds with High Kinase Binding Affinity




Abstract
1. Introduction
2. Results and Discussion
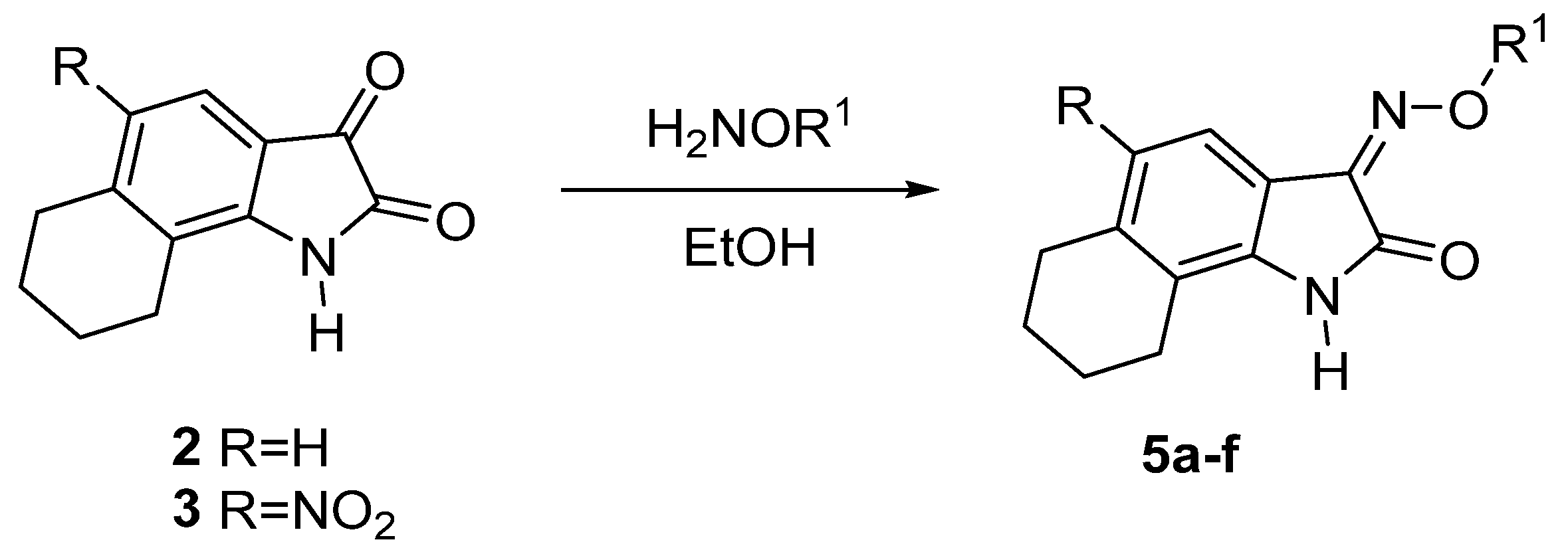
2.1. Chemical Synthesis
2.1.1. Synthesis of NS-102 Precursors
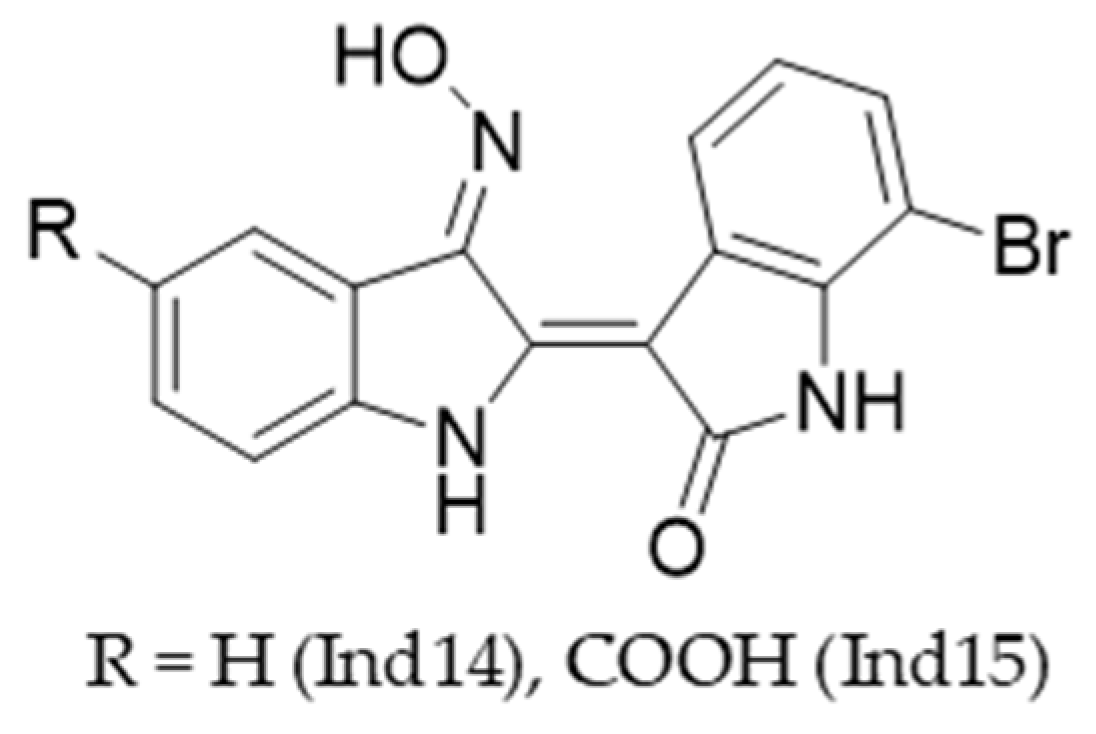
2.1.2. Oximation of Isatin Derivatives
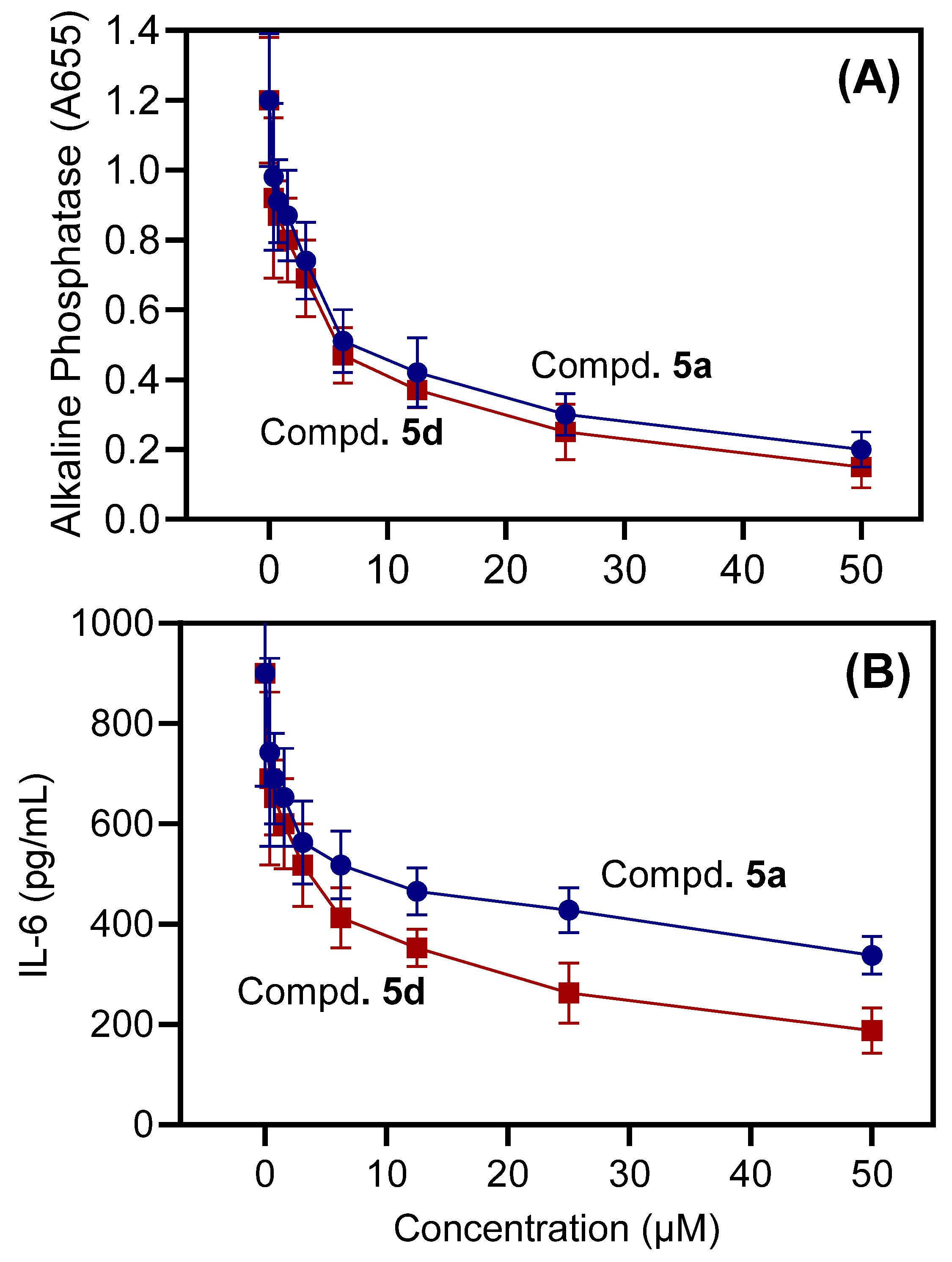
2.2. Biological Activity
2.2.1. Anti-Inflammatory Activity (In Vitro)
2.2.2. Identification of Potential Kinase Targets
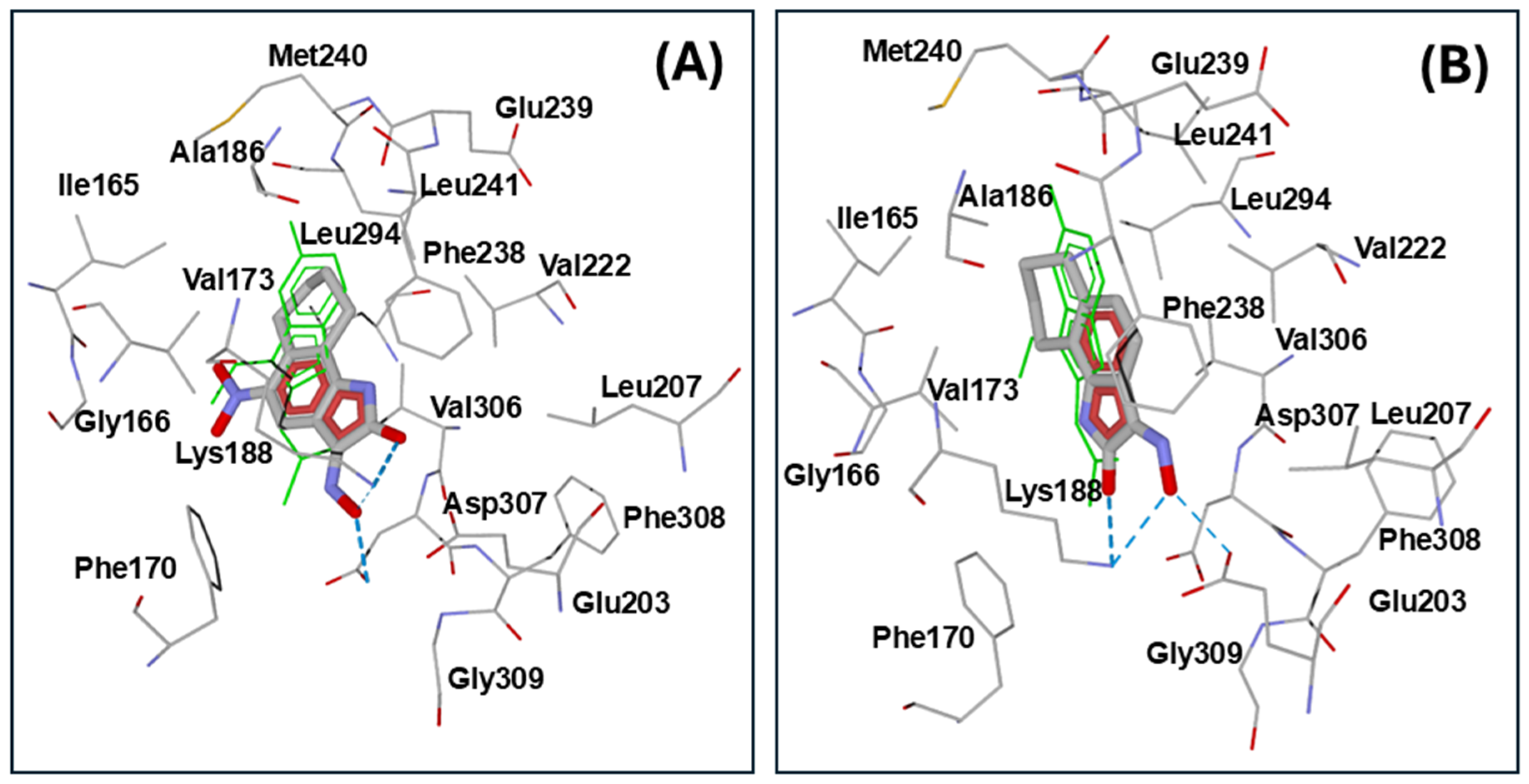
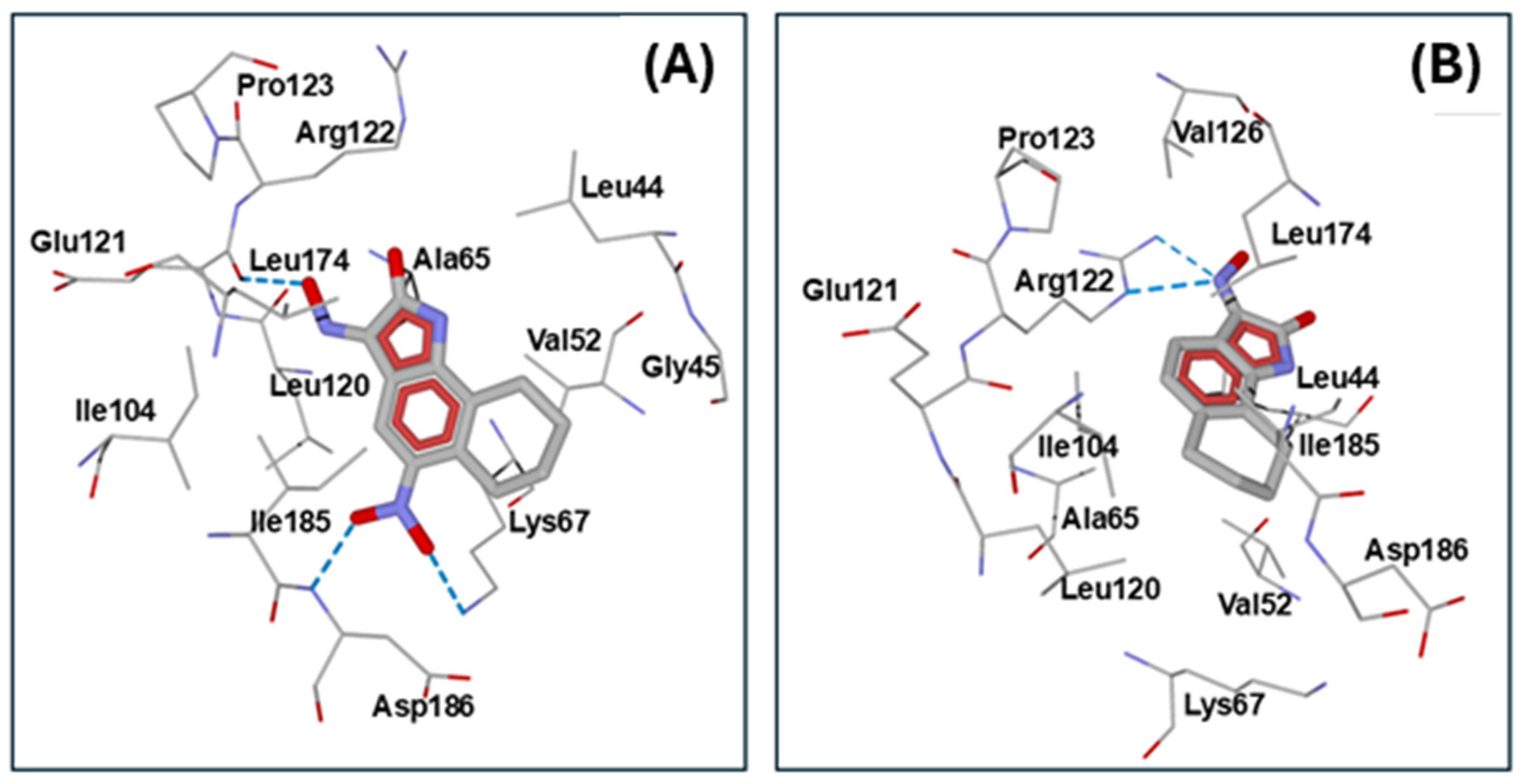
2.3. Molecular Docking
3. Materials and Methods
3.1. Chemistry
3.1.1. Compounds and Reagents
3.1.2. Synthesis
3.2. Biological Assays
3.2.1. Cell Culture
3.2.2. Evaluation of AP-1/NF-κB Activation
3.2.3. Analysis of Cytokine Production
3.2.4. Cytotoxicity Assay
3.2.5. Kinase Profiling and Kd Determination
3.3. Molecular Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Frost, D.; Meechoovet, B.; Wang, T.; Gately, S.; Giorgetti, M.; Shcherbakova, I.; Dunckley, T. Carboline Compounds, Including Harmine, Inhibit DYRK1A and Tau Phosphorylation at Multiple Alzheimer’s Disease-Related Sites. PLoS ONE 2011, 6, e19264. [Google Scholar] [CrossRef] [PubMed]
- Latosińska, M.; Latosińska, J.N. Serine/Threonine Protein Kinases as Attractive Targets for Anti-Cancer Drugs—An Innovative Approach to Ligand Tuning Using Combined Quantum Chemical Calculations, Molecular Docking, Molecular Dynamic Simulations, and Network-like Similarity Graphs. Molecules 2024, 29, 3199. [Google Scholar] [CrossRef] [PubMed]
- Narlik-Grassow, M.; Blanco-Aparicio, C.; Carnero, A. The PIM family of serine/threonine kinases in cancer. Med. Res. Rev. 2013, 34, 136–159. [Google Scholar] [CrossRef]
- Quadri, R.; Sertic, S.; Muzi-Falconi, M. Roles and regulation of Haspin kinase and its impact on carcinogenesis. Cell. Signal. 2022, 93, 110303. [Google Scholar] [CrossRef] [PubMed]
- Nishida-Fukuda, H.; Tokuhiro, K.; Ando, Y.; Matsushita, H.; Wada, M.; Tanaka, H. Evaluation of the antiproliferative effects of the HASPIN inhibitor CHR-6494 in breast cancer cell lines. PLoS ONE 2021, 16, e0249912. [Google Scholar] [CrossRef]
- Lin, C.-I.; Chen, Z.-C.; Chen, C.-H.; Chang, Y.-H.; Lee, T.-C.; Tang, T.-T.; Yang, C.-M.; Tsai, M.-C.; Huang, C.-C.; Yang, T.-W.; et al. Co-inhibition of Aurora A and Haspin kinases enhances survivin blockage and p53 induction for mitotic catastrophe and apoptosis in human colorectal cancer. Biochem. Pharmacol. 2022, 206, 115289. [Google Scholar] [CrossRef]
- Ananthapadmanabhan, V.; Shows, K.H.; Dickinson, A.J.; Litovchick, L. Insights from the protein interaction Universe of the multifunctional Goldilocks kinase DYRK1A. Front. Cell Dev. Biol. 2023, 11, 1277537. [Google Scholar] [CrossRef]
- Kimura, R.; Kamino, K.; Yamamoto, M.; Nuripa, A.; Kida, T.; Kazui, H.; Hashimoto, R.; Tanaka, T.; Kudo, T.; Yamagata, H.; et al. The DYRK1A gene, encoded in chromosome 21 Down syndrome critical region, bridges between-amyloid production and tau phosphorylation in Alzheimer disease. Hum. Mol. Genet. 2006, 16, 15–23. [Google Scholar] [CrossRef]
- Wegiel, J.; Dowjat, K.; Kaczmarski, W.; Kuchna, I.; Nowicki, K.; Frackowiak, J.; Kolecka, B.M.; Wegiel, J.; Silverman, W.P.; Reisberg, B.; et al. The role of overexpressed DYRK1A protein in the early onset of neurofibrillary degeneration in Down syndrome. Acta Neuropathol. 2008, 116, 391–407. [Google Scholar] [CrossRef]
- Meur, S.; Mukherjee, S.; Roy, S.; Karati, D. Role of PIM Kinase Inhibitor in the Treatment of Alzheimers Disease. Mol. Neurobiol. 2024, 61, 10941–10955. [Google Scholar] [CrossRef]
- Tanaka, H.; Matsushita, H.; Tokuhiro, K.; Fukunari, A.; Ando, Y. Ingestion of soybean sprouts containing a HASPIN in-hibitor improves condition in a mouse model of Alzheimer’s disease. Biology 2023, 12, 320. [Google Scholar] [CrossRef]
- Acitelli, E.; Maiorca, C.; Grani, G.; Maranghi, M. Metabolic adverse events of multitarget kinase inhibitors: A systematic review. Endocrine 2023, 81, 16–29. [Google Scholar] [CrossRef]
- Chen, S.; Yang, Y.; Yuan, Y.; Liu, B. Targeting PIM kinases in cancer therapy: An update on pharmacological small-molecule inhibitors. Eur. J. Med. Chem. 2023, 264, 116016. [Google Scholar] [CrossRef] [PubMed]
- Baek, H.S.; Kim, N.; Park, J.W.; Kwon, T.K.; Kim, S. The role of Pim-1 kinases in inflammatory signaling pathways. Inflamm. Res. 2024, 73, 1671–1685. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Fan, X.; Liu, Y.; Ye, D.; Liu, C.; Yang, H.; Su, Z.; Zhang, Y.; Liu, Y. Function and inhibition of DYRK1A: Emerging roles of treating multiple human diseases. Biochem. Pharmacol. 2023, 212, 115521. [Google Scholar] [CrossRef]
- García, E.G.; Varas, P.; González-Naranjo, P.; Ulzurrun, E.; Marcos-Ayuso, G.; Pérez, C.; Páez, J.A.; Insua, D.R.; Santana, S.R.; Campillo, N.E. AI-driven de novo design and development of nontoxic DYRK1A inhibitors. J. Med. Chem. 2025, 68, 10346–10364. [Google Scholar] [CrossRef]
- Kestav, K.; Uri, A.; Lavogina, D. Structure, roles and inhibitors of a mitotic protein kinase Haspin. Curr. Med. Chem. 2017, 24, 2276–2293. [Google Scholar] [CrossRef]
- Corsello, S.M.; Bittker, J.A.; Liu, Z.; Gould, J.; McCarren, P.; Hirschman, J.E.; Johnston, S.E.; Vrcic, A.; Wong, B.; Khan, M.; et al. The Drug Repurposing Hub: A next-generation drug library and information resource. Nat. Med. 2017, 23, 405–408. [Google Scholar] [CrossRef]
- Campillos, M.; Kuhn, M.; Gavin, A.-C.; Jensen, L.J.; Bork, P. Drug target identification using side-effect similarity. Science 2008, 321, 263–266. [Google Scholar] [CrossRef]
- Schepetkin, I.A.; Karpenko, O.S.; Kovrizhina, A.R.; Kirpotina, L.N.; Khlebnikov, A.I.; Chekal, S.I.; Radudik, A.V.; Shybinska, M.O.; Quinn, M.T. Novel tryptanthrin derivatives with selectivity as c-Jun N-terminal kinase (JNK) 3 inhibitors. Molecules 2023, 28, 4806. [Google Scholar] [CrossRef]
- Schepetkin, I.A.; Kovrizhina, A.R.; Stankevich, K.S.; Khlebnikov, A.I.; Kirpotina, L.N.; Quinn, M.T.; Cook, M.J. Design, synthesis and biological evaluation of novel O-substituted tryptanthrin oxime derivatives as c-Jun N-terminal kinase inhibitors. Front. Pharmacol. 2022, 13, 958687. [Google Scholar] [CrossRef] [PubMed]
- Schepetkin, I.A.; Khlebnikov, A.I.; Potapov, A.S.; Kovrizhina, A.R.; Matveevskaya, V.V.; Belyanin, M.L.; Atochin, D.N.; Zanoza, S.O.; Gaidarzhy, N.M.; Lyakhov, S.A.; et al. Synthesis, biological evaluation, and molecular modeling of 11H-indeno-1, 2b-quinoxalin-11-one derivatives and tryptanthrin-6-oxime as c-Jun N-terminal kinase inhibitors. Eur. J. Med. Chem. 2019, 161, 179–191. [Google Scholar] [CrossRef]
- Nie, Z.; Xia, X.; Zhao, Y.; Zhang, S.; Zhang, Y.; Wang, J. JNK selective inhibitor, IQ-1S, protects the mice against lipopolysaccharides-induced sepsis. Bioorganic Med. Chem. 2021, 30, 115945. [Google Scholar] [CrossRef] [PubMed]
- Beauchard, A.; Laborie, H.; Rouillard, H.; Lozach, O.; Ferandin, Y.; Guével, R.L.; Guguen-Guillouzo, C.; Meijer, L.; Besson, T.; Thiéry, V. Synthesis and kinase inhibitory activity of novel substituted indigoids. Bioorganic Med. Chem. 2009, 17, 6257–6263. [Google Scholar] [CrossRef] [PubMed]
- Schepetkin, I.A.; Plotnikov, M.B.; Khlebnikov, A.I.; Plotnikova, T.M.; Quinn, M.T. Oximes: Novel Therapeutics with Anticancer and Anti-Inflammatory Potential. Biomolecules 2021, 11, 777. [Google Scholar] [CrossRef]
- Ishikura, M.; Abe, T.; Choshi, T.; Hibino, S. Simple indole alkaloids and those with a non-rearranged monoterpenoid unit. Nat. Prod. Rep. 2013, 30, 694–752. [Google Scholar] [CrossRef]
- Capó, T.; Rebassa, J.B.; Raïch, I.; Lillo, J.; Badia, P.; Navarro, G.; Reyes-Resina, I. Future perspectives of NMDAR in CNS disorders. Molecules 2025, 30, 877. [Google Scholar] [CrossRef]
- Tripathi, S.; Sharma, Y.; Kumar, D. Exploring New Structures of Kinase Inhibitors and Multitarget Strategies in Alzheimer’s Disease Treatment. Protein Pept. Lett. 2025, 32, 2–17. [Google Scholar] [CrossRef]
- Jakobsen, B.; Tasker, A.; Zimmer, J. Domoic acid neurotoxicity in hippocampal slice cultures. Amino Acids 2002, 23, 37–44. [Google Scholar] [CrossRef]
- Tasker, R.A.; Strain, S.M.; Drejer, J. Selective reduction in domoic acid toxicity in vivo by a novel non-N-methyl-D-aspartate receptor antagonist. Can. J. Physiol. Pharmacol. 1996, 74, 1047–1054. [Google Scholar] [CrossRef]
- Li, Q.; Ma, T.-L.; Qiu, Y.-Q.; Cui, W.-Q.; Chen, T.; Zhang, W.-W.; Wang, J.; Mao-Ying, Q.-L.; Mi, W.-L.; Wang, Y.-Q.; et al. Connexin 36 mediates orofacial pain hypersensitivity through GluK2 and TRPA1. Neurosci. Bull. 2020, 36, 1484–1499. [Google Scholar] [CrossRef] [PubMed]
- Perkinton, M.S.; Sihra, T.S. A high-affinity presynaptic kainate-type glutamate receptor facilitates glutamate exocytosis from cerebral cortex nerve terminals (synaptosomes). Neuroscience 1999, 90, 1281–1292. [Google Scholar] [CrossRef]
- Kamiya, H.; Ozawa, S. Kainate receptor-mediated inhibition of presynaptic Ca2+ influx and EPSP in area CA1 of the rat hippocampus. J. Physiol. 1998, 509, 833–845. [Google Scholar] [CrossRef] [PubMed]
- Munro, G.; Christensen, J.K.; Erichsen, H.K.; Dyhring, T.; Demnitz, J.; Dam, E.; Ahring, P.K. NS383 Selectively Inhibits Acid-Sensing Ion Channels Containing 1a and 3 Subunits to Reverse Inflammatory and Neuropathic Hyperalgesia in Rats. Cns Neurosci. Ther. 2015, 22, 135–145. [Google Scholar] [CrossRef]
- Sun, A.W.; Wu, M.H.; Vijayalingam, M.; Wacker, M.J.; Chu, X.-P. The role of zinc in modulating acid-sensing ion channel function. Biomolecules 2023, 13, 229. [Google Scholar] [CrossRef] [PubMed]
- Wätjen, F.; Nielsen, E.; Drejer, J.; Jensen, L.H. Isatin oximes-A novel series of bioavailable non-NMDA antagonists. Bioorganic Med. Chem. Lett. 1993, 3, 105–106. [Google Scholar] [CrossRef]
- Al-Khuzaie, M.G.A.; Fahad, M.M.; Al-Safi, A.J. Synthesis, reaction and biological importance of isatin derivatives. Biomed. Chem. Sci. 2022, 1, 193–206. [Google Scholar] [CrossRef]
- Zhungietu, G.I.; Rechter, M.A. Isatin and Its Derivatives; Academy of Sciences of the Moldavian SSR, Institute of Chemistry: Chisinau, Moldova, 1977. (In Russian) [Google Scholar]
- Popp, F.D.; McEwen, W.E. Polyphosphoric acids as a reagent in organic chemistry. Chem. Rev. 1958, 58, 321–401. [Google Scholar] [CrossRef]
- Baker, B.R.; Schaub, R.E.; Joseph, J.P.; McEvoy, F.J.; Williams, J.H. An antimalarial alkaloid from hydrangea. XV. Synthesis of 5-, 6-, 7-, and 8-derivatives with two identical substituents. J. Org. Chem. 1952, 17, 149–156. [Google Scholar] [CrossRef]
- Mazhilis, L.I.; Terent’Ev, P.B.; Bolotin, V.A. Mononitration of derivatives of benzisatins. Chem. Heterocycl. Compd. 1989, 25, 50–55. [Google Scholar] [CrossRef]
- Adcock, I.M.; Caramori, G. Cross-talk between pro-inflammatory transcription factors and glucocorticoids. Immunol. Cell Biol. 2001, 79, 376–384. [Google Scholar] [CrossRef] [PubMed]
- Karaman, M.W.; Herrgard, S.; Treiber, D.K.; Gallant, P.; Atteridge, C.E.; Campbell, B.T.; Chan, K.W.; Ciceri, P.; Davis, M.I.; Edeen, P.T.; et al. A quantitative analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2008, 26, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Fabian, M.A.; Biggs, W.H.; Treiber, D.K.; Atteridge, C.E.; Azimioara, M.D.; Benedetti, M.G.; Carter, T.A.; Ciceri, P.; Edeen, P.T.; Floyd, M.; et al. A small molecule-kinase interaction map for clinical kinase inhibitors. Nat. Biotechnol. 2005, 23, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Hoang, V.T.; Nyswaner, K.; Torres-Ayuso, P.; Brognard, J. The protein kinase MAP3K19 phosphorylates MAP2Ks and thereby activates ERK and JNK kinases and increases viability of KRAS-mutant lung cancer cells. J. Biol. Chem. 2020, 295, 8470–8479. [Google Scholar] [CrossRef]
- Ogawa, Y.; Nonaka, Y.; Goto, T.; Ohnishi, E.; Hiramatsu, T.; Kii, I.; Yoshida, M.; Ikura, T.; Onogi, H.; Shibuya, H.; et al. Development of a novel selective inhibitor of the Down syndrome-related kinase DYRK1A. Nat. Commun. 2010, 1, 86. [Google Scholar] [CrossRef] [PubMed]
- Masaki, S.; Kii, I.; Sumida, Y.; Kato-Sumida, T.; Ogawa, Y.; Ito, N.; Nakamura, M.; Sonamoto, R.; Kataoka, N.; Hosoya, T.; et al. Design and synthesis of a potent inhibitor of class 1 DYRK kinases as a suppressor of adipogenesis. Bioorganic Med. Chem. 2015, 23, 4434–4441. [Google Scholar] [CrossRef]
- Cheney, I.W.; Yan, S.; Appleby, T.; Walker, H.; Vo, T.; Yao, N.; Hamatake, R.; Hong, Z.; Wu, J.Z. Identification and structure-activity relationships of substituted pyridones as inhibitors of Pim-1 kinase. Bioorganic Med. Chem. Lett. 2007, 17, 1679–1683. [Google Scholar] [CrossRef]
- Myrianthopoulos, V.; Kritsanida, M.; Gaboriaud-Kolar, N.; Magiatis, P.; Ferandin, Y.; Durieu, E.; Lozach, O.; Cappel, D.; Soundararajan, M.; Filippakopoulos, P.; et al. Novel inverse binding mode of indirubin derivatives yields improved selectivity for DYRK kinases. ACS Med. Chem. Lett. 2013, 4, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Lyskov, S.; Chou, F.-C.; Conchúir, S.; Der, B.S.; Drew, K.; Kuroda, D.; Xu, J.; Weitzner, B.D.; Renfrew, P.D.; Sripakdeevong, P.; et al. Serverification of molecular modeling applications: The Rosetta Online Server that Includes Everyone (ROSIE). PLoS ONE 2013, 8, e63906. [Google Scholar] [CrossRef]
- DeLuca, S.; Khar, K.; Meiler, J. Fully flexible docking of medium sized ligand libraries with Rosetta ligand. PLoS ONE 2015, 10, e0132508. [Google Scholar] [CrossRef]
- Combs, S.A.; DeLuca, S.L.; DeLuca, S.H.; Lemmon, G.H.; Nannemann, D.P.; Nguyen, E.D.; Willis, J.R.; Sheehan, J.H.; Meiler, J. Small-molecule ligand docking into comparative models with Rosetta. Nat. Protoc. 2013, 8, 1277–1298. [Google Scholar] [CrossRef] [PubMed]
- Kothiwale, S.; Mendenhall, J.L.; Meiler, J. BCL::CONF: Small molecule conformational sampling using a knowledge based rotamer library. J. Cheminform. 2015, 7, 47. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Concentration of H2SO4 (%) | Total Yield of Isomers 3 and 4 (%) | Ratio of Products 3 and 4 |
|---|---|---|
| 98 | 10 | 9:1 |
| 75 | 89 | 20:1 |
| Compd. | R | R1 | Yield (%) |
|---|---|---|---|
| 5a | H | H | 96 |
| 5b | H | Me | 93 |
| 5c | H | Et | 89 |
| 5d | NO2 | H | 95 |
| 5e | NO2 | Me | 90 |
| 5f | NO2 | Et | 90 |
| Compd. | AP Production (THP-1 Cells) | IL-6 Production (MonoMac-6 Cells) | Cytotoxicity (THP-1 Cells) | Cytotoxicity (MonoMac-6 Cells) |
|---|---|---|---|---|
| IC50 (µM) | ||||
| 5a | 6.0 ± 1.3 | 48.3 ± 4.4 | N.T. | N.T. |
| 5b | N.A. | N.A. | N.T. | N.T. |
| 5c | N.A. | N.A. | N.T. | N.T. |
| 5d | 5.5 ± 1.4 | 12.6 ± 3.2 | N.T. | N.T. |
| 5e | N.A. | N.A. | N.T. | N.T. |
| 5f | N.A. | N.A. | N.T. | N.T. |
| NS-383 | n.d. | n.d. | 19.2 ± 2.1 | 21.2 ± 2.4 |
| Kinase Target | Inhibition (%) of Ligand Binding by 5d (10 µM) | Compd. 5d | Compd. 5a | NS-383 |
|---|---|---|---|---|
| Kd (µM) | ||||
| DAPK1 | 96.3 | 0.29 ± 0.02 | 13.5 ± 0.7 | N.B. |
| DAPK2 | 98.9 | 0.20 ± 0.042 | 7.7 ± 1.1 | N.B. |
| DAPK3 | 98.5 | 0.59 ± 0.028 | 20.5 ± 3.5 | N.B. |
| DYRK1A | 98.8 | 0.435 ± 0.078 | 0.85 ± 0.12 | N.B. |
| DYRK1B | 96.4 | 0.47 ± 0.02 | N.B. | N.B. |
| Haspin | 99.45 | 0.165 ± 0.021 | 0.61 ± 0.08 | N.B. |
| HIPK1 | 98.5 | 0.52 ± 0.113 | 1.1 ± 0.07 | N.B. |
| HIPK2 | 99.7 | 0.295 ± 0.092 | 0.73 ± 0.64 | N.B. |
| HIPK3 | 100 | 0.135 ± 0.07 | 1.3 ± 0.3 | N.B. |
| IRAK1 | 99.2 | 0.67 ± 0.113 | N.B. | N.B. |
| NEK10 | 100 | 0.66 ± 0.071 | 3.2 ± 1.0 | N.B. |
| PIM1 | 98.2 | 0.122 ± 0.054 | 1.8 ± 0.1 | 14.0 ± 0.7 |
| Kinase (PDB Code) | Compd. | Interface Score * | Hydrogen Bonds ** |
|---|---|---|---|
| DYRK1A (3ANQ) | 5d | −14.11 | Lys188 (carbonyl oxygen, oxime oxygen-weak), Asp307 (oxime OH) |
| 5a | −13.97 | Lys188 (carbonyl oxygen, oxime oxygen), Glu203 (oxime OH), Asp307 (oxime nitrogen-weak) | |
| 5f | −13.50 | Lys188 (NO2 oxygen), Asp307 (NO2 oxygen) | |
| NS-383 | −13.19 | Lys188 (oxime nitrogen), Asp307 (oxime OH), Gly309 (oxime OH-weak) | |
| PIM1 (2OBJ) | 5d | −12.74 | Glu121 (oxime OH), Asp186 (NO2 oxygen), Lys67 (NO2 oxygen) |
| 5a | −12.24 | Arg122 (oxime nitrogen) | |
| 5f | −11.93 | Lys67 (NO2 oxygen), Asp186 (oxime oxygen) | |
| NS-383 | −11.39 | Glu121 (oxime OH) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Uvarov, A.V.; Schepetkin, I.A.; Quinn, M.T.; Khlebnikov, A.I. Tricyclic Isatin Derivatives as Anti-Inflammatory Compounds with High Kinase Binding Affinity. Molecules 2025, 30, 2914. https://doi.org/10.3390/molecules30142914
Uvarov AV, Schepetkin IA, Quinn MT, Khlebnikov AI. Tricyclic Isatin Derivatives as Anti-Inflammatory Compounds with High Kinase Binding Affinity. Molecules. 2025; 30(14):2914. https://doi.org/10.3390/molecules30142914
Chicago/Turabian StyleUvarov, Alexander V., Igor A. Schepetkin, Mark T. Quinn, and Andrei I. Khlebnikov. 2025. "Tricyclic Isatin Derivatives as Anti-Inflammatory Compounds with High Kinase Binding Affinity" Molecules 30, no. 14: 2914. https://doi.org/10.3390/molecules30142914
APA StyleUvarov, A. V., Schepetkin, I. A., Quinn, M. T., & Khlebnikov, A. I. (2025). Tricyclic Isatin Derivatives as Anti-Inflammatory Compounds with High Kinase Binding Affinity. Molecules, 30(14), 2914. https://doi.org/10.3390/molecules30142914