1. Introduction
According to the National Dental and Craniofacial Institute of the United States, it has been found that periodontal disease occurs through tartar, a sticky film of bacteria commonly known as plaque, which accumulates and forms on teeth; periodontal disease has many forms and symptoms, with gingivitis and periodontitis being the most common [
1]. Among them, periodontitis is known as the second most common type of periodontal disease worldwide after tooth decay, and individual oral hygiene problems, smoking, and dietary problems are known to be the main causes of periodontitis, though various other complex problems are also causes [
2]. Periodontitis begins with initial inflammation, and this long-term chronic inflammation affects the bone and bearing capacity of periodontal tissue; it is characterized by pocket spaces created between loose teeth and gums [
3]. Most of the treatments for periodontitis are to physically remove bacterial manipulations and calculus through the use of surgical manual devices or ultrasonic devices, which are successful for most patients, but may not be appropriate for some patients. It can cause severe pain and poor prognosis [
4]. Another treatment option for periodontitis is pharmacological therapy based on host control, such as oral microbes or bacteria, using probiotics and antibacterial agents [
5,
6]. However, although these pharmacological therapies have recently attracted a lot of attention, they have the disadvantage of being ineffective for patients and requiring long-term use. Therefore, studies are being actively conducted to obtain alternatives from safe natural products that are relatively free from these problems [
7,
8].
Among the enzymes involved in the biosynthesis and decomposition of glucocorticoids, 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1) is known to be involved in metabolic diseases including obesity and diabetes and is known to play a role in catalyzing the conversion to active glucocorticoids [
9,
10]. Recent studies have reported clinical results showing that 11β-HSD1 is increased in patients with periodontitis, and in fact, serum and salivary cortisol levels are known to be correlated with chronic periodontitis [
11]. In particular, some patients with periodontitis have metabolic diseases such as diabetes, and it has been reported that diabetic patients have specific polymorphisms or mutations in genes encoding interleukin, which may make inflammatory diseases more severe [
12]. In addition, 11β-HSD1 was known to contribute to the induction of inflammatory gene expression or activation of immunosuppressive genes through binding to the glucocorticoid receptor (GR) and the activation and increase in target receptors such as Angptl4 [
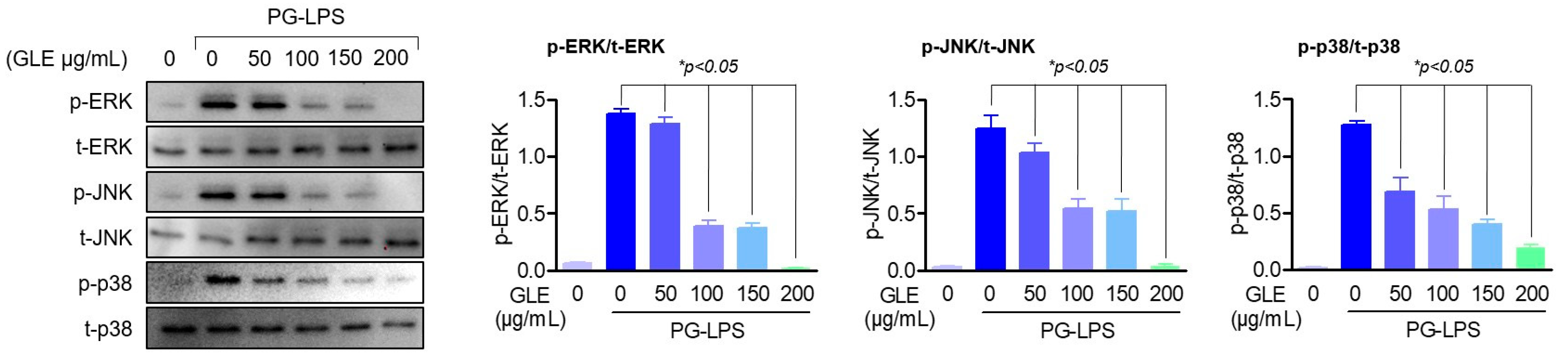
13]; but recent research results have shown that the conversion of cortisol by the increase of 1β-HSD1 is known to amplify inflammatory and oxidative stress responses by activating the NF-κB and MAPK signaling pathways, and conversely, mitogen-activated protein kinase (MAPK) pathways such as p38 and ERK are also known to affect inflammation and cell damage through 11β-HSD1 gene activation [
14]. Given these points, 11β-HSD1 is currently considered a useful therapeutic target not only in metabolic diseases but also in chronic inflammatory diseases, and in research reports on periodontitis; the expression of 11β-HSD1 was detected in oral fibroblasts and keratinocytes, but its role in periodontitis and periodontal ligament is still unclear [
15,
16].
Periodontal ligament (PDL), which is composed of fibroblasts, osteoblasts, and cementoblasts is located between the cementum and the alveolar bone and supports the periodontal connective tissue; it is known to be a very important fibroblast, playing the role of periodontal ligament cells that form the periodontal ligament [
17,
18,
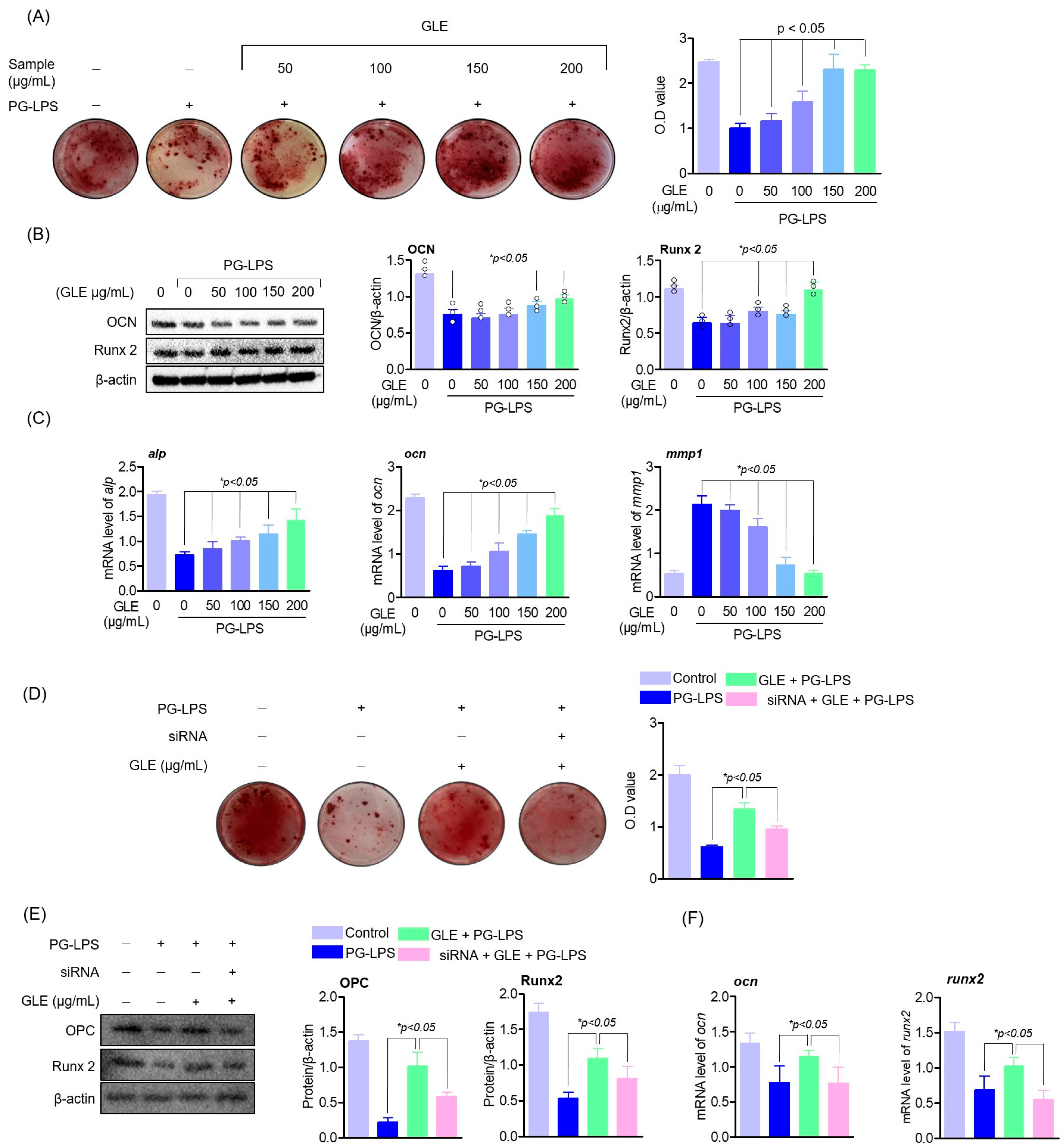
19]. Several previous studies have shown that promoting cell proliferation and osteogenic differentiation of human periodontal ligament cells (HPDLC) to regenerate periodontal tissue has a very positive effect on the treatment of periodontal disease; it has been reported to play a very important role in regulating the length of the periodontal crest or the amount of lost alveolar bone [
20]. Therefore, the recovery of periodontal ligament cells damaged by periodontitis is very important for periodontal health.
Glehnia littoralis Fr. Schmidt ex Miq. is a deep-rooted perennial plant that grows mainly in temperate coastal sand dunes and is classified as a halophyte; it is a medicinal plant that has been cultivated in China for a long time, although it grows in areas with high salt concentrations in soil where general land plants cannot grow [
21,
22].
G. littoralis is called “Beishashen” in traditional Chinese medicine and has traditionally been known to have antibacterial and anti-inflammatory effects, but its direct role in periodontitis has not been known [
23]. The main components of
G. littoralis contain various coumarins and alkaloids, and in addition, they have been studied to have various physiological activities such as antifungal, anti-obesity, and antioxidant effects [
24,
25,
26]. In addition, the lotus leaf of
G. littoralis is mainly used as raw vegetables or herbs such as leafy vegetables and mountain vegetables, and the leaves and stems are used for manufacturing processed products such as makgeolli, rice cakes, tea, and extracts, and it is a familiar food material listed in the Food Codex of South Korea. However, despite the physiological activity and commercial efficiency of
G. littoralis, research is still incomplete. Therefore, in this study, we propose the possibility of
G. littoralis as a periodontitis regulator by evaluating the effect of
G. littoralis extract (GLE) on periodontitis in human periodontal ligament cells stimulated with
Porphyromonas gingivalis-lipopolysaccharides (PG-LPS) and periodontitis-induced in vivo experimental animal model.
3. Discussion
Although various surgical and drug therapies have been implemented to control periodontal disease, they have not been a fundamental treatment strategy for periodontal disease. In addition, many drugs such as antibiotics and non-steroidal anti-inflammatory drugs are currently being used to treat periodontal disease, but side effects occur with long-term use, and they are not new treatment strategies [
27]. Therefore, for fundamental treatment, research is being actively conducted to discover new targets that can control periodontal disease and to explore natural materials that are relatively safe from side effects [
28]. Previously, it was known that chronic periodontitis triggers a hypothalamic–pituitary–adrenal (HPA) axis response, resulting in increased levels of corticosterone (the major glucocorticoid in rodents), especially glucocorticoid hormones such as 11β-HSD1 [
29,
30]. However, more research has been conducted on metabolic diseases including visceral fat obesity, dyslipidemia, insulin resistance, and hypertension rather than periodontitis. It has been reported that glucocorticoids play a role in experimental periodontitis, but their role in the periodontal inflammation situation and periodontal tissue has not yet been elucidated [
31]. Therefore, in this study, we aimed to evaluate the effect of 11β-HSD1 regulation of GLE in periodontal inflammation.
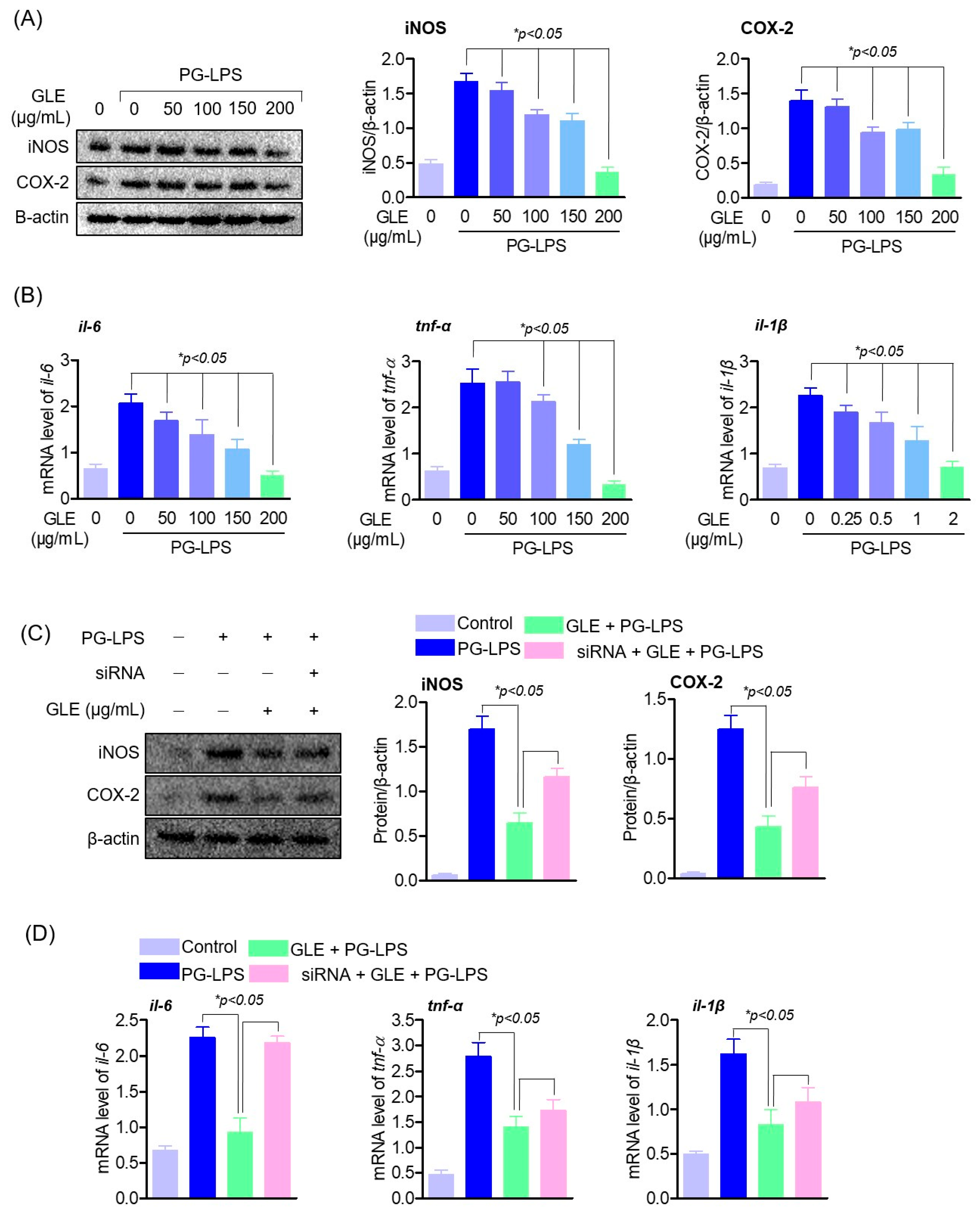
Periodontitis is a disease caused by inflammation in the periodontium, a tissue that mainly supports teeth. In the process of periodontitis, chemokines such as CXCL-8, CXCL-1, and CCL-5 and cytokines such as TNF-α, IL-1β, and IL-6 are present, due to the action of these multi-factors, it progresses to severe periodontitis with loss of periodontal support structures [
32,
33]. In the inflammatory process of periodontitis, the activation of 11β-HSD1 enhances cytokines, oxidative stress, apoptosis, and mitochondrial damage through the NF-κB and MAPK signaling pathways, and further aggravates the lesions of periodontitis [
34]. In particular, treatment with U0126, an ERK inhibitor, has been reported to directly down-regulate 11β-HSD1 protein expression in airway smooth muscle cells [
35]; the results of this study suggest that GLE induces a decrease in 11β-HSD1 by inhibiting the phosphorylation of MAPK proteins. Also, it has been known that the regulation of these inflammatory cytokines plays an important role in periodontal inflammation as well as alveolar bone formation. Among them, cytokines such as TNF-α, IL-1β, and IL-6 are highly expressed in patients with periodontitis and promote bone resorption by directly or indirectly supporting not only inflammation but also soft tissue degradation, lymphocyte promotion, and osteoclast formation [
36]. In particular, it was found that IL-6 is present in the apical end and produced by a subpopulation of neutrophils residing in the apical lesion and can directly induce osteoclast formation and bone resorption in the periodontal tissue [
37,
38]. Thus, this study showed that GLE effectively down-regulated the expression of pro-inflammatory cytokines and mediators, including IL-6, in experimental periodontitis-inducing models both in vitro and in vivo, and that this anti-inflammatory effect of GLE restored the lost osteoblastic differentiation capacity of HPDL cells. These results suggest the possibility that GLE may not only regulate 11β-HSD1 but also the activity of upstream signal regulators MAPK, and ultimately appears to have an effect of protecting periodontal tissue by regulating periodontal inflammation.
Generally, the production of ROS stimulates cell proliferation and differentiation in various cell lines, but excessive ROS production overwhelms the antioxidant defense system, causing cell damage and periodontal destruction due to oxidative stress [
39]. The production of these ROS is directly or indirectly related to tooth soft tissue destruction and bone resorption and is known to be related to increased expression of pro-inflammatory cytokines. Therefore, studies on the protective effects of periodontitis and alveolar bone destruction through the regulation of nuclear factor erythroid-2-related factor 2 (Nrf2) have recently been reported [
40,
41]. In addition, recent studies have shown that the inhibition of oxidative stress due to the knockout of 11β-HSD1 can have a therapeutic effect on myocardial dysfunction caused by sepsis; 11β-HSD1 is recognized as a regulator of various diseases as a target for controlling not only inflammation but also oxidative stress [
42]. The results of this study showed that the regulatory effect of GLE’s ROS regulation and antioxidant enzyme proteins (SOD, CAT) was reversed by silencing 11β-HSD1, this suggests that GLE exerts an antioxidant effect through regulating 11β-HSD1 from oxidative stress generated by periodontitis. Additionally, these results demonstrate an unknown relationship between 11β-HSD1 and oxidative stress in the periodontitis process.
Thus, in this study, GLE suppressed the expression level of pro-inflammatory cy-tokines in PG-LPS-induced human periodontal ligament cells and periodontitis-induced experimental animal models, and induced osteoblast differentiation in human periodontal ligament cells as well as specific osteoblast differentiation. The expression levels of markers ALP and OPN were increased. In addition, it restored the amount of alveolar bone along with the lost periodontal tissue in two experimental animal models inducing periodontitis and showed a therapeutic effect on inflammatory infiltration. These results suggest that GLE may play an important role in the recovery of tissue damage caused by early periodontitis by restoring alveolar bone loss, which can be found in chronic severe periodontitis, and that it will act together with the result of suppressing inflammatory infiltration due to the accumulation of inflammatory cells. Also, in the context of periodontal inflammatory lesions, GLE has been shown to act as an effective regulator of ROS production. These results suggest that GLE protects lost periodontal tissue by suppressing periodontal inflammation, suggesting a novel biological activity of GLE that has not been previously reported.
Although in this study, the in vitro study results suggest that the periodontitis regulating effect of GLE is due to 11β-HSD1, its direct effect in in vivo animal models should be further elucidated. In addition, 11β-HSD1 has been known as a major pathological target of inflammatory responses and metabolic diseases such as diabetes and obesity, and, in particular, diabetes and periodontitis are known to show a close interaction [
12]. Therefore, the relationship of 11β-HSD1 between these diseases or the role of 11β-HSD1 in diabetic periodontitis still needs to be further studied.
4. Materials and Methods
4.1. Sample Preparation and Extraction
Dried G. littoralis F. Schmidit ex Miq leaves were harvested from Yeongdeok-gun, Gyeongsangbuk-do, Republic of Korea and extracted twice with 20-fold volume of 70% ethanol at 85 ± 5 °C for 6 h. The extract was passed through a 50-mesh filter and evaporated to obtain an extract containing about 20% solid contents (or 20% brix) in vacuo at 60 °C. The product was spray-dried with dextrin and yielded 30 ± 5%. The dried powder was used in experiments.
4.2. Component Analysis of GLE
GLE powder was dissolved at a concentration of 10 mg/mL in 70% Ethanol and filtrated through a 0.2 um polyvinylidene fluoride (PVDF) membrane filter. Standard of chlorogenic acid (C3878; Sigma-Aldrich, St. Louis, MO, USA) and rutin (PHL89270; Phytolab, Vestenbergsgreuth, Middle Franconia, Germany) was dissolved in 70% ethanol and used for analysis. The GLE was analyzed using high-performance liquid chromatography (Agilent Technologies 1260 Infinity II system, Santa Clara, CA, USA), and a Inertsil® ODS-3 column (4.6 × 250 mm, 5 µm, GL Sciences, Torrance, CA, USA) was used. The temperature was maintained at 30 °C, with an injection volume of 10 uL at a flow rate of 1.0 mL/min. The mobile phase was composed of acetonitrile (A) and water containing 0.1% trifluoroacetic acid (B). The gradient elution conditions were as follows: initial 0–10 min A:B (14:86, v/v), 12 min A:B (20:80), 22 min A:B (30:70), 25 min A:B (100:0), 30 min A:B (100:0), and 35 min A:B (14:86). The detection wavelength was 326 nm.
4.3. Reagents and Antibodies
For cell culture, Minimum Essential Medium-Alpha (α-MEM), fetal bovine serum (FBS), and trypsin-ethylene diamine tetra acetic acid (EDTA) culture reagents were purchased from Wellgene (Gyeongsan-si, Republic of Korea). In addition, 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide (MTT) for cell viability analysis was purchased from Amresco Inc (Cleveland, OH, USA), and for anti-inflammatory effect evaluation, TNF-α, IL-6, and IL-1β ELISA (enzyme-linked immunosorbent assay) kits were purchased from R&D system (Minneapolis, MN, USA) and analyzed according to the manufacturer’s instructions. PG-LPS used as a stimulus was purchased from InvivoGen (San Diego, CA, USA). For protein analysis, mouse monoclonal antibodies iNOS, COX-2, 11β-HSD1, GR, ANGPTL4, OCN, RUNX2, SOD, and CAT secondary antibodies were purchased from Santa Cruz Biotechnology Inc. (Dallas, TX, USA). Hybond ECL Nitrocellular membrane (Amersham Pharmacia Biotech Inc., Piscataway, NJ, USA) was purchased. Alizarin Red S, ascorbic acid, dexamethasone, and β-glycerophosphate were purchased from Sigma-Aldrich (St. Louis, MO, USA). 11β-HSD1 siRNA plasmid was purchased from Santa Cruz Biotechnology Inc. (Dallas, TX, USA). Opti-MEM and Lipofectamine RNAiMAX reagents were purchased from Invitrogen (Carlsbad, CA, USA).
4.4. Cell Culture
Human periodontal ligament cells (HPDL) were provided and used by Kyungpook National University (Daegu, Republic of Korea) and were reviewed and approved by the Kyungpook National University Institutional Review Board (KNU 2017-78). Isolation of cells was obtained from donor third molars as previously reported [
16]. Briefly, HPDL cell culture was performed as follows: PDL was carefully separated from the root surface, and then cultured with 3 mg/mL type 1 collagenase (Worthington Biochem, Freehold, NJ, USA) and 4 mg/mL dispase (Roche, Mannheim, Germany) solution for 1 h. PDL samples from different individuals were pooled, and single-cell suspensions were obtained by passing the cells through a 70 μm strainer (Falcon, BD Labware, Franklin Lakes, NJ, USA), and then the single-cell suspensions (1 × 10
4 cells) were seeded onto 10 cm culture plates. After that, the culture medium was cultured in α-MEM supplemented with 10% (
v/v) fetal bovine serum (FBS) and 1% penicillin/streptomycin (Gibco BRL, Grand Island, NY, USA), and the cells were maintained at 37 °C in a humidified atmosphere of 5% CO
2.
4.5. Cell Viability and Confluency
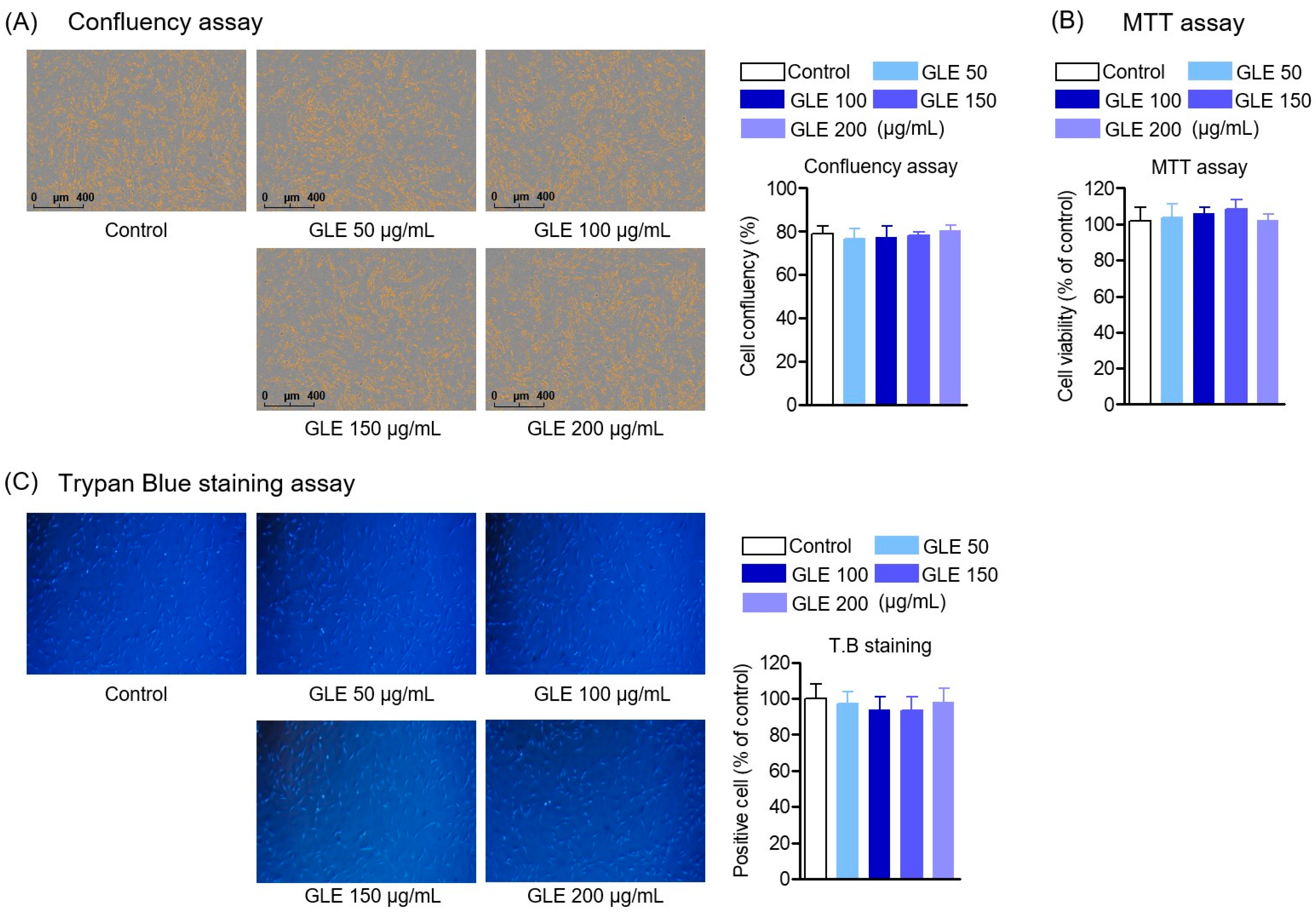
To investigate cell viability, HPDL cells were seeded in a 96-well plate at a density of 5 × 102 cells/well, and cultured for 24 h, followed by treatment with indicated concentrations of GLE in 7 days. After that, cell counting was performed using the Incucyte® Live-Cell Analysis System (Göttingen, Germany). For cell counting, cell counts in the stained control group were converted to percentages and averaged for the GLE treatment group. In addition, cells were cultured in the same way and treated with GLE, and 5 mg/mL MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide) was added to 50 μL of cell suspension and cultured for 4 h. The medium was removed and 100 μL dimethyl sulfoxide (DMSO) was added. The absorbance of the dissolved formazan crystals was measured at 540 nm with a microplate reader (Tecan Trading AG) (Männedorf, Switzerland). Similarly, trypan blue (Sigma-Aldrich, St. Louis, MO, USA) staining was performed to count the number of positive cells, the number of cells in the control group was converted to a percentage, and the number in the GLE treatment group was averaged.
4.6. ROS Production
Reactive oxygen species (ROS) was measured using H2DCFDA (Sigma-Aldrich, St. Louis, MO, USA) and after treatment with GLE at the indicated concentrations. To determine intracellular ROS levels, HPDL cells were cultured in 6-well plates for 24 h, then treated with all concentrations of GLE (50, 100, 150, and 200 μg/mL), and then treated with 10 μg/mL PG-LPS and incubated for 2 h under humidified conditions (37 °C, 5% CO2). After incubation, medium containing 10 μM DCF-DA was treated to each well and incubated under humidified conditions (37 °C, 5% CO2). Cells were then rinsed twice with PBS and measured using the Incucyte® Live-Cell Analysis System (Sartorius, Göttingen, Germany).
4.7. Alizarin Red S Staining (Mineralization Assay)
After seeding HPDL cells at 1 × 10
4 cells/well in a 6-well plate, osteogenesis was induced for 14 days in an osteoblast differentiation medium (containing 50 µg/mL ascorbic acid, 0.1 µM dexamethasone, and 10 mM β-glycerophosphate). Afterwards, the cells were fixed with 4% polyformaldehyde for 30 min and stained with 0.1% Alizarin Red S at room temperature for 1 h to stain the calcified nodules, and then observed under an optical microscope (Nikon, Tokyo, Japan) to photograph the calcified nodules. To quantify the amount of calcium accumulation in the calcified nodules, the calcium was dissolved in 10% cetylpyridinium chloride (CPC) and the absorbance was measured at 560 nm using a multifunctional microplate reader (M1000 Pro, TECAN, Männedorf, Switzerland), referring to the method of a previously reported study [
43].
4.8. Fluorescein Isothiocyanate (FITC) Staining
To confirm knockout of 11-βHSD1 in HPDL cells, fluorescein isothiocyanate (FITC) analysis was performed after transfection. After transfection, cells were fixed by treating with 4% formaldehyde solution for 30 min and then cultured overnight at 4 °C using 11β-HSD1 antibody (Santa Cruz Biotechnology Inc., cat. no. sc-518168, Dallas, TX, USA). The following staining was performed. Afterwards, cells were photographed using the Incucyte® Live-Cell Analysis System.
4.9. Detection of mRNA Levels by Real-Time Quantitative PCR
Total RNA was extracted using TRIzol/chloroform reagent (Bioneer, Daejeon, Republic of Korea) according to the manufacturer’s instructions, and reverse transcription of RNA was performed using the RT PreMix kit (Enzynomics, Daejeon, Republic of Korea). cDNA was then amplified with SYBR Premix Ex Taq (Takara, Tokyo, Japan) and 40 cycles of 50 °C for 2 min, 95 °C for initial denaturation for 10 min, 95 °C for 15 s, and 60 °C for annealing for 30 s were performed. Each target mRNA analyzed was normalized to the cycle threshold (Ct) value of GAPDH as a housekeeping gene. Primers for each gene used in this study were provided by Bioneer Co., Ltd. (Daejeon, Republic of Korea), and the sequences of the primers are shown in
Table 2.
4.10. Western Blot Analysis
The expression levels of each target protein were analyzed using Western blot analysis, and the experimental method is as follows. HPDL cells were washed with PBS and lysed in RIPA lysis buffer (50 mM Tris pH 8.0, 150 mM NaCl, 0.02% sodium azide, 0.2% SDS, 1 mM PMFS, 10 μL/mL aprotinin, 1% igapel 630 (Sigma-Aldrich, St. Louis, MO, USA)), and protein samples were extracted and centrifuged at 23,000× g for 15 min. The protein supernatant was quantified using a protein assay kit (Bradford Reagent, Sigma-Aldrich, St. Louis, MO, USA). Equal amounts of protein (20 μg) were then separated by 12% SDS-PAGE and transferred to PVDF membranes. The membranes were washed with 5% (w/v) skim milk for 1 h at room temperature. After blocking, the membranes were rinsed with TBST buffer for 1 h and then incubated with primary antibodies overnight at 4 °C. The membranes incubated with primary antibodies were washed with Tris-buffered saline (TBS-T) buffer containing Tween 20 and then incubated with secondary antibodies. Protein bands were detected using Enhanced Chemiluminescence (ECL) Western blotting detection reagents (Thermo Fisher Scientific, Waltham, MA, USA). Band images were scanned with a ChemiDoc XRS+ system (Bio-Rad, Hercules, CA, USA). The detected bands were analyzed and quantified using Image J version 1.54i software (National Institutes of Health, Bethesda, MD, USA).
4.11. Serum Cytokines ELISA
Each inflammatory cytokine expression was analyzed according to the instructions of the kit manufacturer, R&D Systems (Minneapolis, MN, USA). After the experiment was completed, blood samples were collected from the veins of the mice. The samples were then centrifuged and centrifuged at a low temperature of 4 °C. Serum was separated. The separated blood crystals were sealed and stored at −20 °C until analysis. Serum collected from mice in each group was used in an enzyme-linked immunosorbent assay (ELISA) kit (according to the manufacturer’s instructions). The analysis was performed using a 96-well plate (R&D system, NE, Minneapolis, MN, USA) as follows: For each cytokine analysis, each antibody was incubated in a 96-well plate, and then each well was washed with washing buffer. After that, the diluent was added and incubated at room temperature for 1 h, then the sample was treated and incubated at room temperature for 3 h. After that, a second wash was performed and treated with Streptavi-din-HRP B solution and incubated for 30 min. Afterwards, each substrate solution was placed in a well plate, incubated at room temperature, protected from light, and analyzed at 540 nm wavelength using a plate reader (Tecan Trading AG) (Männedorf, Switzerland).
4.12. Transfection with siRNA
HPDL cells were transfected with Opti-MEM and Lipofectamine RNAiMAX reagent (Invitrogen, Carlsbad, CA, USA) were mixed and transfected with 50 nM 11β-HSD1 siRNA (Santa Cruz Biotechnology Inc., cat. no. sc-41377, Dallas, TX, USA), and the cells were transfected for 48 h, after that, they were used in the experiment.
4.13. Animals
All animal experimental procedures were approved by the Animal Care Committee of Chungnam National University (202308-CNU-334) and were performed in accordance with the “Guidelines for Animal Use” established by the Animal Care Committee of Chungnam National University (Daejeon, Republic of Korea). Briefly, the experimental animals and their housing environment were described. Eight-week-old male Spra-gue-Dawley rats were purchased from Samtako Inc. (Osan, Republic of Korea) and were maintained in the Community Animal Laboratory of Chungnam National University (Daejeon, Republic of Korea). They were isolated and acclimated in stainless steel wire cages (260 W × 350 D × 210 H mm) for 1 week before the experiment, and the housing environment was maintained at a temperature of 21.0–23.1 °C and a relative humidity of 30–50%. In addition, the lighting time was 12 h/day (7:00 a.m.–7:00 p.m.), and the illuminance was 150–300 lux. The feed used was solid feed for laboratory animals (Harlan Laborato-ries, Inc., Greenfield, IN, USA), and the solid feed was placed in a feeding trough so that the animals could consume it freely. The drinking water was filtered using a filter sterilizer and then irradiated with ultraviolet rays from Cheongju City tap water and supplied freely using an automatic watering device.
4.14. Ligature-Induced Periodontitis Inflammation Model
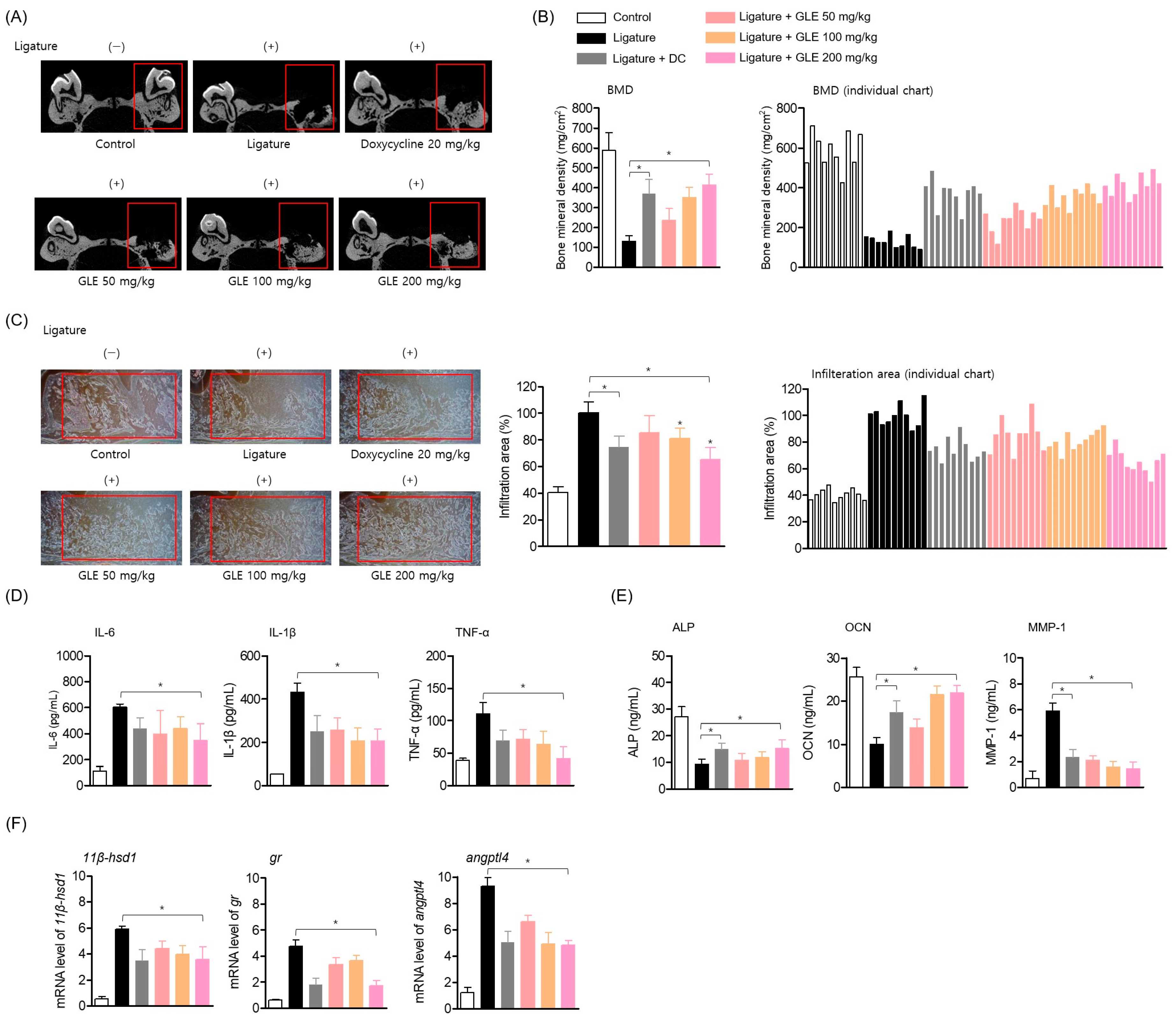
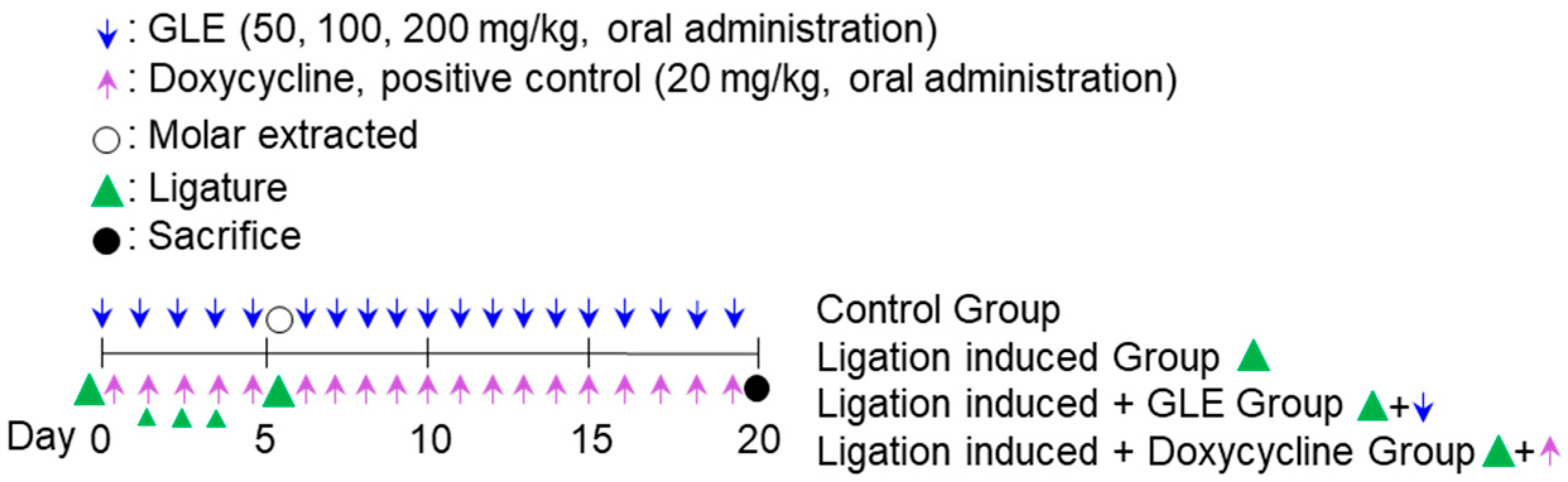
In the experimental model of ligature-induced periodontitis, GLE was administered orally. Eight-week-old male Sprague-Dawley rats (each group
n = 10, total of 60 rats, each group: control, ligation-induced group, ligation + doxycycline 20 mg/kg, ligation + oral administration of GLE 50 mg/kg, ligation + oral administration of GLE 100 mg/kg, ligation + oral administration of GLE 200 mg/kg) were anesthetized by CO
2 in-halation. Except for the control group, the maxillary right first molar was fixed and ligated with a non-absorbable silk knot for 5 days. The ligation site was checked daily to determine whether periodontitis was induced, and ad libitum food was administered. During the ligation period of 5 days, GLE and doxycycline, a positive control, were administered orally at the indicated concentrations to each group. On the 5th day after ligation, the silk knot was removed, the first molar was extracted, and hemostasis was performed on the periodontal pocket formation site. After that, GLE and doxycycline were administered orally to each group for 14 days, and the oral administration was continued for a total of 19 days including the ligation period. All experimental animals were sacrificed on the 20th day after the end of the experiment, and the maxillary cranial bones were extracted (
Scheme 1).
4.15. PG-LPS Induced Periodontal Inflammation
For periodontitis induction using PG-LPS, 8-week-old SD rats were divided into each group (each group
n = 10, total 60, each group: control group, PG-LPS-only induced group, PG-LPS + doxycycline 20 mg/kg, PG-LPS + GLE 50 mg/kg, PG-LPS + GLE 100 mg/kg, PG-LPS + GLE 200 mg/kg). After that, each group except the control group was anesthetized by CO
2 inhalation and PG-LPS was injected daily between the first and second molars for 5 days, and at the same time, GLE at the indicated dose and doxycycline as a positive control were orally administered for 5 days. After the completion of PG-LPS induction for 5 days, GLE at the indicated dose and doxycycline as a positive control were orally administered to each group for 14 days. Similarly, GLE and doxycycline were administered for a total of 19 days, and after the end of the experiment, the experimental animals were sacrificed, and the maxillary skull was extracted. Similarly, GLE and doxycycline were administered for a total of 19 days, and at the end of the experiment on the 20th day, the experimental animals were sacrificed, and the maxillary skulls were removed (
Scheme 2).
4.16. Micro-CT Imaging and Analysis
Micro-CT analysis was performed on sacrificed experimental animals, and the experimental methods and conditions are as follows. Bone mineral density analysis (pixel size 10 μM) was performed using tube current (160 μA), tube voltage (90 kVp), imaging time (180 s), pixel size (10 μM), and FOV (rectangular volume of alveolar bone (400 μm width × 500 μm thickness × 1400 μm height) next to the mesial and distal roots) from the accumulated maxillary skull. In addition, the raw data obtained from the coronal section by changing direction were loaded into CTAn, and the images were scanned to calculate the BMD based on the above threshold. In addition, the alveolar bone excluding the root of the cemento-enamel junction was set as the region of interest (ROI) using the alveolar bone interpolation method, and VGStudio MAX 1.2.1 software (Volume Graphics, GmbH, Heidelberg, Germany) was used for ROI analysis. Data were analyzed by mean ± SD and one-way analysis of variance in SPSS Statistics 19.0 software (Armonk, NY, USA).
4.17. Histological Staining
To perform hematoxylin-eosin (H&E) staining on periodontal tissues obtained from the extracted maxillary skull, the extracted periodontal tissues were fixed in 10% formalin, embedded in paraffin, cut into 5 μm sections, and mounted on slides. H&E staining was then performed on the fixed slides. Tissue infiltration was observed from the stained slides using a fluorescence Olympus IX microscope 71-F3 2PH (Tokyo, Japan), and the same region of interest was set at the periodontal extraction site of all samples and compared with the ligation-induced group. The area of white soft tissue was expressed as a percentage compared to the control group.
4.18. Measurements of CEJ-ABC
The area from the CEJ (cement-enamel junction) between the fixed maxillary second and third molars to the alveolar bone ridge (ABC) was set as the region of interest for analysis, and the linear areas measured were the buccal palate, palatine palate, mesio-buccal, and the mesial-palatal areas were selected as measurement areas. Then, the distance from the furthest root of the second and third molars to the top of the teeth was measured (in millimeters, measured only in the PG-LPS-induced model).
4.19. Statistics
Each experiment was independently repeated three times for reproducibility, and the results were expressed as the mean ± standard deviation (S.D.). Statistical analysis was performed using SPSS Statistics 19.0 software (Armonk, NY, USA), and differences between groups were analyzed using one-way analysis of variance (ANOVA) followed by Tukey’s test or Student’s t-test. Statistical significance was determined as p-value < 0.05.



 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}