
Selective Air Oxidation of Bis- and Trisphosphines Adsorbed on Activated Carbon Surfaces


Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
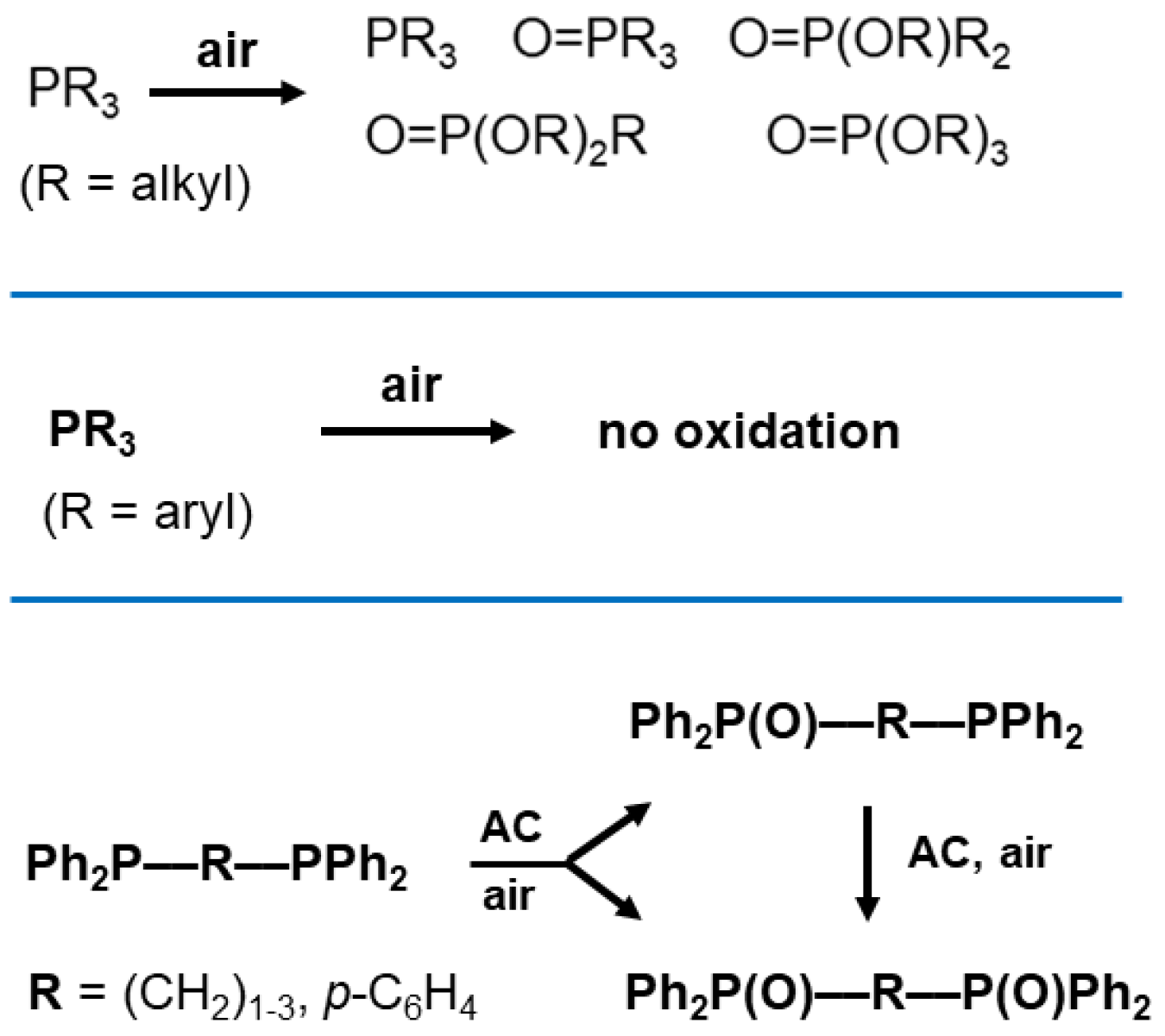
1. Introduction
2. Results and Discussion
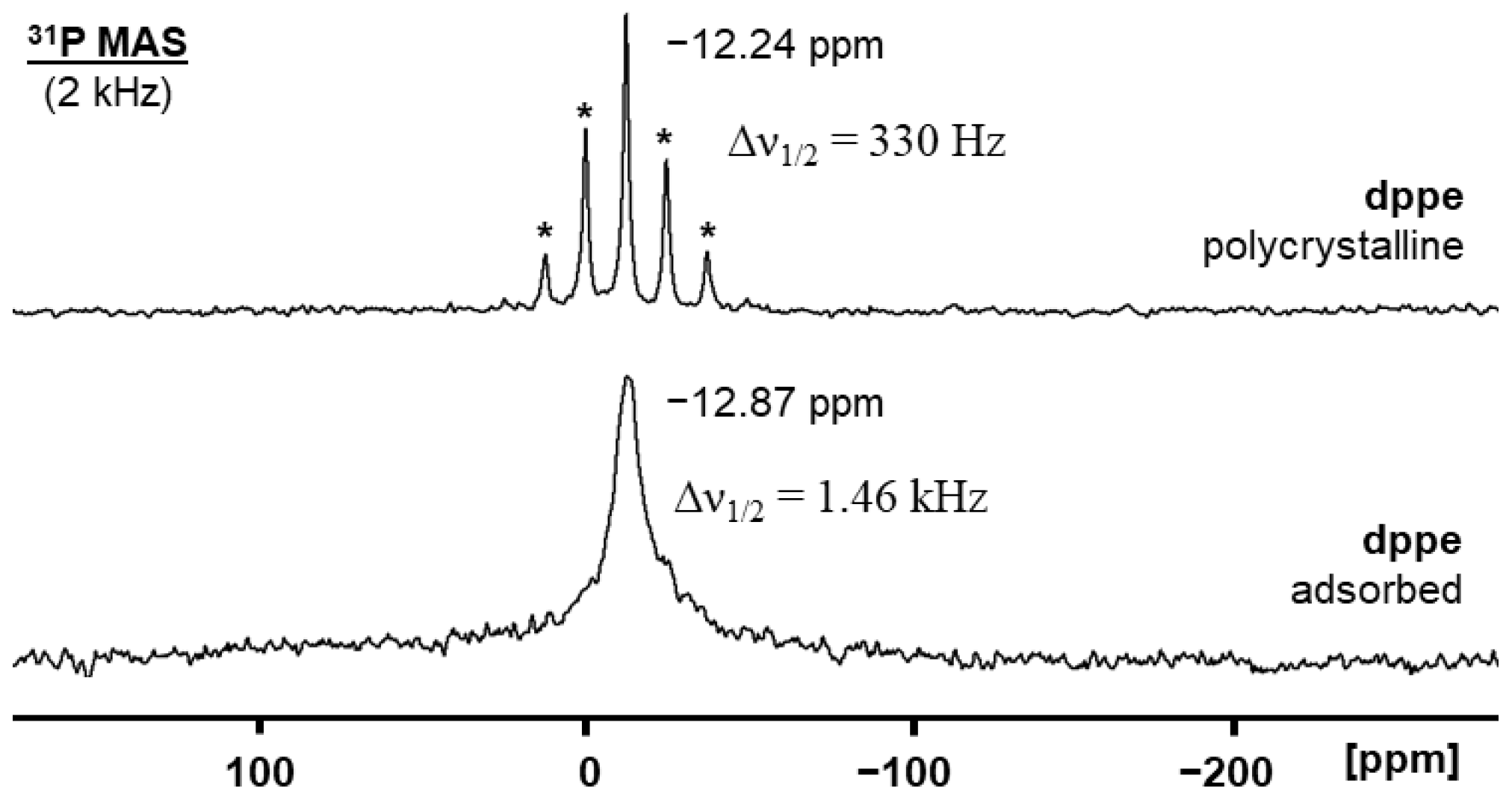
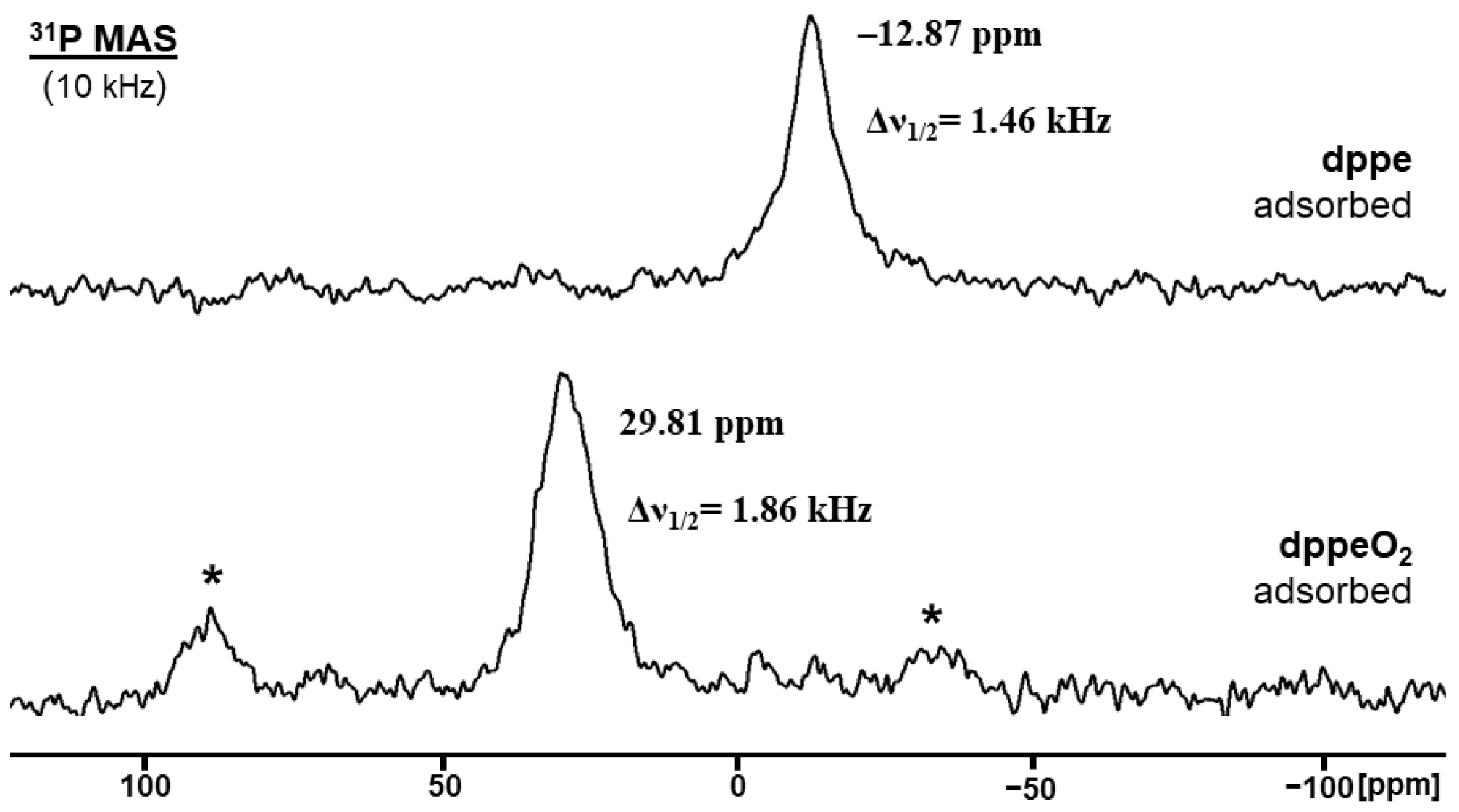
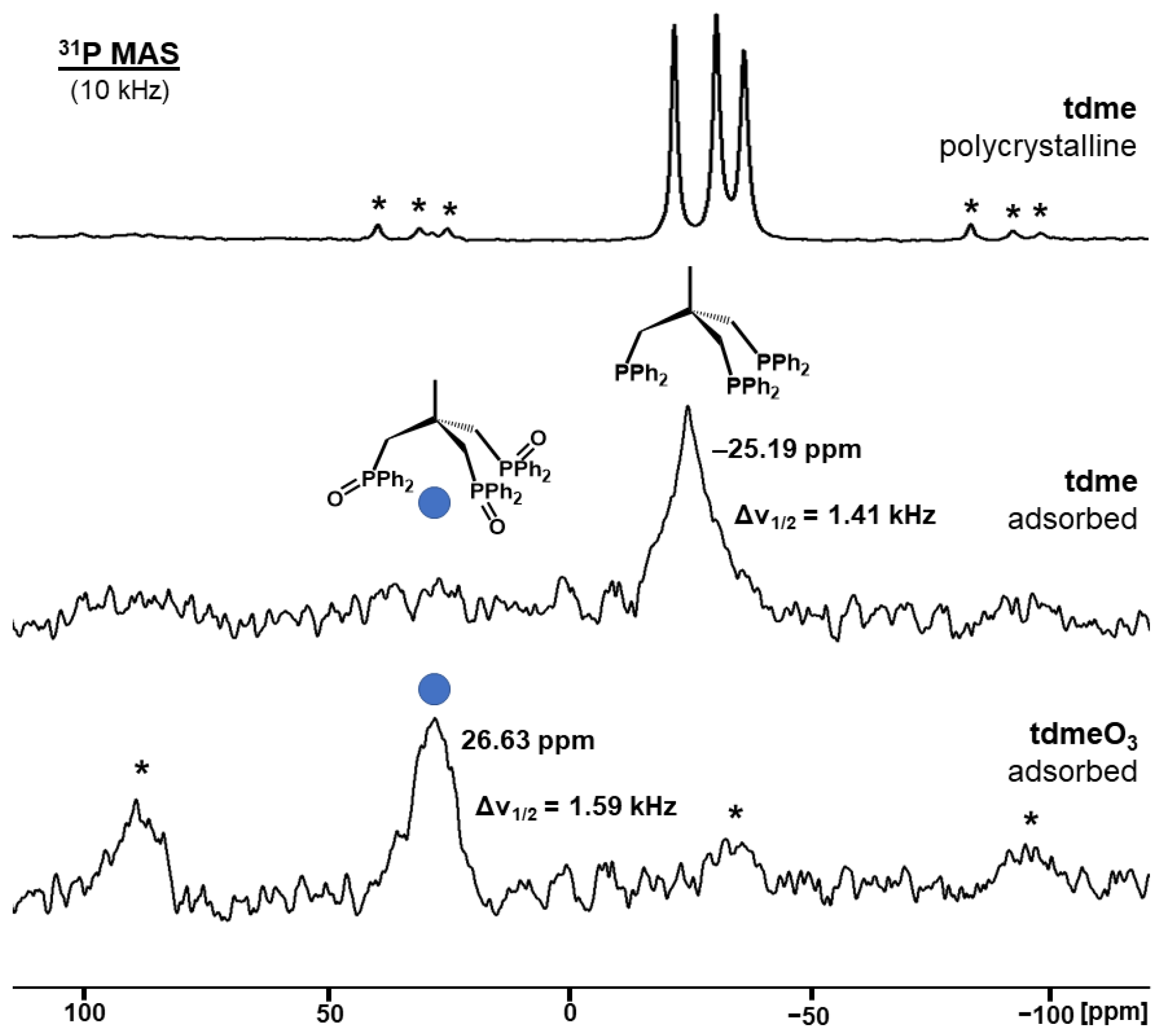
2.1. Solid-State NMR of Adsorbed and Oxidized Bis- and Trisphosphines on AC Surfaces
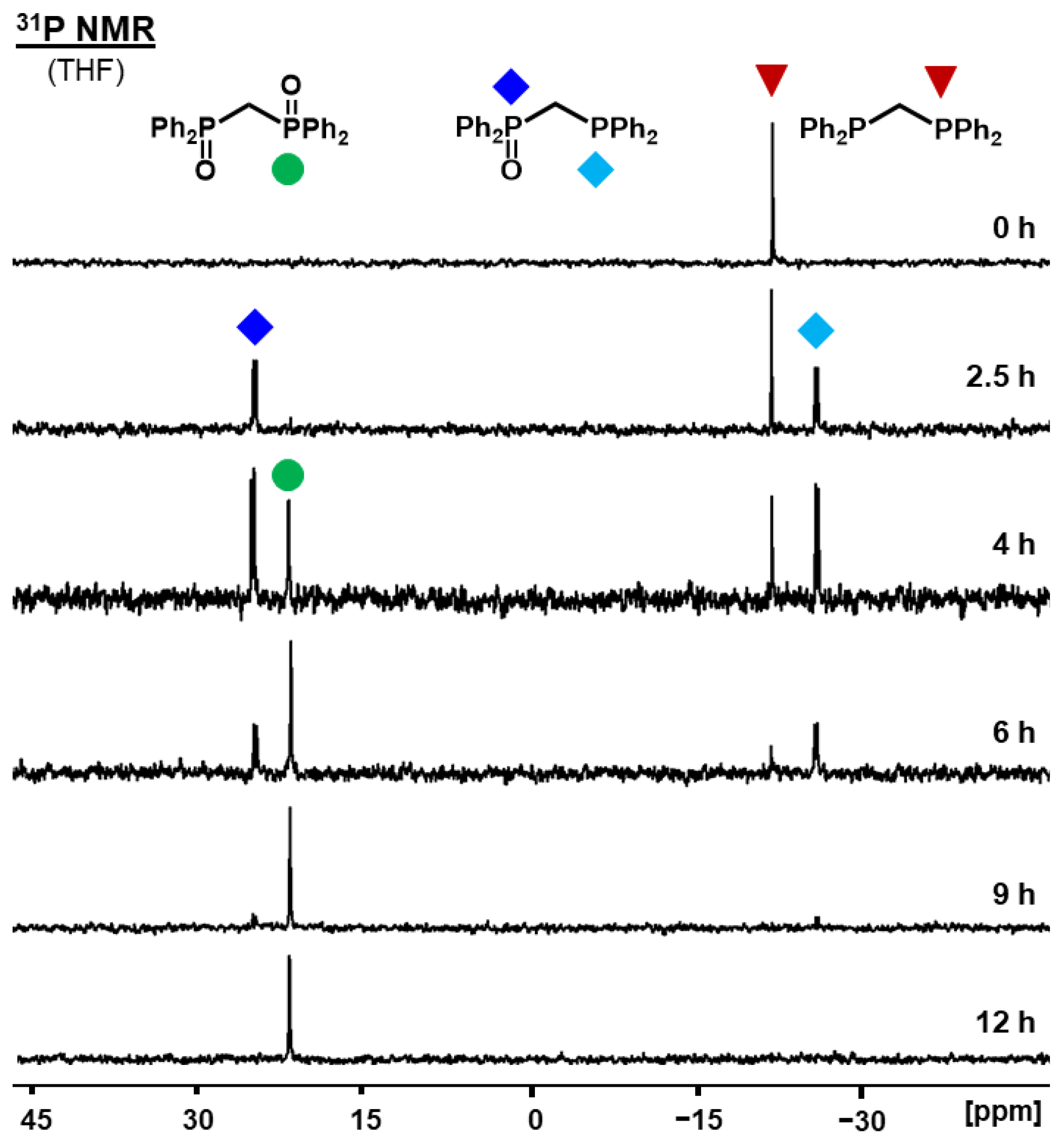
2.2. Monitoring the Oxidation of Phosphines on the AC Surface
2.2.1. Methodology for Monitoring the Reaction
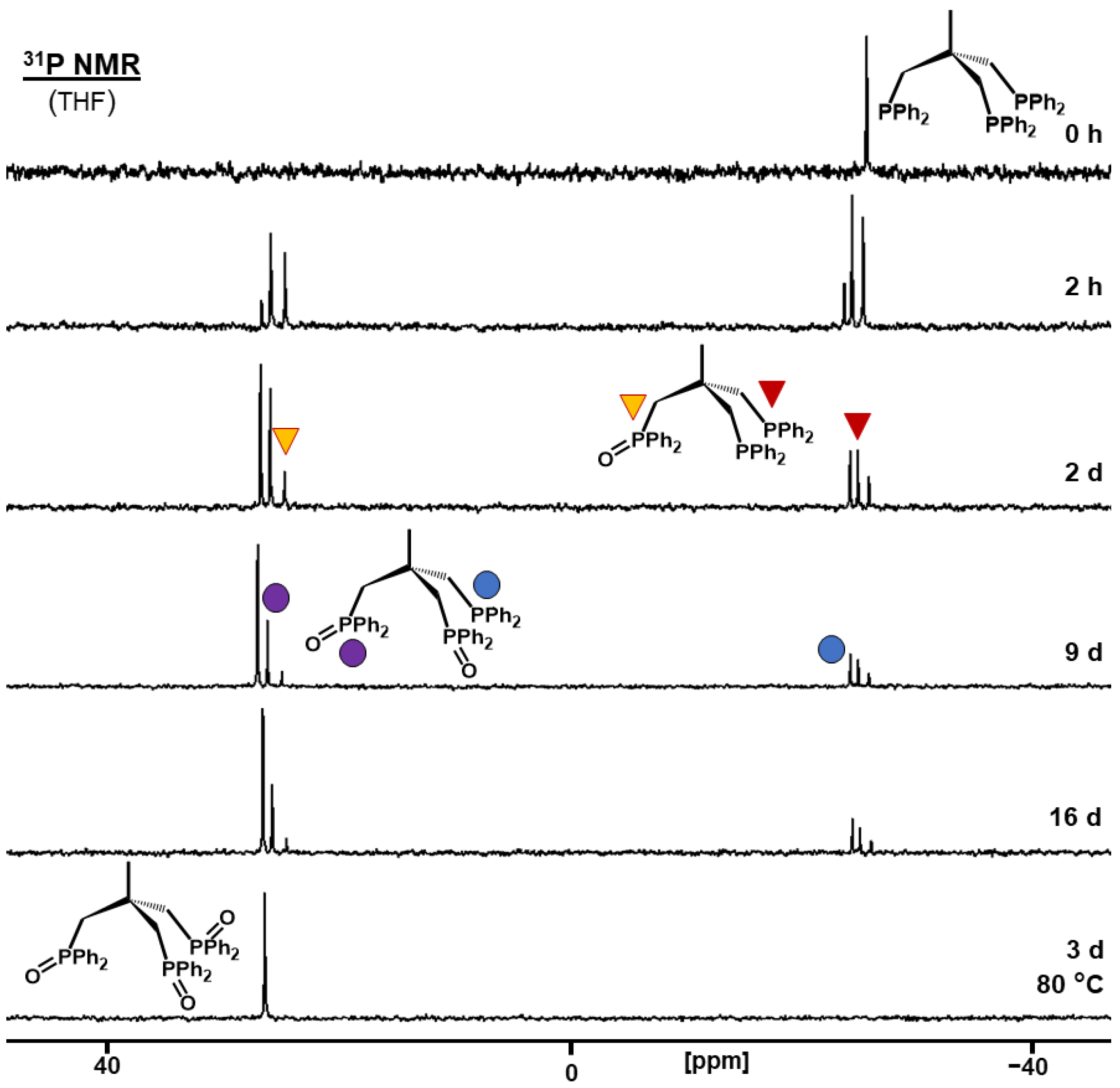
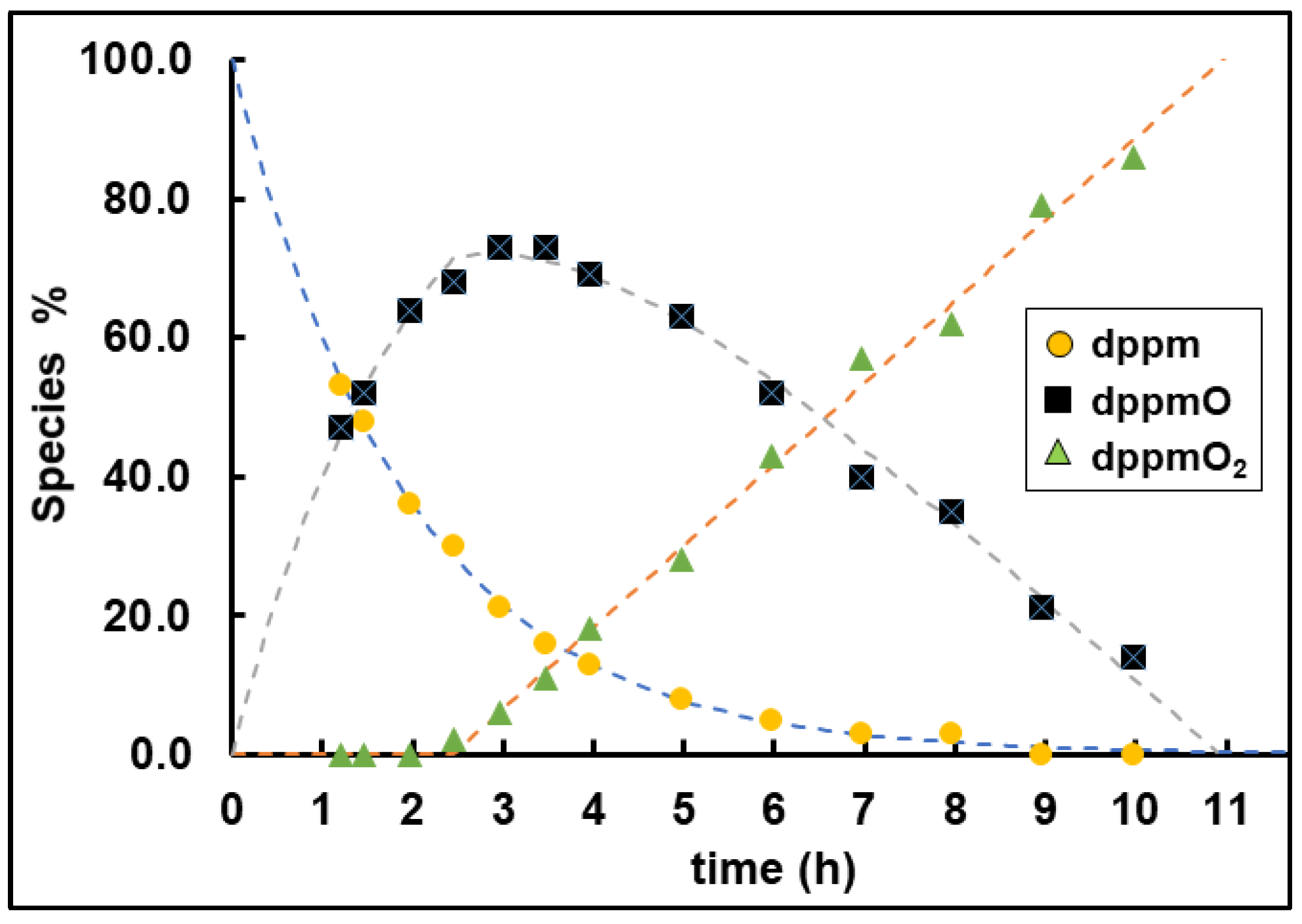
2.2.2. Oxidation of Bis- and Trisphosphines Adsorbed on AC
2.3. Factors Influencing Phosphine Oxidation
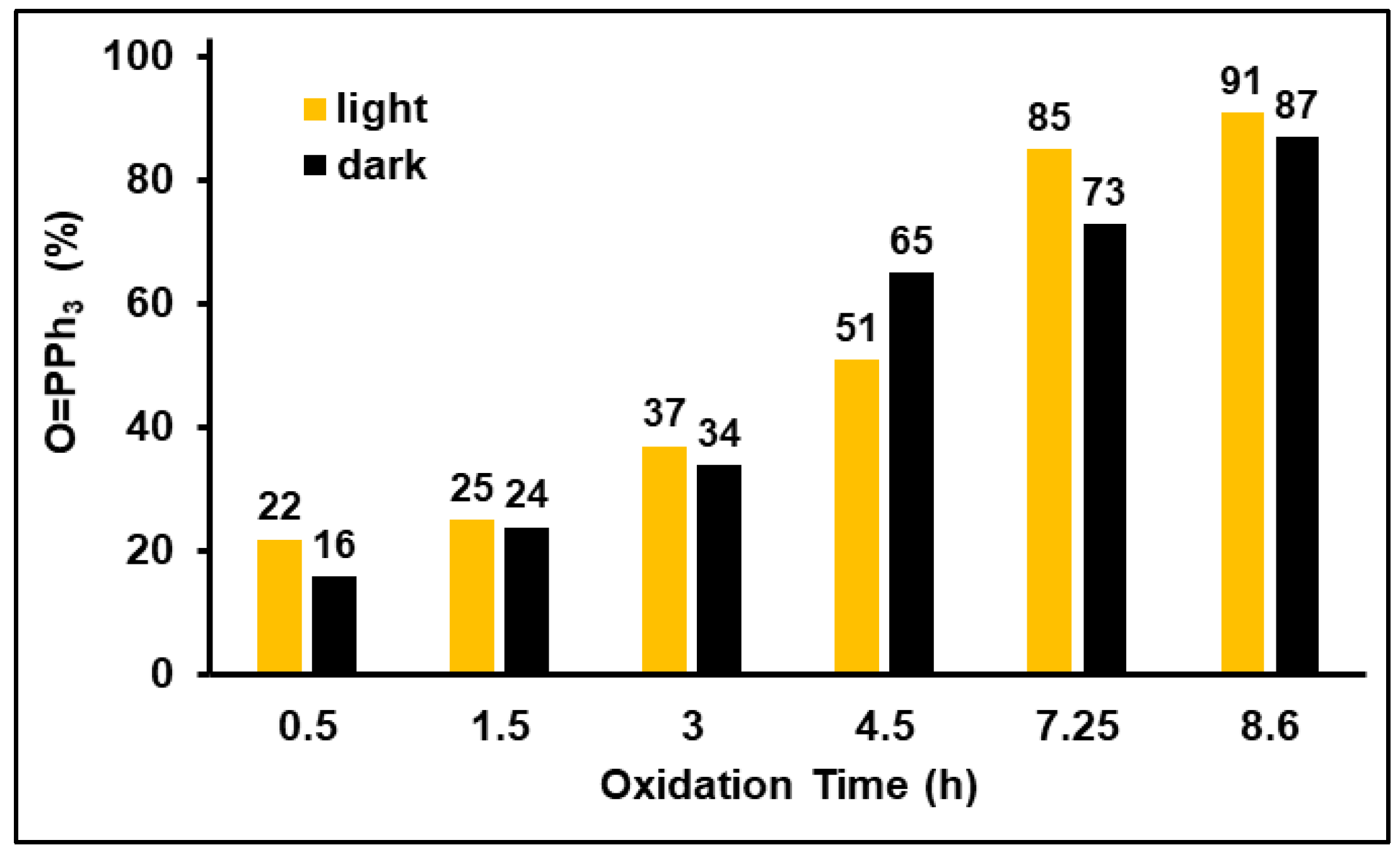
2.3.1. Effect of Light on Phosphine Oxidation
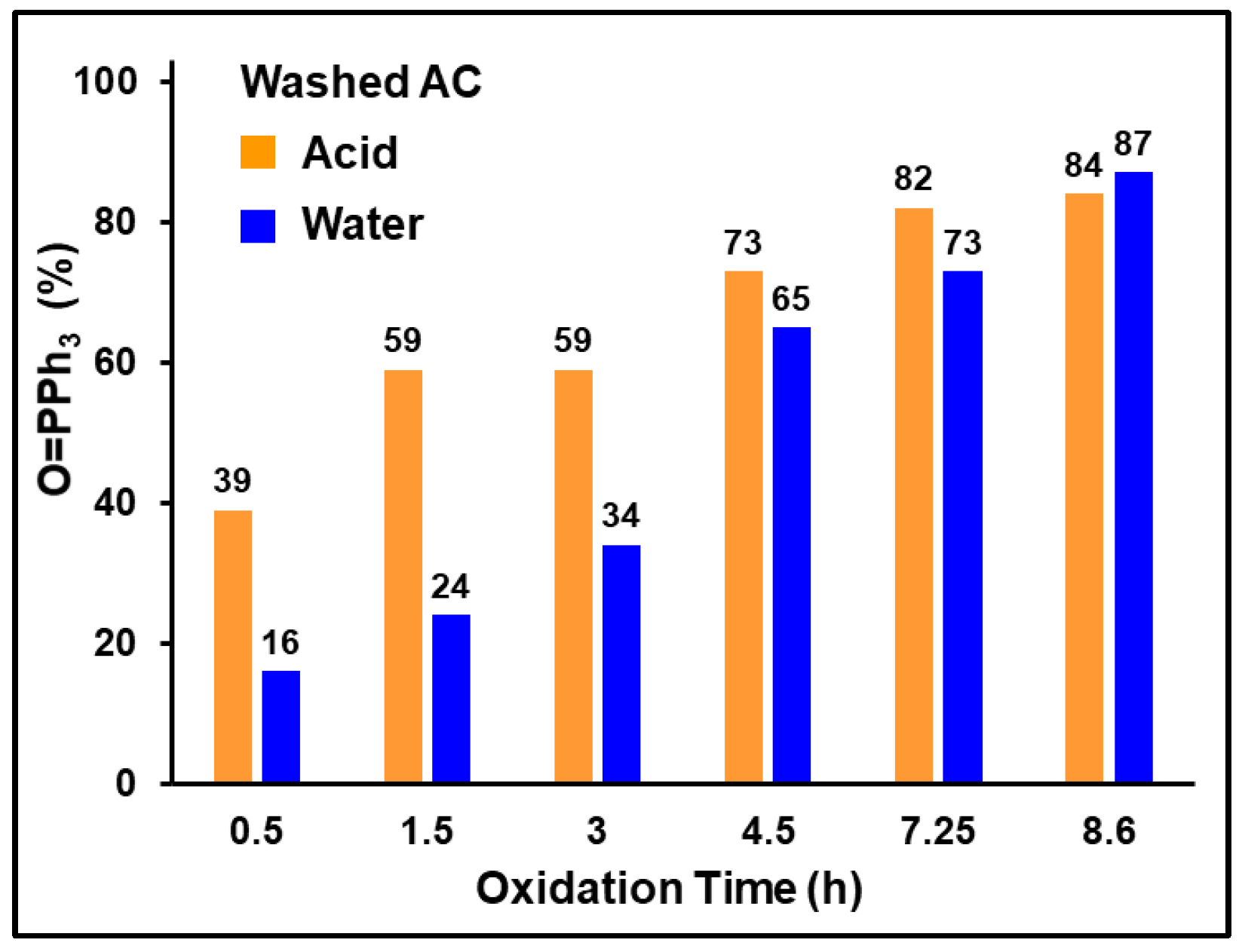
2.3.2. Effect of AC Acid Treatment on Phosphine Oxidation
2.3.3. Effect of Temperature on Phosphine Oxidation
2.3.4. Oxidation of PPh3 in the Presence of AIBN
2.4. Phosphine Oxide Recovery
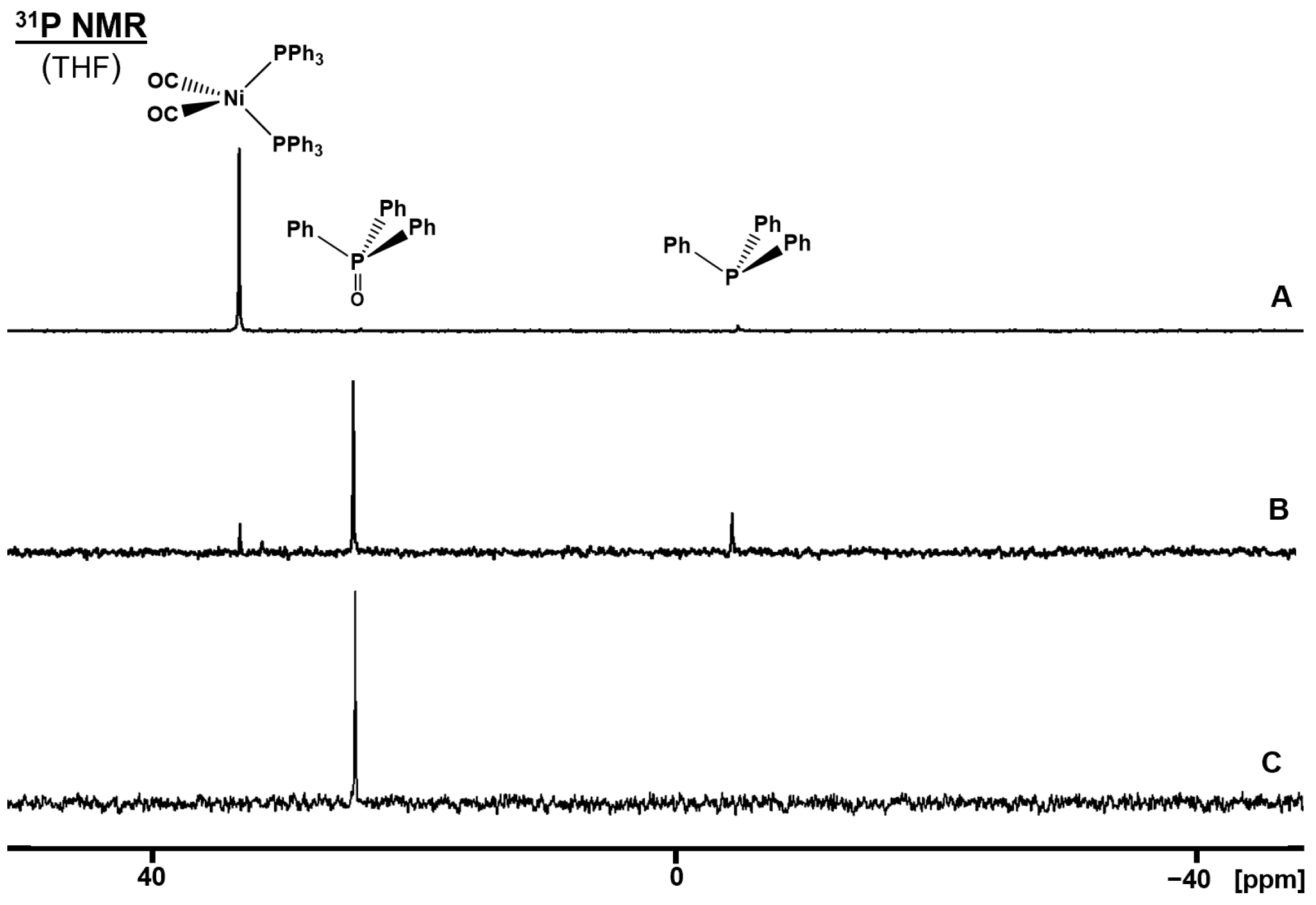
2.5. Oxidation of (CO)2Ni(PPh3)2 on the AC Surface
2.6. Mechanistic Considerations
3. Conclusions
4. Experimental Section
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pourhakkak, P.; Taghizadeh, A.; Taghizadeh, M.; Ghaedi, M.; Haghdoust, S. Fundamentals of Adsorption Technology. In Ad-sorption: Fundamental Processes and Applications; Interface Science and Technology; Ghaedi, M., Ed.; Academic Press: London, UK, 2021; Volume 33, pp. 1–70. [Google Scholar]
- Michałowska, A.; Jędrzejewski, K.; Kudelski, A. Influence of the Co-Adsorbed Ions on the Surface-Enhanced Raman Scattering Spectra of Dopamine Adsorbed on Nanostructured Silver. Materials 2022, 15, 5972. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Li, H.; He, Y.; Cheng, P.; Zhang, Y.-Q.; Feng, B.; Li, H.; Wu, K.; Chen, L. Condensation and asymmetric amplification of chirality in achiral molecules adsorbed on an achiral surface. Nat. Commun. 2023, 14, 2100. [Google Scholar] [CrossRef]
- Hoefler, J.C.; Yang, Y.; Blümel, J. Adsorption of solid phosphines on silica and implications for catalysts on oxide surfaces. New, J. Chem. 2023, 47, 21190–21198. [Google Scholar] [CrossRef]
- Wyrick, J.; Wang, X.; Namboodiri, P.; Kashid, R.V.; Fei, F.; Fox, J.; Silver, R. Enhanced Atomic Precision Fabrication by Adsorption of Phosphine into Engineered Dangling Bonds on H–Si Using STM and DFT. ACS Nano 2022, 16, 19114–19123. [Google Scholar] [CrossRef] [PubMed]
- Wilmsmeyer, A.R.; Gordon, W.O.; Davis, E.D.; Mantooth, B.A.; Lalain, T.A.; Morris, J.R. Multifunctional ultra-high vacuum apparatus for studies of the interactions of chemical warfare agents on complex surfaces. Rev. Sci. Instrum. 2014, 85, 014101. [Google Scholar] [CrossRef]
- Zhang, X.; Gao, B.; Creamer, A.E.; Cao, C.; Li, Y. Adsorption of VOCs onto engineered carbon materials: A review. J. Hazard. Mater. 2017, 338, 102–123. [Google Scholar] [CrossRef]
- Pui, W.K.; Yusoff, R.; Aroua, M.K. A review on activated carbon adsorption for volatile organic compounds (VOCs). Rev. Chem. Eng. 2018, 35, 649–668. [Google Scholar] [CrossRef]
- Chiang, Y.; Chiang, P.; Chang, E. Evaluations of the physicochemical characterizations of activated carbons. J. Environ. Sci. Health Part A 1998, 33, 1437–1463. [Google Scholar] [CrossRef]
- Harris, R.K.; Thompson, T.V.; Norman, P.R.; Pottage, C. Adsorption competition onto activated carbon, studied by magic-angle spinning NMR. J. Chem. Soc. Faraday Trans. 1996, 92, 2615–2618. [Google Scholar] [CrossRef]
- Wang, X.; Ning, P.; Shi, Y.; Jiang, M. Adsorption of low concentration phosphine in yellow phosphorus off-gas by impregnated activated carbon. J. Hazard. Mater. 2009, 171, 588–593. [Google Scholar] [CrossRef]
- Cluff, K.J.; Blümel, J. Adsorption of Ferrocene on Carbon Nanotubes, Graphene, and Activated Carbon. Organometallics 2016, 35, 3939–3948. [Google Scholar] [CrossRef]
- Zhang, E.; Wu, Y.-C.; Shao, H.; Klimavicius, V.; Zhang, H.; Taberna, P.-L.; Grothe, J.; Buntkowsky, G.; Xu, F.; Simon, P.; et al. Unraveling the Capacitive Charge Storage Mechanism of Nitrogen-Doped Porous Carbons by EQCM and ssNMR. J. Am. Chem. Soc. 2022, 144, 14217–14225. [Google Scholar] [CrossRef] [PubMed]
- Colabella, J.; Stall, R.; Sorenson, C. The adsorption and subsequent oxidation of AsH3 and PH3 on activated carbon. J. Cryst. Growth 1988, 92, 189–195. [Google Scholar] [CrossRef]
- Mikhalovsky, S.V.; Zaitsev, Y.P. Catalytic Properties of Activated Carbons, I. Gas-Phase Oxidation of Hydrogen Sulphide. Carbon 1997, 35, 1367–1374. [Google Scholar] [CrossRef]
- Aguilar, C.; García, R.; Soto-Garrido, G.; Arraigada, R. Catalytic oxidation of aqueous methyl and dimethylamines by activated carbon. Top. Catal. 2005, 33, 201–206. [Google Scholar] [CrossRef]
- Sousa, J.P.; Pereira, M.F.; Figueiredo, J.L. Modified activated carbon as catalyst for NO oxidation. Fuel Process. Technol. 2012, 106, 727–733. [Google Scholar] [CrossRef]
- Fang, Z.; Yu, X.; Tu, S. Catalytic oxidation of NO on activated carbons. Energy Procedia 2019, 158, 2366–2371. [Google Scholar] [CrossRef]
- Zhu, J.; Li, G.; Wang, Q.; Zhou, Y.; Wang, J. Engineering Surface Groups of Commercially Activated Carbon for Benzene Hydroxylation to Phenol with Dioxygen. Ind. Eng. Chem. Res. 2019, 58, 20226–20235. [Google Scholar] [CrossRef]
- Kawabata, H.; Hayashi, M. Benzylic oxygenation of alkylarenes with molecular oxygen in the presence of activated carbon. Tetrahedron Lett. 2004, 45, 5457–5459. [Google Scholar] [CrossRef]
- Hayashi, M. Oxidation using activated carbon and molecular oxygen system. Chem. Rec. 2008, 8, 252–267. [Google Scholar] [CrossRef]
- Hoefler, J.C.; Jackson, D.; Blümel, J. Surface-Assisted Selective Air Oxidation of Phosphines Adsorbed on Activated Carbon. Inorg. Chem. 2024, 63, 9275–9287. [Google Scholar] [CrossRef] [PubMed]
- Shenderovich, I.G.; Limbach, H.-H. Solid State NMR for Nonexperts: An Overview of Simple but General Practical Methods. Solids 2021, 2, 139–154. [Google Scholar] [CrossRef]
- Schmidt-Rohr, K.; Spiess, H. Multidimensional Solid-State NMR and Polymers; Academic Press Inc.: San Diego, CA, USA, 1994; ISBN 0-12-626630-1. [Google Scholar]
- Döller, S.; Gutmann, T.; Hoffmann, M.; Buntkowsky, G. A Case Study on the Influence of Hydrophilicity on the Signal Enhancement by Dynamic Nuclear Polarization. Solid-State NMR. 2022, 122, 101829. [Google Scholar] [CrossRef]
- Bonfante, S.; Lorber, C.; Lynam, J.M.; Simonneau, A.; Slattery, J.M. Metallomimetic C-F activation catalysis by simple phosphines. J. Am. Chem. Soc. 2024, 146, 2005–2014. [Google Scholar] [CrossRef]
- Etter, M.C.; Baures, P.W. Triphenylphosphine oxide as a crystallization aid. J. Am. Chem. Soc. 1988, 110, 639–640. [Google Scholar] [CrossRef]
- Chrzanowski, J.; Krasowska, D.; Drabowicz, J. Synthesis of optically active tertiary phosphine oxides: A historical overview and the latest advances. Heteroat. Chem. 2018, 29, e21476. [Google Scholar] [CrossRef]
- Fletcher, M.D. Organophosphorus Reagents; Murphy, P.J., Ed.; Oxford University Press: London, UK, USA, 2004; pp. 171–214. [Google Scholar]
- Adams, H.; Collins, R.C.; Jones, S.; Warner, C.J.A. Enantioselective Preparation of P-Chiral Phosphine Oxides. Org. Lett. 2011, 13, 6576–6579. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.S.; Goyal, N.; Herbage, M.A.; Sieber, J.D.; Qu, B.; Xu, Y.; Zhibin, L.; Reeves, J.T.; Desrosiers, J.-N.; Ma, S.; et al. Efficient Asymmetric Synthesis of P-Chiral Phosphine Oxides via Properly Designed and Activated Benzoxazaphosphinine-2-oxide Agents. J. Am. Chem. Soc. 2013, 135, 2474–2477. [Google Scholar] [CrossRef]
- Swamy, K.C.K.; Kumar, N.N.B.; Balaraman, E.; Kumar, K.V.P.P. Mitsunobu and Related Reactions: Advances and Applica-tions. Chem. Rev. 2009, 109, 2551–2651. [Google Scholar] [CrossRef]
- Dembinski, R. Recent Advances in the Mitsunobu Reaction: Modified Reagents and the Quest for Chromatography-Free Sep-aration. Eur. J. Org. Chem. 2004, 2004, 2763–2772. [Google Scholar] [CrossRef]
- Beddoe, R.H.; Andrews, K.G.; Magné, V.; Cuthbertson, J.D.; Saska, J.; Shannon-Little, A.L.; Shanahan, S.E.; Sneddon, H.F.; Denton, R.M. Redox-neutral organocatalytic Mitsunobu reactions. Science 2019, 365, 910–914. [Google Scholar] [CrossRef]
- Zheng, A.; Liu, S.-B.; Deng, F. 31P NMR Chemical Shifts of Phosphorus Probes as Reliable and Practical Acidity Scales for Solid and Liquid Catalysts. Chem. Rev. 2017, 117, 12475–12531. [Google Scholar] [CrossRef]
- Zasukhin, D.S.; Kasyanov, I.A.; Kolyagin, Y.G.; Bulygina, A.I.; Kharas, K.C.; Ivanova, I.I. Evaluation of Zeolite Acidity by 31P MAS NMR Spectroscopy of Adsorbed Phosphine Oxides: Quantitative or Not? ACS Omega 2022, 7, 12318–12328. [Google Scholar] [CrossRef] [PubMed]
- Kharel, S.; Cluff, K.J.; Bhuvanesh, N.; Gladysz, J.A.; Blümel, J. Structures and Dynamics of Secondary and Tertiary Al-kylphosphine Oxides Adsorbed on Silica. Chem. Asian J. 2019, 14, 2704–2711. [Google Scholar] [CrossRef]
- Bewick, N.A.; Arendt, A.; Li, Y.; Szafert, S.; Lis, T.; Wheeler, K.A.; Young, J.; Dembinski, R. Synthesis and Solid-State Structure of (4-Hydroxy-3,5-diiodophenyl)phosphine Oxides. Dimeric Motifs with the Assistance of O-H···O=P Hydrogen Bonds. Curr. Org. Chem. 2015, 19, 469–474. [Google Scholar] [CrossRef]
- Burke, N.J.; Burrows, A.D.; Mahon, M.F.; Warren, J.E. Hydrogen bond network structures based on sulfonated phosphine ligands: The effects of complex geometry, cation substituents and phosphine oxidation on guanidinium sulfonate sheet formation. Inorganica Chim. Acta 2006, 359, 3497–3506. [Google Scholar] [CrossRef]
- Joshi, R.; Pasilis, S.P. The effect of tributylphosphate and tributyl phosphine oxide on hydrogen bonding interactions between water and the 1-ethyl-3-methylimidazolium cation in 1-ethyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide. J. Mol. Liq. 2015, 209, 381–386. [Google Scholar] [CrossRef]
- Tupikina, E.Y.; Bodensteiner, M.; Tolstoy, P.M.; Denisov, G.S.; Shenderovich, I.G. P═O Moiety as an Ambidextrous Hydrogen Bond Acceptor. J. Phys. Chem. C 2018, 122, 1711–1720. [Google Scholar] [CrossRef]
- Begimova, G.U.; Tupikina, E.Y.; Yu, V.K.; Denisov, G.S.; Bodensteiner, M.; Shenderovich, I.G. Effect of Hydrogen Bonding to Water on the 31P Chemical Shift Tensor of Phenyl- and Trialkylphosphine Oxides and α-Amino Phosphonates. J. Phys. Chem. C 2016, 120, 8717–8729. [Google Scholar] [CrossRef]
- Hilliard, C.R.; Bhuvanesh, N.; Gladysz, J.A.; Blümel, J. Synthesis, purification, and characterization of phosphine oxides and their hydrogen peroxide adducts. Dalton Trans. 2011, 41, 1742–1754. [Google Scholar] [CrossRef]
- Arp, F.F.; Bhuvanesh, N.; Blümel, J. Hydrogen peroxide adducts of triarylphosphine oxides. Dalton Trans. 2019, 48, 14312–14325. [Google Scholar] [CrossRef] [PubMed]
- Ashirov, R.; Todorovic, M.; Bhuvanesh, N.; Blümel, J. Hydrogen-Bonded Di(hydroperoxy)alkane Adducts of the Type Cy3P=O·(HOO)2CHR (R = Alkyl). Molecules 2025, 30, 329. [Google Scholar] [CrossRef]
- Barder, T.E.; Buchwald, S.L. Rationale Behind the Resistance of Dialkylbiaryl Phosphines toward Oxidation by Molecular Ox-ygen. J. Am. Chem. Soc. 2007, 129, 5096–5101. [Google Scholar] [CrossRef] [PubMed]
- Kendall, A.J.; Salazar, C.A.; Martino, P.F.; Tyler, D.R. Direct Conversion of Phosphonates to Phosphine Oxides: An Improved Synthetic Route to Phosphines Including the First Synthesis of Methyl JohnPhos. Organometallics 2014, 33, 6171–6178. [Google Scholar] [CrossRef]
- Zhang, D.; Celaje, J.A.; Agua, A.; Doan, C.; Stewart, T.; Bau, R.; Selke, M. Photooxidation of Mixed Aryl and Biarylphosphines. Org. Lett. 2010, 12, 3100–3103. [Google Scholar] [CrossRef]
- Stewart, B.; Harriman, A.; Higham, L.J. Predicting the Air Stability of Phosphines. Organometallics 2011, 30, 5338–5343. [Google Scholar] [CrossRef]
- Buckler, S.A. Autooxidation of Trialkylphosphines. Org. Biol. Chem. 1962, 84, 3093–3097. [Google Scholar]
- Duncan, T.M. A Compilation of Chemical Shift Anisotropies; Farragut Press: Chicago, IL, USA, 1990. [Google Scholar]
- Pelizzi, C.; Pelizzi, G. Crystal structures of two forms of 1,2-bis(diphenylphosphino)ethane. Struct. Sci. 1979, 35, 1785–1790. [Google Scholar] [CrossRef]
- Davies, J.A.; Dutremez, S. Solid state 31P NMR spectroscopic studies of tertiary phosphines and their complexes. Coord. Chem. Rev. 1992, 114, 61–103. [Google Scholar] [CrossRef]
- Harris, R.K.; Merwin, L.H.; Hägele, G. Solid-state nuclear magnetic resonance study of a series of phosphonic and phosphinic acids. J. Chem. Soc. Faraday Trans. Phys. Chem. Condens. Phases 1989, 85, 1409–1423. [Google Scholar] [CrossRef]
- Bogza, M.; Oeser, T.; Blümel, J. Synthesis, structure, immobilization and solid-state NMR of new dppp- and tripod-type chelate linkers. J. Organomet. Chem. 2005, 690, 3383–3389. [Google Scholar] [CrossRef]
- Mealli, C. Structure of 1,1,1-tris[(diphenylphosphino)methyl]ethane (triphos). Acta Crystallogr. Sect. B Struct. Crystallogr. Cryst. Chem. 1982, 38, 1040–1043. [Google Scholar] [CrossRef]
- Sommer, J.; Yang, Y.; Rambow, D.; Blümel, J. Immobilization of Phosphines on Silica: Identification of Byproducts via 31P CP/MAS Studies of Model Alkyl-, Aryl-, and Ethoxyphosphonium Salts. Inorg. Chem. 2004, 43, 7561–7563. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Holland, G.P.; Solomon, V.C.; Zimmermann, H.; Schiffenhaus, S.; Amin, S.A.; Buttry, D.A.; Yarger, J.L. NMR Characterization of Ligand Binding and Exchange Dynamics in Triphenylphosphine-Capped Gold Nanoparticles. J. Phys. Chem. C 2009, 113, 16387–16393. [Google Scholar] [CrossRef]
- Yang, Y.; Beele, B.; Blümel, J. Easily Immobilized Di- and Tetraphosphine Linkers: Rigid Scaffolds that Prevent Interactions of Metal Complexes with Oxide Supports. J. Am. Chem. Soc. 2008, 130, 3771–3773. [Google Scholar] [CrossRef] [PubMed]
- Tada, N.; Cui, L.; Okubo, H.; Miura, T.; Itoh, A. An Efficient Synthesis of gem-Dihydroperoxides with Molecular Oxygen and Anthracene under Light Irradiation. Adv. Synth. Catal. 2010, 352, 2383–2386. [Google Scholar] [CrossRef]
- Yasui, S.; Tojo, S.; Majima, T. Reaction of Triarylphosphine Radical Cations Generated from Photoinduced Electron Transfer in the Presence of Oxygen. J. Org. Chem. 2005, 70, 1276–1280. [Google Scholar] [CrossRef]
- Cheng, H.; Wartelle, L.H.; Klasson, K.T.; Edwards, J.C. Solid-state NMR and ESR studies of activated carbons produced from pecan shells. Carbon 2010, 48, 2455–2469. [Google Scholar] [CrossRef]
- Qiongfen, Y.; Ming, L.; Ping, N.; Honghong, Y.; Xiaolong, T. Characterization of Metal Oxide-modified Walnut-shell Activated Carbon and Its Application for Phosphine Adsorption: Equilibrium, Regeneration, and Mechanism Studies. J. Wuhan. Univ. Technol-Mater. Sci. Ed. 2019, 34, 487–495. [Google Scholar]
- Puziy, A.M.; Poddubnaya, O.I.; Socha, R.P.; Gurgul, J.; Wisniewski, M. XPS and NMR studies of phosphoric acid activated carbons. Carbon 2008, 46, 2113–2123. [Google Scholar] [CrossRef]
- Heidarinejad, Z.; Dehghani, M.H.; Heidari, M.; Javedan, G.; Ali, I.; Sillanpää, M. Methods for preparation and activation of activated carbon: A review. Environ. Chem. Lett. 2020, 18, 393–415. [Google Scholar] [CrossRef]
- Piette, H.L. Chemical Applications of EPR. In NMR and EPR Spectroscopy; NMR-EPR Staff of Varian Associates, Ed.; Pergamon Press: London, UK, 1960; pp. 207–223. [Google Scholar]
- Guenther, J.; Reibenspies, J.; Blümel, J. Synthesis and Characterization of Tridentate Phosphine Ligands Incorporating Ethox-ysilane Groups and Long Alkyl Chains for Immobilizing Molecular Rhodium Catalysts. Mol. Catal. 2019, 479, 110629. [Google Scholar] [CrossRef]
- Kimura, M.; Miyamoto, I. Discovery of the Activated-Carbon Radical AC+ and the Novel Oxidation Reactions Comprising the AC/AC+ Cycle as a Catalyst in an Aqueous Solution. Bull. Chem. Soc. Jpn. 1994, 67, 2357–2360. [Google Scholar] [CrossRef]
- Beran, S.; Dubský, J.; Slanina, Z. Quantum chemical study of oxygen adsorption on graphite: I. Molecular orbital study of adsorption and migration of molecular oxygen on graphite. Surf. Sci. 1979, 79, 39–52. [Google Scholar] [CrossRef]
- Chen, S.G.; Yang, R.T.; Kapteijn, F.; Moulijn, J.A. A new surface oxygen complex on carbon: Toward a unified mechanism for carbon gasification reactions. Ind. Eng. Chem. Res. 1993, 32, 2835–2840. [Google Scholar] [CrossRef]
- Montoya, A.; Gil, J.O.; Mondragon, F.; Truong, T.N. Oxygen Adsorption on Nitrogen Containing Carbon Surfaces. Fuel Chem. Div. Preprints 2002, 47, 424–425. [Google Scholar]
- Horký, F.; Franz, R.; Bruhn, C.; Pietschnig, R. A General Strategy for Increasing the Air Stability of Phosphines Including Primary Phosphines. Chem. A Eur. J. 2023, 29, e202302518. [Google Scholar] [CrossRef]
- Grushin, V.V. Synthesis of Hemilabile Phosphine-Phosphine Oxide Ligands via the Highly Selective Pd-Catalyzed Mono-oxidation of Bidentate Phosphines: Scope, Limitations, and Mechanism. Organometallics 2001, 20, 3950–3961. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shakeri, E.; Hoefler, J.C.; Blümel, J. Selective Air Oxidation of Bis- and Trisphosphines Adsorbed on Activated Carbon Surfaces. Molecules 2025, 30, 2737. https://doi.org/10.3390/molecules30132737
Shakeri E, Hoefler JC, Blümel J. Selective Air Oxidation of Bis- and Trisphosphines Adsorbed on Activated Carbon Surfaces. Molecules. 2025; 30(13):2737. https://doi.org/10.3390/molecules30132737
Chicago/Turabian StyleShakeri, Ehsan, John C. Hoefler, and Janet Blümel. 2025. "Selective Air Oxidation of Bis- and Trisphosphines Adsorbed on Activated Carbon Surfaces" Molecules 30, no. 13: 2737. https://doi.org/10.3390/molecules30132737
APA StyleShakeri, E., Hoefler, J. C., & Blümel, J. (2025). Selective Air Oxidation of Bis- and Trisphosphines Adsorbed on Activated Carbon Surfaces. Molecules, 30(13), 2737. https://doi.org/10.3390/molecules30132737