
A Case of Competitive Aromatization vs. Sigmatropic [1,5]-Hydrogen Atom Migration in a 1,2,4-Cyclohexatriene Intermediate Derived from a Bis-Enyne Cyclization


Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
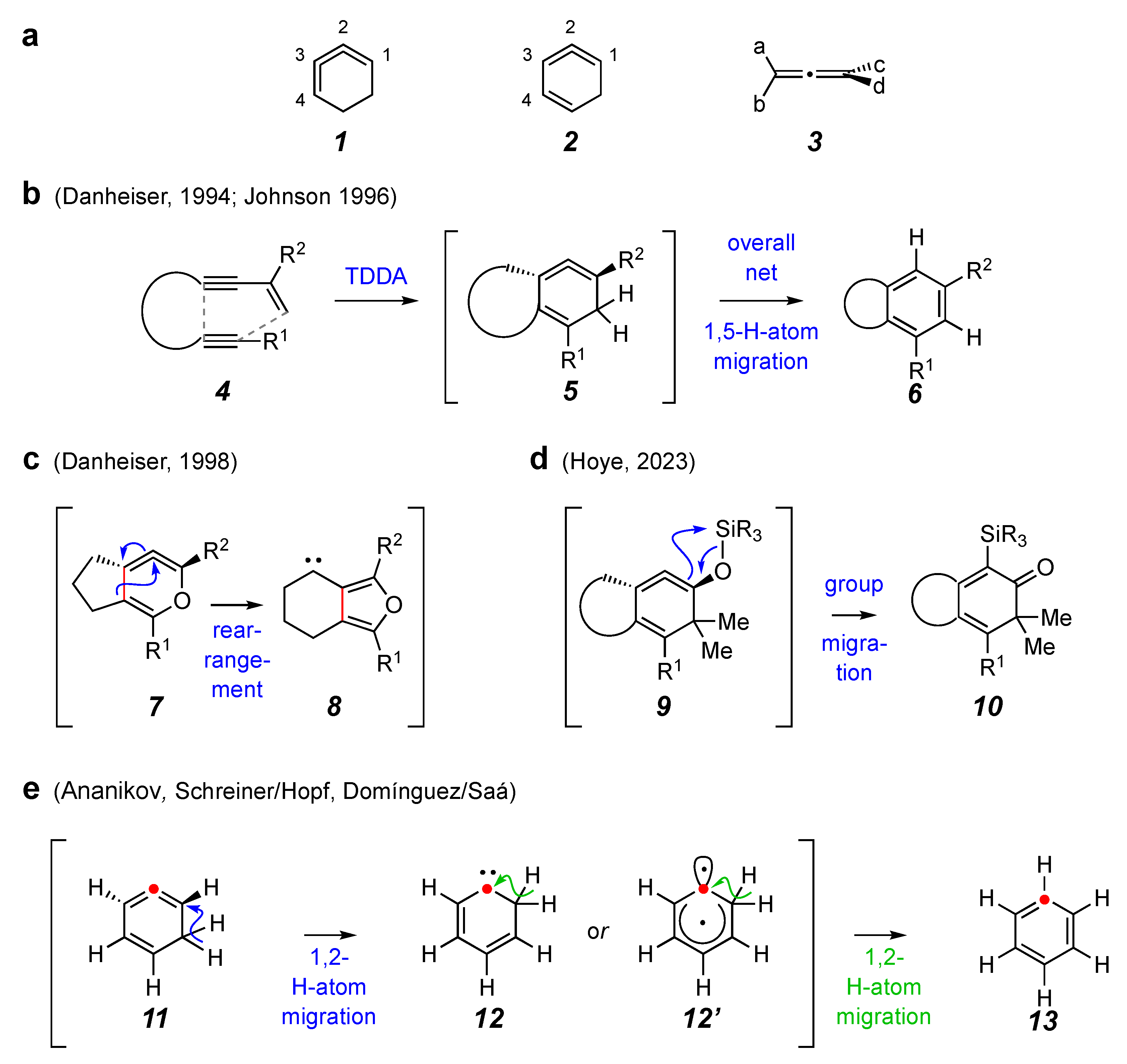
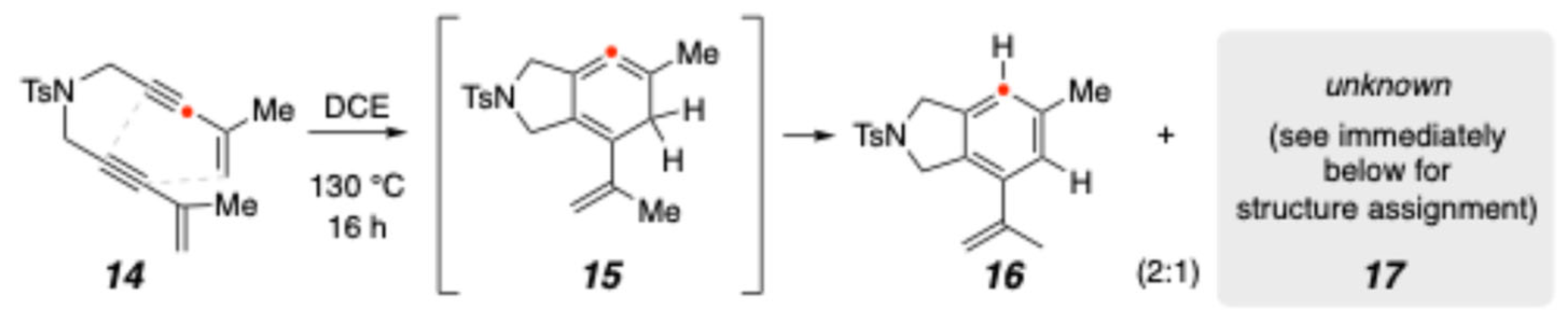
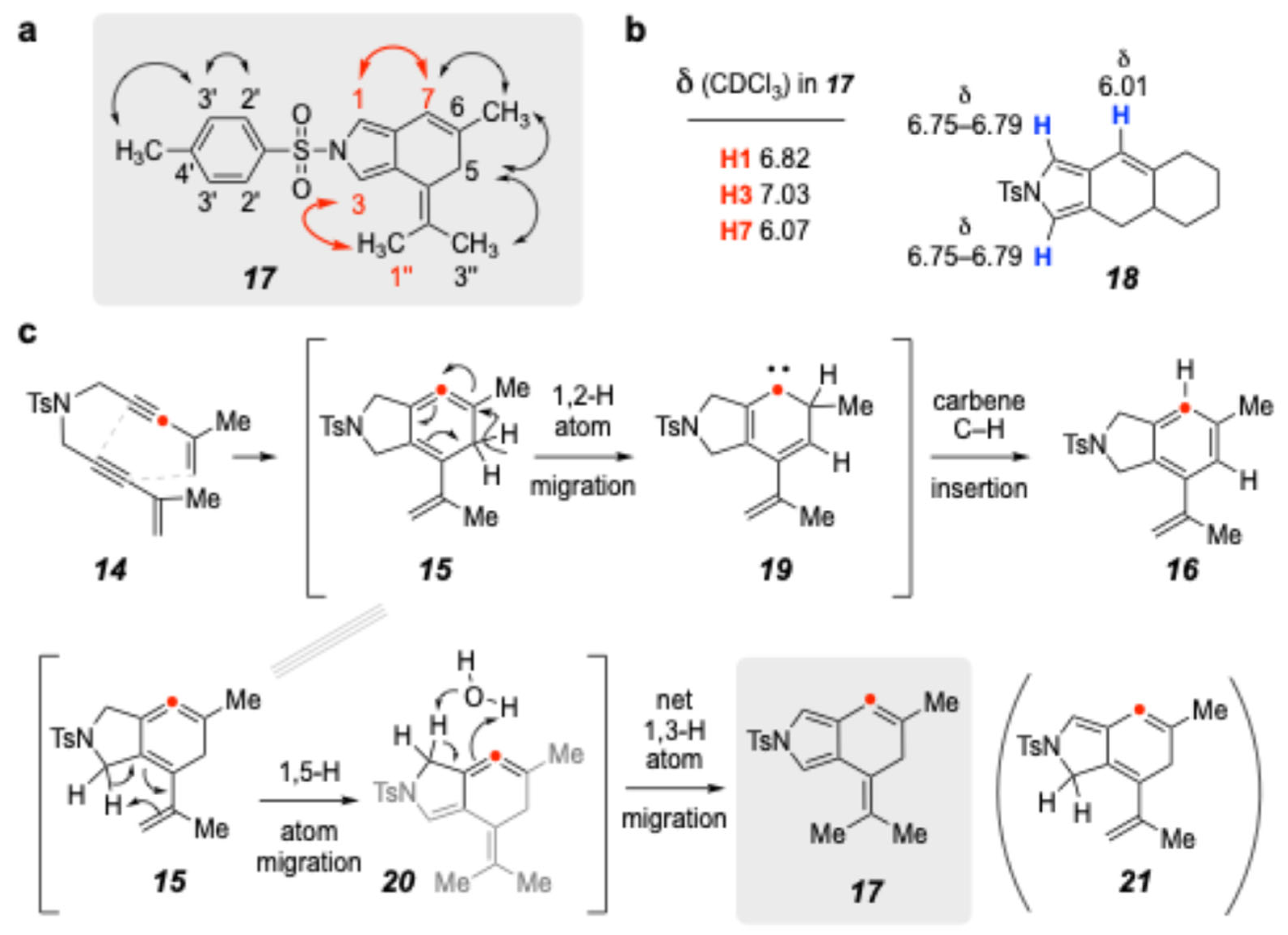
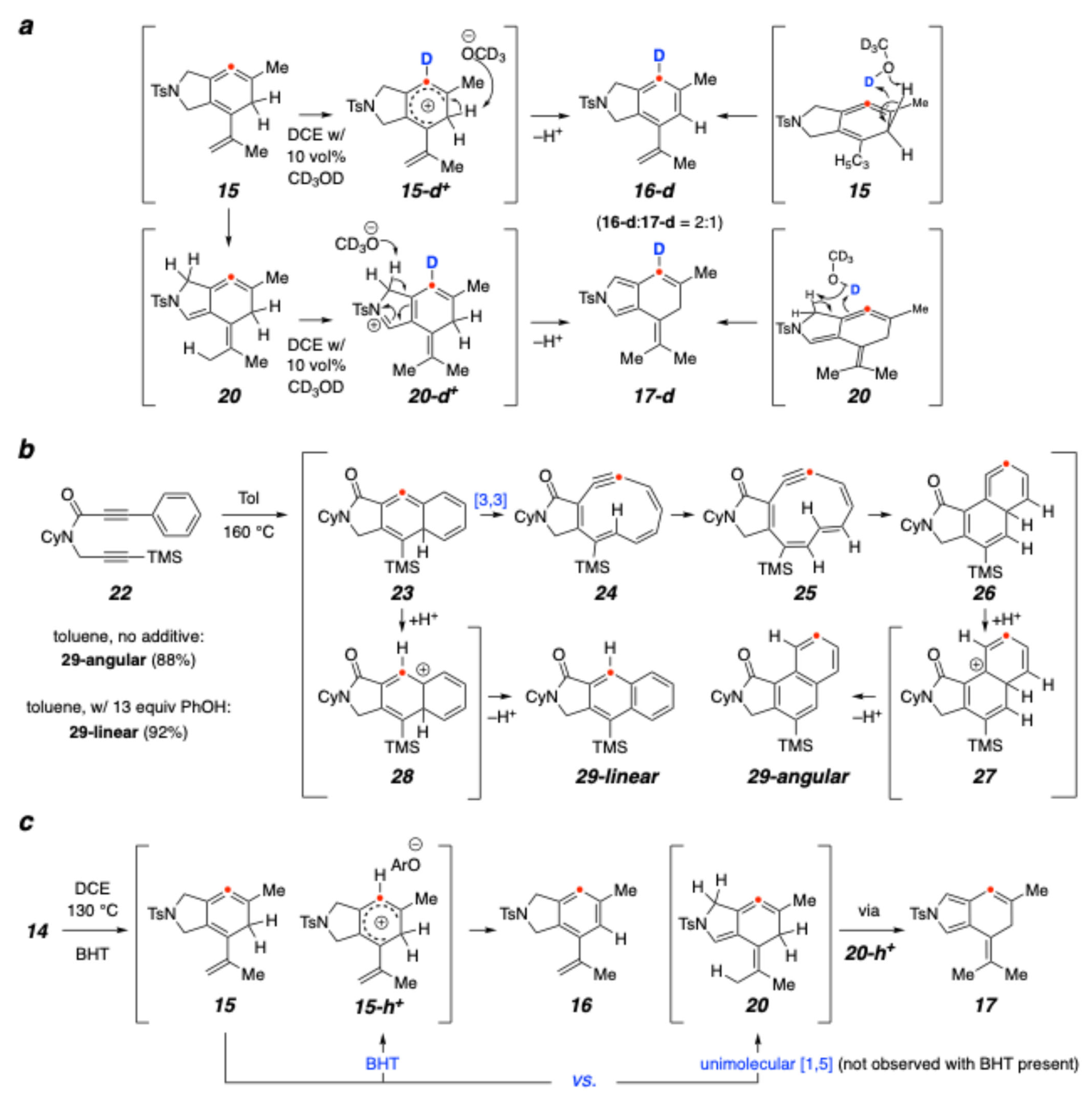
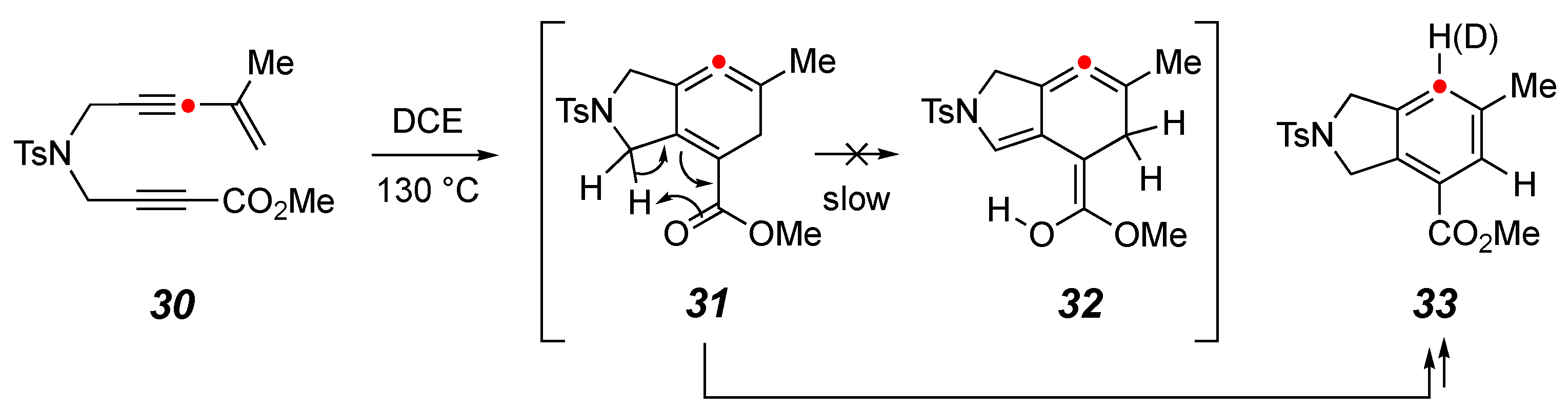
2. Results and Discussion
3. Experimental Section
3.1. Chemistry
3.1.1. General Experimental Protocols
3.1.2. Synthesis of Precursors 14 and 30 (Structures of Intermediates Not Specifically Shown in Any of Figure 1, Figure 2, Figure 3, Figure 4 and Figure 5 Are Given S#s Here)
3.1.3. Synthesis of Products of the TDDA Reactions
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| ATR | attenuated total reflectance |
| BHT | butylated hydroxytoluene |
| DCE | dichloroethane |
| DCM | dichloromethane |
| ESI | electrospray ionization |
| EtOAc | ethyl acetate |
| GCMS | gas chromatography–mass spectrometry |
| Hex | hexanes |
| IR | infrared |
| mp | melting point |
| MPLC | medium pressure liquid chromatography |
| NMR | nuclear magnetic resonance |
| NOEs | nuclear Overhauser effects |
| NOESY | nuclear Overhauser effect spectroscopy |
| NTs | N-tosyl |
| SI | supporting information |
| TDDA | tetradehydro-Diels–Alder |
| THF | tetrahydrofuran |
| TLC | thin layer chromatography |
References
- Shakespeare, W.C.; Johnson, R.P. 1,2,3-Cyclohexatriene and cyclohexen-3-yne: Two new highly strained C6H6 isomers. J. Am. Chem. Soc. 1990, 112, 8578–8579. [Google Scholar] [CrossRef]
- Christl, M.; Braun, M.; Müller, G. 1,2,4-Cyclohexatriene, an isobenzene, and bicyclo[4.4.0]deca-1,3,5,7,8-pentaene, an isonaphthalene: Generation and trapping reactions. Angew. Chem. Int. Ed. Engl. 1992, 31, 473–476. [Google Scholar] [CrossRef]
- Kelleghan, A.V.; Bulger, A.S.; Witkowski, D.C.; Garg, N.K. Strain-promoted reactions of 1,2,3-cyclohexatriene and its derivatives. Nature 2023, 618, 748–754. [Google Scholar] [CrossRef] [PubMed]
- Witkowski, D.C.; Turner, D.W.; Bulger, A.S.; Houk, K.N.; Garg, N.K. Synthesis and reactivity of nitrogen-containing strained cyclic 1,2,3-trienes. Nat. Synth 2025. [Google Scholar] [CrossRef]
- Daoust, K.J.; Hernandez, S.M.; Konrad, K.M.; Mackie, I.D.; Winstanley, J., Jr.; Johnson, R.P. Strain estimates for small-ring cyclic allenes and butatrienes. J. Org. Chem. 2006, 71, 5708–5714. [Google Scholar] [CrossRef]
- Angus, R.O., Jr.; Johnson, R.P. Small-ring cyclic cumulenes: The structure and energetics of cyclic butatrienes and the synthesis of 1,2,3-cyclononatriene. J. Org. Chem. 1984, 49, 2880–2883. [Google Scholar] [CrossRef]
- Wessig, P.; Müller, G. The dehydro-Diels-Alder reaction. Chem. Rev. 2008, 108, 2051–2063. [Google Scholar] [CrossRef]
- Hoye, T.R.; Baire, B.; Niu, D.; Willoughby, P.H.; Woods, B.P. The hexadehydro-Diels-Alder reaction. Nature. 2012, 490, 208–212. [Google Scholar] [CrossRef]
- Danheiser, R.L.; Gould, A.E.; de la Pradilla, R.F.; Helgason, A.L. Intramolecular [4 + 2] cycloaddition reactions of conjugated enynes. J. Org. Chem. 1994, 59, 5514–5515. [Google Scholar] [CrossRef]
- Burrell, R.C.; Daoust, K.J.; Bradley, A.Z.; DiRico, K.J.; Johnson, R.P. Strained cyclic cumulene intermediates in Diels-Alder cycloaddition of enynes and diynes. J. Am. Chem. Soc. 1996, 118, 4218–4219. [Google Scholar] [CrossRef]
- Wills, M.S.B.; Danheiser, R.L. Intramolecular [4 + 2] cycloaddition reactions of conjugated ynones. Formation of polycyclic furans via the generation and rearrangement of strained heterocyclic allenes. J. Am. Chem. Soc. 1998, 120, 9378–9379. [Google Scholar] [CrossRef]
- Xu, Q.; Hoye, T.R. A cascade of strain-driven events converting benzynes to alkynylbenzocyclobutenes to 1,3-dien-5-ynes to cyclic allenes to benzocyclohexadienones. J. Am. Chem. Soc. 2024, 146, 6438–6443. [Google Scholar] [CrossRef] [PubMed]
- Ananikov, V.P. Ab initio study of the mechanisms of intermolecular and intramolecular [4 + 2] cycloaddition reactions of conjugated enynes. J. Phys. Org. Chem. 2001, 14, 109–121. [Google Scholar] [CrossRef]
- Prall, M.; Krüger, A.; Schreiner, P.R.; Hopf, H. The cyclization of parent and cyclic hexa-1,3-dien-5-ynes—A combined theoretical and experimental study. Chem. Eur. J. 2001, 7, 4386–4394. [Google Scholar] [CrossRef]
- Rodríguez, D.; Navarro-Vázquez, A.; Castedo, L.; Domínguez, D.; Saá, C. Cyclic allene intermediates in intramolecular dehydro Diels-Alder reactions: Labeling and theoretical cycloaromatization studies. J. Org. Chem. 2003, 68, 1938–1946. [Google Scholar] [CrossRef]
- Watson, W. On byproducts and side products. Org. Process Res. Dev. 2012, 16, 1877. [Google Scholar] [CrossRef]
- Tokimizu, Y.; Wieteck, M.; Rudolph, M.; Oishi, S.; Fujii, N.; Hashmi, A.S.K.; Ohno, H. Dual gold catalysis: A novel synthesis of bicyclic and tricyclic pyrroles from N-propargyl ynamides. Org. Lett. 2015, 17, 604–607. [Google Scholar] [CrossRef]
- Zanardi, M.M.; Sarotti, A.M. Sensitivity analysis of DP4+ with the probability distribution terms: Development of a universal and customizable method. J. Org. Chem. 2021, 86, 8544–8548. [Google Scholar] [CrossRef]
- Smadja, W. Electrophilic addition to allenic derivatives: Chemo-, regio-, and stereochemistry and mechanisms. Chem. Rev. 1983, 83, 263–320. [Google Scholar] [CrossRef]
- Sharma, R.K.; Shoulders, B.A.; Gardner, P.D. Oxymercuration of allenes. J. Org. Chem. 1967, 32, 241–244. [Google Scholar] [CrossRef]
- Rodríguez, D.; Navarro-Vázquez, A.; Castedo, L.; Domínguez, D.; Saá, C. Strained intermediates in intramolecular dehydro Diels-Alder reactions: Rearrangement of cyclic allenes via 1,2-dehydro[10]annulenes. J. Am. Chem. Soc. 2001, 123, 9178–9179. [Google Scholar] [CrossRef] [PubMed]
- Olmstead, W.N.; Margolin, Z.; Bordwell, F.G. Acidities of water and simple alcohols in dimethyl sulfoxide solution. J. Org. Chem. 1980, 45, 3295–3299. [Google Scholar] [CrossRef]
- Bordwell, F.G.; McCallum, R.J.; Olmstead, W.N. Acidities and hydrogen bonding of phenols in dimethyl sulfoxide. J. Org. Chem. 1984, 49, 1424–1427. [Google Scholar] [CrossRef]
- Hoye, T.R.; Hanson, P.R.; Vyvyan, J.R. A Practical guide to first-order multiplet analysis in 1H NMR spectroscopy. J. Org. Chem. 1994, 59, 4096–4103. [Google Scholar] [CrossRef]
- Hoye, T.R.; Zhao, H. A method for easily determining coupling constant values: An addendum to “A practical guide to first-order multiplet analysis in 1H NMR spectroscopy”. J. Org. Chem. 2002, 67, 4014–4016. [Google Scholar] [CrossRef]
- Parisot, W.; Haddad, M.; Phansavath, P.; Lefèvre, G.; Ratovelomanana-Vidal, V. A versatile, functional group-tolerant, and bench-stable iron precatalyst for building arene and triazine rings by [2+2+2] cycloadditions. Chem. Eur. J. 2024, 30, e202400096. [Google Scholar] [CrossRef]
- Trost, B.M.; Rudd, M.T. Ruthenium-catalyzed cycloisomerizations of diynols. J. Am. Chem. Soc. 2005, 127, 4763–4776. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, R.; Xu, Q.; Hoye, T.R. A Case of Competitive Aromatization vs. Sigmatropic [1,5]-Hydrogen Atom Migration in a 1,2,4-Cyclohexatriene Intermediate Derived from a Bis-Enyne Cyclization. Molecules 2025, 30, 2610. https://doi.org/10.3390/molecules30122610
Tang R, Xu Q, Hoye TR. A Case of Competitive Aromatization vs. Sigmatropic [1,5]-Hydrogen Atom Migration in a 1,2,4-Cyclohexatriene Intermediate Derived from a Bis-Enyne Cyclization. Molecules. 2025; 30(12):2610. https://doi.org/10.3390/molecules30122610
Chicago/Turabian StyleTang, Rong, Qian Xu, and Thomas R. Hoye. 2025. "A Case of Competitive Aromatization vs. Sigmatropic [1,5]-Hydrogen Atom Migration in a 1,2,4-Cyclohexatriene Intermediate Derived from a Bis-Enyne Cyclization" Molecules 30, no. 12: 2610. https://doi.org/10.3390/molecules30122610
APA StyleTang, R., Xu, Q., & Hoye, T. R. (2025). A Case of Competitive Aromatization vs. Sigmatropic [1,5]-Hydrogen Atom Migration in a 1,2,4-Cyclohexatriene Intermediate Derived from a Bis-Enyne Cyclization. Molecules, 30(12), 2610. https://doi.org/10.3390/molecules30122610