3. Materials and Methods
General remarks. All non-aqueous reactions were performed under an inert atmosphere of argon or nitrogen. Inert gases were passed over a U-tube of silica and activated 3 Å molecular sieves. Photochemical reactions were carried out in a dedicated apparatus consisting of an aluminum cooling plate (LWH 160 × 100 × 25 mm) with 6 OSRAM® (Munich, Germany) Oslon SSL 80 LDCQ7P (nominal 450 nm, royal blue) LEDs mounted in series powered by a MeanWell® (New Taipei City, Taiwan) LPC-20-700 constant current power supply (700 mA) and a water-cooled aluminum vessel holder. The vessel holder (LWH 170 × 110 × 38 mm) holds the sealed vials 10 mm over the LEDs in a fixed position while cooling both the LED plate and the vial, keeping the temperature of the latter below 20 °C. Both the LED plate and the water-cooled vessel were custom-built, while the vials were purchased from Wicom International Co. (Heppenheim, Germany) (WIC43005, 5 mL crimp top vial 38.5 × 22.0 mm; WIC44510 20 mm crimp caps with 3.0 mm PTFE septum). For reactions performed under a controlled temperature, the vessel holder was connected to a thermostat Lab. Companion (Daejeon, Republic of Korea) RW-0525G and the temperature was 25 °C or 35 °C. Reactions were stirred magnetically and monitored by thin-layer chromatography (TLC). Anhydrous solvents were purchased by Merck (Darmstadt, Germany). The solvents used for the isocyanide insertion were further degassed with Ar for 20 min. Analytical TLC was performed using MERCK Silica Gel 60 F254 0.25 mm TLC glass plates and visualized by ultraviolet light (UV, 254 nm). TLC plates were also stained by dipping and heating with cerium ammonium molybdate (CAM, Hanessian’s stain, 1 g Ce(SO4)2·4H2O in 31 mL of H2SO4 and 469 mL of H2O), KMnO4 (1.5 g KMnO4, 10 g K2CO3, 1 mL of 10% aq. NaOH in 200 mL of H2O), and 20% H2SO4 in ethanol (spraying and heating). Rf values were measured after an elution of 7–9 cm. After extractions, the aqueous phases were always re-extracted three times with the appropriate organic solvent, and the organic extracts were always dried over Na2SO4 and filtered before evaporation to dryness. Concentration under reduced pressure was performed by rotavapor at 40 °C. Chromatographic purification was performed as flash chromatography (FC) on MERCK Geduran® Si 60 (40–63 µm) under positive pressure, and the crude mixtures were loaded as solid absorbed on silica or as a solution in the eluent. Purified compounds were dried further under high vacuum (~10−2 torr). Petroleum ether (PE) is 40–60 °C. Nuclear Magnetic Resonance (NMR) spectra were recorded on VARIAN MERCURY (Palo Alto, CA, USA) (300 MHz 1H and 75 MHz 13C) or on Jeol JNM-ECZ400R (Akishima, Tokyo, Japan) (400 MHz 1H and 100 MHz 13C). Chemical shifts (δ) are reported in parts per million (ppm) using tetramethylsilane (TMS, 0.00 ppm) or the residual solvent signal (CDCl3 at 77.16 ppm for 13C) as internal standards. The data are reported as follows: chemical shift, integration, multiplicity (indicated as s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; and combinations thereof), coupling constants (J) in Hertz (Hz), and assignation (when possible). Peak assignments were performed with the aid of gCOSY (gradient correlation spectroscopy), gHSQC (gradient heteronuclear single-quantum correlation spectroscopy), and gHMBC (gradient heteronuclear multiple-bond correlation spectroscopy). All NMR spectra were analyzed using the software MestReNova (Santiago de Compostela, Spain) (Mestrelab Research® v. 14.2). Fourier transform infrared (FTIR) spectra were recorded on a Perkin Elmer Spectrum 65 (Perkin Elmer, Waltham, MA, USA) instrument, equipped with a universal attenuated total reflectance (ATR) sampling accessory. IR peaks are reported as decreasing wavenumber (ῡ, cm−1). All spectra were elaborated using the software Spectrum (PerkinElmer® v.10.03) and processed with a 10% smooth algorithm. Melting points were determined with an electrothermal apparatus Büchi (Flawil, Switzerland) B-535. The thermal program used is 1 °C/min. High-Performance Liquid Chromatography (HPLC) analysis was performed on Agilent (Palo Alto, CA, USA) HP 1100 equipped with a DAD detector (220 nm) and column C6 PHENYLIC RP column (150 × 3 mm, 3 μ) at 25 °C with flow = 0.34 mL/min and gradient H2O/CH3CN, (A = H2O, B = CH3CN), 0 min A = 55%, 30 min B = 40%. Gas Chromatography–Mass Spectrometry (GC-MS) analysis was performed on Shimadzu (Kyoto, Japan) GC−MS-QP2010 SE with an AOC-20i Plus auto-injector mounting an Avantor (Radnor, PA, USA) Hichrom HI-5 MS column (internal diameter 0.25 mm, film thickness 0.25 mm, length 30 m). A Mass Analyzer Metal quadrupole mass filter with pre-rods was used and the ionization was obtained through Electronic Impact (EI) operating with an ionization potential of 70 eV. He was used as carrier gas with the following details: flow = 1.0 mL/min, Vinj = 1 µL, and a split ratio of 1:10. Samples were prepared as EtOAc solutions at 100 ppm: solvent delay = 2.5 min, mass range 35–400, Tinj = 250 °C, T detector = 250 °C. Three thermal methods were employed: Method A: Tin = 100 °C, tin = 3 min, gradient = 25 °C/min, Tfinal = 300 °C; Method B: Tin = 70 °C, tin = 3 min, gradient = 25 °C/min, Tfinal = 300 °C; and Method C: Tin = 120 °C, tin = 3 min, gradient = 25 °C/min, Tfinal = 300 °C. The analysis was processed using the software GCMS LabSolution Shimadzu (v. 4.52) and the data are reported as follows: retention time (tR, min), m/z (threshold 3%), and relative abundance. High-Resolution Mass Spectrometer (HRMS) analyses were carried out on a Waters (Milford, MA, USA) Synapt G2 QToF mass spectrometer. MS signals were acquired in ESI positive ionization mode.
Isocyanide insertion general procedure, condition A. In a photochemical vial, the iodide (1 equiv) and DMAP (6 mol%) were added and then Ar was fluxed through a septum. Then, degassed anhydrous toluene (0.3 M), Et3N (1.7 equiv), isocyanide (1.1 equiv), Pd(PPh3)4 (6 mol%), and bidistilled water (40 equiv) were added. The septum was replaced by the cap and crimped. The vial was placed in the photochemical plate and stirred for 48 h. The crude mixture was diluted with s.s. (saturated solution) NH4Cl, extracted with Et2O, washed with brine, dried (Na2SO4), and evaporated to dryness. The crude product was purified by FC on silica.
Isocyanide insertion general procedure, condition B. In a photochemical vial, the iodide (1 equiv) and DMAP (6 mol%) were added and then Ar was fluxed through a septum. Then, degassed anhydrous toluene (0.3 M), Et3N (1.7 equiv), isocyanide (2.2 equiv), Pd(PPh3)4 (6 mol%), and bidistilled water (40 equiv) were added. The septum was replaced by the cap and crimped. The vial was placed in the photochemical plate and stirred for 48 h at 25 °C. The crude mixture was diluted with s.s. NH4Cl, extracted with Et2O, washed with brine, dried (Na2SO4), and evaporated to dryness. The crude product was purified by FC on silica.
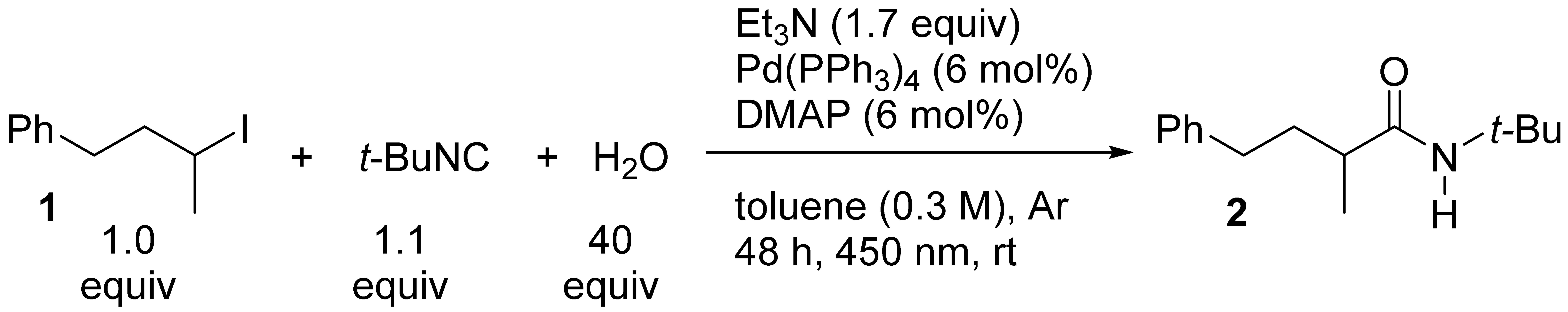
N-(tert-Butyl)-2-methyl-4-phenylbutanamide (2). Following the general procedure (condition B), the mixture of (3-iodobutyl)benzene 1 (100 mg, 0.384 mmol), DMAP (2.8 mg, 0.0231 mmol), Pd(PPh3)4 (26.7 mg, 0.0231 mmol), Et3N (91 µL, 0.653 mmol), t-butyl isocyanide (96 µL, 0.845 mmol), and bidistilled H2O (277 µL, 15.36 mmol) in dry toluene (1.3 mL) was reacted. After extraction, the crude product was purified by silica FC (PE/Et2O 8:2), delivering 2 as a pale-yellow solid (66 mg, 74% yield). Rf 0.29 (PE/Et2O 8:2, UV (weak), CAM (weak)). 1H NMR (400 MHz, CDCl3, 27 °C) δ 7.31–7.27 (m, 2H, 2×CHarom), 7.24–7.11 (m, 3H, 3×CHarom), 5.18 (s, 1H, NH), 2.66 (ddd, J = 14.3, 9.2, 5.3 Hz, 1H, PhCHH), 2.61–2.51 (m, 1H, PhCHH), 2.08–1.91 (m, 2H, PhCH2CHH + CH), 1.71–1.64 (m, 1H, PhCH2CHH), 1.36 (s, 9H, C(CH3)3), 1.12 (d, J = 6.6 Hz, 3H, CH3). 13C NMR (101 MHz, CDCl3, 27 °C) δ 175.6 (C=O), 142.0 (Cq Ar), 128.6 (2×CHarom), 128.5 (2×CHarom), 126.0 (p-CHarom), 51.2 (C(CH3)3), 41.6 (CH), 35.9 (PhCH2CH2), 33.7 (PhCH2), 29.0 (C(CH3)3), 18.2 (CH3). GC-MS: tR = 8.665 min (Method A); m/z (%): 39 (7), 41 (24), 42 (9), 44 (5), 51 (3), 55 (6), 56 (6), 57 (37), 58 (29), 65 (13), 69 (4), 72 (8), 73 (100), 74 (20), 77 (4), 86 (3), 91 (67), 92 (7), 100 (5), 104 (3), 114 (3), 115 (3), 129 (96), 130 (8). IR ῡ: 3309, 3068, 3029, 2969, 2926, 1642, 1544, 1496, 1481, 1453, 1389, 1360, 1286, 1259, 1225, 1094, 1068, 1033, 948, 928, 888, 790, 762, 742, 719, 695, 672. HRMS (ESI+): calcd. for C15H24NO [M+H]+ 234.1852, found 234.1857.
N-(tert-Butyl)cyclohexanecarboxamide (3). Following the general procedure (condition A), the mixture of cyclohexyl iodide (101 mg, 0.48 mmol), DMAP (3.5 mg, 0.0288 mmol), Pd(PPh3)4 (33.3 mg, 0.0288 mmol), Et3N (114 µL, 0.816 mmol), t-butyl isocyanide (60 µL, 0.528 mmol), and bidistilled H2O (277 µL, 15.36 mmol) in dry toluene (1.6 mL) was reacted. After extraction, the crude product was purified by silica FC (PE/AcOEt 9:1), delivering 3 as pale-yellow oil (42 mg, 48% yield). Rf 0.24 (PE/AcOEt 9:1, 20% H2SO4 in EtOH). 1H NMR (300 MHz, CDCl3, 27 °C) δ 5.21 (s, 1H, NH), 1.94 (tt, J = 11.6, 3.4 Hz, 1H, CH), 1.86–1.72 (m, 2H, CH2), 1.70–1.63 (m, 2H, CH2), 1.49–1.36 (m, 2H, CH2), 1.33 (s, 9H, C(CH3)3), 1.30–1.15 (m, 4H, 2×CH2). 13C NMR (75 MHz, CDCl3, 27 °C) δ 175.7 (C=O), 50.9 (C(CH3)3), 46.4 (CH), 30.0 (CH2), 29.9 (CH2), 29.0 (C(CH3)3), 25.9 (3×CH2). GC-MS: tR = 6.644 min (Method A); m/z (%): 39 (15), 40 (3), 41 (47), 42 (14), 43 (4), 44 (3), 53 (5), 54 (5), 55 (45), 56 (18), 57 (52), 58 (100), 59 (12), 72 (15), 81 (3), 82 (3), 83 (36), 84 (3), 93 (3), 98 (5), 111 (9), 112 (6), 115 (5), 128 (57), 129 (5), 142 (5), 168 (4), 183 (16) [M]+. HRMS (ESI+): calcd. for C11H22NO [M+H]+ 184.1696, found 184.1704.
N-(tert-butyl)-4-phenylbutanamide (4). Following the general procedure (condition B), the mixture of (3-iodopropyl)benzene (S3) (100 mg, 0.406 mmol), DMAP (3.0 mg, 0.0244 mmol), Pd(PPh3)4 (28.2 mg, 0.0244 mmol), Et3N (96 µL, 0.691 mmol), t-butyl isocyanide (101 µL, 0.894 mmol), and bidistilled H2O (293 µL, 16.3 mmol) in dry toluene (1.4 mL) was reacted. After extraction, the crude product was purified by silica FC (PE/Et2O 7:3), delivering 4 as a pale-yellow solid (71 mg, 80% yield). Rf 0.20 (PE/Et2O 7:3, UV, CAM). M.p. 68.8–70.2 °C. 1H NMR (400 MHz, CDCl3, 27 °C) δ 7.32–7.24 (m, 2H, 2×CHarom), 7.22–7.16 (m, 3H, 3×CHarom), 5.20 (bs, 1H, NH), 2.64 (t, J = 7.4 Hz, 2H, PhCH2), 2.08 (t, J = 7.3 Hz, 2H, CH2CO), 1.94 (p, J = 7.4 Hz, 2H, CH2CH2CH2), 1.34 (s, 9H, C(CH3)3). 13C NMR (101 MHz, CDCl3, 27 °C) δ 172.1 (C=O), 141.8 (Cq Ar), 128.7 (2×CHarom), 128.5 (2×CHarom), 126.0 (p-CHarom), 51.2 (C(CH3)3), 36.9 (CH2CO), 35.2 (PhCH2), 29.0 (C(CH3)3), 27.2 (CH2CH2CH2). GC-MS: tR = 8.705 min (Method A); m/z (%): 39 (9), 41 (23), 42 (11), 43 (4), 44 (4), 51 (4), 55 (12), 56 (6), 57 (26), 58 (51), 59 (100), 60 (40), 65 (12), 72 (5), 77 (5), 78 (3), 91 (43), 92 (4), 100 (10), 104 (3), 105 (3), 115 (77), 116 (6), 117 (4), 129 (3), 147 (5), 219 (7) [M]+. IR ῡ: 3308, 3070, 3030, 2966, 2931, 2864, 1665, 1639, 1546, 1497, 1453, 1421, 1390, 1360, 1281, 1227, 1158, 1133, 1081, 1033, 965, 923, 907, 843, 759, 738, 698, 667, 622. HRMS (ESI+): calcd. for C14H22NO [M+H]+ 220.1696, found 220.1699.
(3r,5r,7r)-N-(tert-Butyl)adamantane-1-carboxamide (6). Following the general procedure (condition B), the mixture of 1-iodoadamantane (100 mg, 0.381 mmol), DMAP (2.8 mg, 0.0229 mmol), Pd(PPh3)4 (26.5 mg, 0.0229 mmol), Et3N (90 µL, 0.648 mmol), t-butyl isocyanide (95 µL, 0.839 mmol), and bidistilled H2O (275 µL, 15.3 mmol) in dry toluene (1.3 mL) was reacted. After extraction, the crude product was purified by silica FC (PE/Et2O 8:2), delivering 6 as a pale-yellow solid (18 mg, 20% yield). 1H NMR (400 MHz, CDCl3, 27 °C) δ 5.37 (bs, 1H, NH), 2.03 (bs, 3H, 3×CH), 1.83–1.78 (m, 6H, 3×2-CH2), 1.76–1.65 (m, 6H, 3×4-CH2), 1.33 (s, 9H, C(CH3)3). 13C NMR (101 MHz, CDCl3, 27 °C) δ 177.5 (C=O), 50.7 (C(CH3)3), 41.0 (C-CO), 39.5 (3×2-CH2), 36.7 (3×4-CH2), 29.0 (C(CH3)3), 28.3 (3×CH). GC-MS: tR = 8.980 min (Method A); m/z (%): 39 (5), 41 (17), 42 (6), 53 (4), 55 (6), 56 (4), 57 (12), 58 (4), 65 (3), 67 (8), 77 (9), 79 (21), 80 (3), 81 (5), 91 (9), 92 (3), 93 (19), 94 (3), 107 (10), 135 (100), 136 (15), 163 (8), 180 (11), 193 (3), 220 (13), 235 (18) [M]+. IR ῡ: 3327, 2974, 2958, 2900, 2850, 1636, 1533, 1470, 1445, 1387, 1359, 1343, 1328, 1287, 1267, 1228, 1178, 1106, 1090, 1045, 981, 944, 925, 913, 816, 746, 673, 635. HRMS (ESI+): calcd. for C15H26NO [M+H]+ 236.2009, found 236.2017.
N-(tert-Butyl)-3,3-dimethyl-4-phenylbutanamide (7). Following the general procedure (condition A), the mixture of (3-iodo-2,2-dimethylpropyl)benzene (S8) (100 mg, 0.365 mmol), DMAP (2.7 mg, 0.0219 mmol), Pd(PPh3)4 (25.3 mg, 0.0219 mmol), Et3N (85 µL, 0.620 mmol), t-butyl isocyanide (45 µL, 0.402 mmol), and bidistilled H2O (263 µL, 14.6 mmol) in dry toluene (1.2 mL) was reacted. After extraction, the crude product was purified by silica FC (PE/Et2O 8:2), delivering 7 as a pale-yellow solid (39 mg, 43% yield). Rf 0.24 (PE/Et2O 7:3, UV, CAM). M.p. 72.2–73.7 °C. 1H NMR (400 MHz, CDCl3, 27 °C) δ 7.30–7.24 (m, 2H, 2×CHarom), 7.23–7.16 (m, 3H, 3×CHarom), 5.20 (bs, 1H, NH), 2.66 (s, 2H, PhCH2), 1.94 (s, 2H, CH2CO), 1.35 (s, 9H, C(CH3)3), 1.01 (s, 6H, C(CH3)2). 13C NMR (101 MHz, CDCl3, 27 °C) δ 171.2 (C=O), 138.9 (Cq Ar), 131.0 (2×CHarom), 127.8 (2×CHarom), 126.1 (p-CHarom), 51.3 (C(CH3)3)), 49.0 (CH2CO), 48.5 (PhCH2), 34.6 (C(CH3)2), 28.9 (C(CH3)3), 27.4 (C(CH3)2). GC-MS: tR = 8.980 min (Method A); m/z (%): 39 (8), 41 (28), 42 (9), 43 (3), 44 (4), 55 (10), 56 (10), 57 (88), 58 (38), 59 (74), 60 (36), 65 (11), 72 (3), 83 (3), 91 (50), 92 (8), 100 (17), 105 (4), 115 (100), 116 (8), 117 (5), 132 (3), 133 (4), 247 (3) [M]+. IR ῡ: 3311, 3064, 3030, 2966, 2925, 1637, 1607, 1543, 1492,1452, 1389, 1354, 1329, 1317, 1290, 1265, 1225, 1182, 1145, 1114, 1073, 1031, 991, 934, 897, 791, 754, 713, 702, 685, 613. HRMS (ESI+): calcd. for C16H26NO [M+H]+ 248.2009, found 248.2003.
4-Phenyl-N-(2,4,4-trimethylpentan-2-yl)butanamide (8). Following the general procedure (condition A), the mixture of (3-iodopropyl)benzene (S3) (100 mg, 0.406 mmol), DMAP (3.0 mg, 0.0244 mmol), Pd(PPh3)4 (28.2 mg, 0.0244 mmol), Et3N (96 µL, 0.691 mmol), 1,1,3,3-tetramethylbutyl isocyanide (78 µL, 0.447 mmol), and bidistilled H2O (293 µL, 16.3 mmol) in dry toluene (1.4 mL) was reacted. After extraction, the crude product was purified by silica FC (PE/Et2O 7:3), delivering 8 as a pale-yellow solid (80 mg, 72% yield). Rf 0.31 (PE/Et2O 7:3, UV (weak), CAM (weak)). 1H NMR (400 MHz, CDCl3, 27 °C) δ 7.33–7.24 (m, 2H, 2×CHarom), 7.23–7.15 (m, 3H, 3×CHarom), 5.17 (bs, 1H, NH), 2.65 (t, J = 7.5 Hz, 2H, PhCH2), 2.08 (t, J = 7.6 Hz, 2H, CH2CO), 2.00–1.88 (m, 2H, CH2CH2CH2), 1.74 (s, 2H, CH2C(CH3)3), 1.39 (s, 6H, C(CH3)2), 0.99 (s, 9H, C(CH3)3). 13C NMR (101 MHz, CDCl3, 27 °C) δ 171.8 (C=O), 141.8 (Cq Ar), 128.6 (2×CHarom), 128.5 (2×CHarom), 126.0 (p-CHarom), 55.2 (C(CH3)2), 51.6 (CH2C(CH3)3), 37.2 (CH2CO), 35.3 (PhCH2), 31.8 (C(CH3)3), 31.6 (C(CH3)3), 29.4 (C(CH3)2), 27.1 (CH2CH2CH2). GC-MS: tR = 10.110 min (Method A); m/z (%): 39 (5), 41 (19), 42 (7), 43 (7), 55 (13), 56 (4), 57 (29), 58 (100), 59 (48), 60 (4), 65 (6), 72 (6), 77 (3), 91 (32), 92 (3), 97 (13), 100 (3), 112 (3), 114 (12), 115 (4), 129 (3), 147 (11), 164 (8), 171 (9), 204 (9), 218 (4), 275 (2) [M]+. IR ῡ: 3295, 3083, 3028, 2949, 2866, 1639, 1545, 1477, 1453, 1387, 1361, 1348, 1315, 1279, 1260, 1229, 1159, 1142, 1080, 1053, 1030, 969, 929, 908, 857, 809, 743. HRMS (ESI+): calcd. for C18H30NO [M+H]+ 276.2322, found 276.2327.
2-Methyl-4-phenyl-N-(2,4,4-trimethylpentan-2-yl)butanamide (9). Following the general procedure (condition A), the mixture of (3-iodobutyl)benzene 1 (100 mg, 0.384 mmol), DMAP (2.8 mg, 0.0231 mmol), Pd(PPh3)4 (26.7 mg, 0.0231 mmol), Et3N (91 µL, 0.653 mmol), 1,1,3,3-tetramethylbutyl isocyanide (74 µL, 0.422 mmol), and bidistilled H2O (277 µL, 15.36 mmol) in dry toluene (1.3 mL) was reacted. After extraction, the crude product was purified by silica FC (PE/Et2O 8:2), delivering 9 as a pale-yellow solid (69 mg, 62% yield). Rf 0.27 (PE/Et2O 9:1, UV (weak), CAM (weak)). 1H NMR (400 MHz, CDCl3, 27 °C) δ 7.32–7.23 (m, 2H, 2×CHarom), 7.22–7.13 (m, 3H, 3×CHarom), 5.18 (s, 1H, NH), 2.71–2.50 (m, 2H, PhCH2), 2.09–1.90 (m, 2H, CH + PhCH2CHH), 1.81 (d, J = 14.9 Hz, 1H, CHHC(CH3)3), 1.70–1.58 (m, 2H, CHHC(CH3)3 + PhCH2CHH), 1.42 (s, 3H, CH3CCH3), 1.39 (s, 3H, CH3CCH3), 1.11 (d, J = 6.7 Hz, 3H, CHCH3), 0.99 (s, 9H, C(CH3)3). 13C NMR (101 MHz, CDCl3, 27 °C) δ 175.2 (C=O), 142.1 (Cq Ar), 128.6 (2×CHarom), 128.5 (2×CHarom), 126.0 (p-CHarom), 55.3 (CH3CCH3), 52.2 (CH2C(CH3)3), 41.8 (CH), 35.9 (PhCH2CH2), 33.7 (PhCH2), 31.8 (C(CH3)3), 31.7 (C(CH3)3), 29.33 (CH3CCH3), 29.26 (CH3CCH3), 18.0 (CHCH3). GC-MS: tR = 10.085 min (Method A), m/z (%): 39 (4), 41 (20), 42 (7), 43 (8), 44 (3), 55 (12), 56 (5), 57 (40), 58 (100), 59 (4), 65 (9), 69 (7), 72 (8), 73 (77), 74 (9), 77 (3), 91 (88), 92 (8), 97 (15), 112 (4), 114 (14), 115 (3), 133 (8), 161 (13), 178 (9), 185 (28), 186 (3), 218 (8), 232 (5). IR ῡ: 3306, 3071, 2948, 2870, 1639, 1546, 1496, 1475, 1453, 1387, 1363, 1287, 1260, 1228, 1178, 1155, 1093, 1055, 1033, 956, 928, 903, 883, 861, 807, 785, 766, 742, 723, 693, 679. HRMS (ESI+): calcd. for C19H32NO [M+H]+ 290.2478, found 290.2470.
N-Cyclohexyl-2-methyl-4-phenylbutanamide (11). Following the general procedure (condition A), the mixture of (3-iodobutyl)benzene 1 (100 mg, 0.384 mmol), DMAP (2.8 mg, 0.0231 mmol), Pd(PPh3)4 (26.7 mg, 0.0231 mmol), Et3N (91 µL, 0.653 mmol), c-Hex isocyanide (53 µL, 0.422 mmol), and bidistilled H2O (277 µL, 15.36 mmol) in dry toluene (1.3 mL) was reacted. After extraction, the crude product was purified by silica FC (PE/Et2O 6:4), delivering 11 as a pale-yellow solid (41 mg, 41% yield). Rf 0.28 (PE/Et2O 6:4, UV (weak), CAM (weak)). 1H NMR (400 MHz, CDCl3, 27 °C) δ 7.31–7.27 (m, 2H, 2×CHarom), 7.22–7.15 (m, 3H, 3×CHarom), 5.23 (bd, J = 7.5 Hz, 1H, NH), 3.86–3.74 (m, 1H, CH c-Hex), 2.66 (ddd, J = 14.7, 9.4, 5.6 Hz, 1H, PhCHH), 2.56 (ddd, J = 13.8, 8.9, 7.0 Hz, 1H, PhCHH), 2.15–2.05 (m, 1H, CHCH3), 2.04–1.86 (m, 3H, PhCH2CHH + 2×CHH c-Hex), 1.76–1.58 (m, 4H, PhCH2CHH + 2×CHH c-Hex + CHH c-Hex), 1.45–1.30 (m, 2H, 2×CHH c-Hex), 1.22–1.04 (m, 3H, 2×CHH c-Hex + CHH c-Hex), 1.15 (d, J = 6.8 Hz, 3H, CH3). 13C NMR (101 MHz, CDCl3, 27 °C) δ 175.2 (C=O), 141.9 (Cq.), 128.5 (2×CHarom), 128.5 (2×CHarom), 126.0 (p-CHarom), 48.0 (CH c-Hex), 41.0 (CHCH3), 35.9 (PhCH2CH2), 33.6 (PhCH2), 33.5 (2-CH2 c-Hex), 33.3 (6-CH2 c-Hex), 25.6 (CH2 c-Hex), 25.0 (2×CH2 c-Hex), 18.2 (CH3). GC-MS: tR = 10.590 min (Method A); m/z (%): 39 (6), 41 (16), 42 (3), 43 (6), 44 (5), 54 (4), 55 (21), 56 (13), 57 (4), 65 (10), 67 (6), 69 (4), 72 (4), 73 (11), 74 (100), 75 (4), 77 (3), 79 (3), 82 (5), 83 (6), 91 (70), 92 (6), 98 (5), 104 (3), 126 (11), 155 (64), 156 (7). IR ῡ (cm−1): 3295, 3032, 2931, 2853, 2221, 1634, 1542, 1494, 1446, 1389, 1366, 1348, 1300, 1269, 1247, 1233, 1171, 1152, 1122, 1101, 1076, 1048, 1031, 973, 944, 891, 865, 846, 786, 749, 724, 697, 648. HRMS (ESI+): calcd. for C17H26NO [M+H]+ 260.2009, found 260.2004.
2-Methyl-4-phenyl-N-(p-tolyl)butanamide (14). Following the general procedure (condition A), the mixture of (3-iodobutyl)benzene 1 (100 mg, 0.384 mmol), DMAP (2.8 mg, 0.0231 mmol), Pd(PPh3)4 (26.7 mg, 0.0231 mmol), Et3N (91 µL, 0.653 mmol), 1-isocyano-4-methylbenzene (49.5 mg, 0.422 mmol), and bidistilled H2O (277 µL, 15.36 mmol) in dry toluene (1.3 mL) was reacted. After extraction, the crude product was purified by silica FC (PE/Et2O 7:3), delivering 14 as a pale-yellow oil (7 mg, 7% yield). Rf 0.22 (PE/Et2O 7:3, UV, CAM). 1H NMR (400 MHz, CDCl3, 27 °C) δ 7.43–7.38 (m, 2H, para-syst. 2×CHarom), 7.33–7.27 (m, 2H, 2×CHarom), 7.23–7.17 (m, 3H, 3×CHarom), 7.15–7.11 (m, 2H, para-syst. 2×CHarom), 7.00 (bs, 1H, NH), 2.73 (ddd, J = 14.5, 9.0, 5.8 Hz, 1H, PhCHH), 2.64 (dt, J = 14.2, 7.5 Hz, 1H, PhCHH), 2.38–2.23 (m, 4H, CH + CH3-C6H4), 2.16–2.05 (m, 1H, PhCH2CHH), 1.85–1.73 (m, 1H, PhCH2CHH), 1.26 (d, J = 6.8 Hz, 3H, CHCH3). 13C NMR (100 MHz, CDCl3, 27 °C) δ 174.4 (C=O), 141.7 (Cq.), 135.5 (Cq.), 134.0 (Cq.), 129.6 (para-syst. 2×CHarom), 128.6 (2×CHarom), 128.6 (2×CHarom), 126.2 (CHarom), 120.0 (para-syst. 2×CHarom), 41.8 (CH), 35.8 (PhCH2CH2), 33.6 (PhCH2), 21.0 (CH3-C6H4), 18.2 (CHCH3). HRMS (ESI+): calcd. for C18H22NO [M+H]+ 268.1696, found 268.1704.
N-(2,6-Dimethylphenyl)-2-methyl-4-phenylbutanamide (15). Following the general procedure (condition B), the mixture of (3-iodobutyl)benzene 1 (100 mg, 0.384 mmol), DMAP (2.8 mg, 0.0231 mmol), Pd(PPh3)4 (26.7 mg, 0.0231 mmol), Et3N (91 µL, 0.653 mmol), 2,6-dimethylphenyl isocyanide (111 mg, 0.845 mmol), and bidistilled H2O (277 µL, 15.36 mmol) in dry toluene (1.3 mL) was reacted. After extraction, the crude product was purified by silica FC (PE/Et2O 7:3), delivering 15 as a pale-yellow solid (71 mg, 66% yield). Rf 0.28 (PE/Et2O 7:3, UV (weak), CAM (weak)). 1H NMR (400 MHz, CDCl3, 27 °C) δ 7.33–7.27 (m, 2H, 2×CHarom of Ph), 7.24–7.18 (m, 3H, 3×CHarom of Ph), 7.12–7.06 (m, 3H, 3×CHarom of dimethylphenyl), 6.68 (s, 1H, NH amide), 2.84–2.64 (m, 2H, PhCH2), 2.54–2.40 (m, 1H, CH), 2.24 (s, 6H, 2×CH3 of dimethylphenyl), 2.23–2.09 (m, 1H, PhCH2CHH), 1.86–1.73 (m, 1H, PhCH2CHH), 1.33 (d, J = 6.9 Hz, 3H, CHCH3). 13C NMR (101 MHz, CDCl3, 27 °C) δ 174.5 (C=O), 141.9 (Cq. of Ph), 135.5 (2×Cq.-CH3), 133.9 (Cq.-NH), 128.6 (2×CHarom of Ph), 128.6 (2×CHarom of Ph), 128.4 (2×CHarom of dimethylphenyl), 127.4 (CHarom of dimethylphenyl), 126.1 (CHarom of Ph), 41.3 (CH), 36.0 (PhCH2CH2), 33.9 (PhCH2CH2), 18.7 (2×CH3 of dimethylphenyl), 18.7 (CHCH3). GC-MS: tR = 11.415 min (Method A); m/z (%): 39 (4), 41 (4), 51 (3), 55 (3), 65 (11), 77 (10), 78 (3), 79 (5), 91 (100), 92 (9), 103 (4), 104 (3), 105 (5), 106 (6), 120 (27), 121 (35), 122 (4), 133 (3), 148 (34), 149 (4), 177 (38), 178 (5), 281 (1) [M]+. IR ῡ (cm−1): 3226, 3027, 2967, 2926, 2850, 1669, 1648, 1602, 1526, 1496, 1474, 1453, 1430, 1367, 1265, 1234, 1206, 1162, 1122, 1090, 1052, 1033, 945, 844, 774, 765, 742, 729, 697, 648. HRMS (ESI+): calcd. for C19H24NO [M+H]+ 282.1852, found 282.1849.
N-Cyclohexyl-3,3-dimethyl-4-phenylbutanamide (16). Following the general procedure (condition A), the mixture of (3-iodo-2,2-dimethylpropyl)benzene S8 (91.8 mg, 0.335 mmol), DMAP (2.4 mg, 0.0201 mmol), Pd(PPh3)4 (23.2 mg, 0.0201 mmol), Et3N (19 µL, 0.569 mmol), c-Hex isocyanide (46 µL, 0.368 mmol), and bidistilled H2O (241 µL, 13.4 mmol) in dry toluene (1.2 mL) was reacted. After extraction, the crude product was purified by silica FC (PE/Et2O 7:3), delivering 16 as a pale-yellow solid (18 mg, 20% yield). Rf 0.23 (PE/Et2O 7:3, UV, CAM). 1H NMR (400 MHz, CDCl3, 27 °C) δ 7.26 (t, J = 3.6 Hz, 2H, 2×CHarom), 7.24–7.18 (m, 3H, 2×CHarom), 5.22 (bd, J = 7.3 Hz, 1H, NH), 3.79 (tdt, J = 12.1, 8.1, 3.9 Hz, 1H, CH c-Hex), 2.67 (s, 2H, PhCH2), 1.99 (s, 2H, CH2CO), 1.97–1.88 (m, 2H, 2×CHH c-Hex), 1.70 (dt, J = 13.2, 3.4 Hz, 2H, 2×CHH c-Hex), 1.61 (dt, J = 12.7, 3.6 Hz, 1H, CHH c-Hex), 1.41–1.34 (m, 2H, 2×CHH c-Hex), 1.16–1.07 (m, 3H, CHH c-Hex + 2×CHH c-Hex), 1.01 (s, 6H, 2×CH3). 13C NMR (101 MHz, CDCl3, 27 °C) δ 170.8 (C=O), 138.9 (Cq.), 131.0 (2×CHarom), 127.9 (2×CHarom), 126.1 (CHarom), 48.5 (PhCH2), 48.3 (CH2CO), 48.2 (CH c-Hex), 34.6 (Cq.-(CH3)2), 33.5 (2×CH2 c-Hex), 27.4 (2×CH3), 25.7 (CH2 c-Hex), 25.0 (2×CH2 c-Hex). GC-MS: tR = 13.960 min (Method A); m/z (%): 41 (15), 43 (6), 55 (26), 56 (14), 57 (20), 59 (5), 60 (100), 65 (5), 67 (5), 82 (7), 83 (36), 91 (36), 92 (5), 98 (8), 126 (8), 141 (97), 142 (9). IR ῡ (cm−1): 3267, 3076, 3026, 2929, 2853, 1632, 1552, 1489, 1465, 1448, 1389, 1368, 1346, 1259, 1229, 1181, 1152, 1143, 1124, 1071, 1028, 994, 892, 846, 798, 763, 720, 701, 630. HRMS (ESI+): calcd. for C18H28NO [M+H]+ 274.2165, found 274.2171.
N-Cyclohexyl-2-methyl-4-(thiophen-2-yl)butanamide (21). Following the general procedure (condition B), the mixture of 2-(3-iodobutyl)thiophene 20 (100 mg, 0.376 mmol), DMAP (2.8 mg, 0.0226 mmol), Pd(PPh3)4 (26.4 mg, 0.0226 mmol), Et3N (89 µL, 0.639 mmol), t-butyl isocyanide (103 µL, 0.827 mmol), and bidistilled H2O (271 µL, 15.04 mmol) in dry toluene (1.3 mL) was reacted. After extraction, the crude product was purified by silica FC (PE/Et2O 7:3), delivering 21 as a pale-yellow solid (64 mg, 64% yield). Rf 0.28 (PE/Et2O 7:3, UV, CAM). 1H NMR (400 MHz, CDCl3, 27 °C) δ 7.12 (dd, J = 5.1, 1.1 Hz, 1H, 5-CH thiophene), 6.92 (dd, J = 5.1, 3.4 Hz, 1H, 4-CH thiophene), 6.82–6.75 (m, 1H, 3-CH thiophene), 5.30 (s, 1H, NH), 3.80 (dddd, J = 14.9, 10.8, 8.3, 3.9 Hz, 1H, CH c-Hex), 2.93–2.73 (m, 2H, CH2CH2CH), 2.21–2.10 (m, 1H CH2CH), 2.05 (dtd, J = 14.4, 8.2, 5.7 Hz, 1H, CHHCH), 1.98–1.86 (m, 2H, 2×CHH c-Hex), 1.79–1.57 (m, 4H, CHHCH + 2×CHH c-Hex + CHH c-Hex), 1.44–1.30 (m, 2H, 2×CHH c-Hex), 1.23–1.03 (m, 3H, 2×CHH c-Hex + CHH c-Hex), 1.15 (d, J = 6.7 Hz, 3H, CH3). 13C NMR (101 MHz, CDCl3, 27 °C) δ 175.0 (C=O), 144.8 (Cq.), 126.9 (4-CH thiophene), 124.5 (3-CH thiophene), 123.2 (5-CH thiophene), 48.1 (CH c-Hex), 40.6 (CH), 36.1 (CH2CH), 33.5 (2-CH2 c-Hex), 33.3 (6-CH2 c-Hex), 27.7 (CH2CH2CH), 25.7 (CH2 c-Hex), 25.0 (2×CH2 c-Hex), 18.1 (CH3). GC-MS: tR = 10.620 min (Method A); m/z (%): 39 (5), 41 (15), 42 (3), 43 (4), 44 (6), 45 (7), 53 (7), 54 (3), 55 (18), 56 (8), 57 (5), 67 (6), 69 (5), 72 (4), 73 (10), 74 (100), 75 (4), 82 (5), 83 (3), 97 (36), 98 (6), 110 (3), 126 (10), 155 (45), 156 (4), 265 (1) [M]+. IR ῡ (cm−1): 3282, 3099, 2931, 2853, 2221, 1636, 1544, 1440, 1387, 1372, 1346, 1303, 1271, 1245, 1234, 1225, 1181, 1151, 1099, 1075, 1030, 976, 945, 892, 854, 823, 796, 698, 614. HRMS (ESI+): calcd. for C15H24NOS [M+H]+ 266.1573, found 266.1576.
N-(tert-Butyl)-2-methyl-4-(thiophen-2-yl)butanamide (22). Following the general procedure (condition B), the mixture of 2-(3-iodobutyl)thiophene 20 (100 mg, 0.376 mmol), DMAP (2.8 mg, 0.0226 mmol), Pd(PPh3)4 (26.4 mg, 0.0226 mmol), Et3N (89 µL, 0.639 mmol), t-butyl isocyanide (93 µL, 0.827 mmol), and bidistilled H2O (271 µL, 15.04 mmol) in dry toluene (1.3 mL) was reacted. After extraction, the crude product was purified by silica FC (PE/Et2O 7:3)m, delivering 22 as a pale-yellow solid (76 mg, 69% yield). Rf 0.38 (PE/Et2O 7:3, UV, CAM). 1H NMR (400 MHz, CDCl3, 27 °C) δ 7.11 (dd, J = 5.1, 1.1 Hz, 1H, 5-CH thiophene), 6.92 (dd, J = 5.1, 3.4 Hz, 1H, 4-CH thiophene), 6.82–6.75 (m, 1H, 3-CH thiophene), 5.26 (bs, 1H, NH), 2.96–2.69 (m, 2H, CH2CH2CH), 2.15–1.96 (m, 2H, CHHCH + CH), 1.70 (dtd, J = 13.3, 7.8, 5.0 Hz, 1H, CHHCH), 1.36 (s, 9H, C(CH3)3), 1.12 (d, J = 6.6 Hz, 3H, CH3). 13C NMR (101 MHz, CDCl3, 27 °C) δ 175.4 (C=O), 144.8 (Cq.), 126.9 (4-CH thiophene), 124.5 (3-CH thiophene), 123.2 (5-CH thiophene), 51.2 (C(CH3)3), 41.1 (CH), 36.2 (CH2CH), 29.0 (C(CH3)3), 27.7 (CH2CH2CH), 18.1 (CH3). GC-MS: tR = 8.720 min (Method A); m/z (%): 39 (3), 41 (11), 42 (3), 44 (3), 45 (6), 53 (5), 55 (3), 56 (3), 57 (17), 58 (10), 69 (3), 72 (8), 73 (100), 74 (18), 97 (30), 98 (3), 100 (5), 110 (3), 114 (3), 129 (82), 130 (7), 239 (4) [M]+. IR ῡ (cm−1): 3302, 3072, 2964, 2925, 2870, 1641, 1543, 1479, 1450, 1440, 1389, 1359, 1310, 1294, 1262, 1229, 1185, 1140, 1082, 1065, 1038, 933, 890, 852, 829, 781, 762, 748, 691, 679, 670, 649, 615. HRMS (ESI+): calcd. for C13H22NOS [M+H]+ 240.1417, found 240.1427.
N-(tert-Butyl)-4-(4-methoxyphenoxy)-2-methylbutanamide (23). Following the general procedure (condition B), the mixture of 1-(3-Iodobutoxy)-4-methoxybenzene S12 (89.7 mg, 0.293 mmol), DMAP (2.1 mg, 0.0176 mmol), Pd(PPh3)4 (20.3 mg, 0.0176 mmol), Et3N (69 µL, 0.498 mmol), t-butyl isocyanide (36 µL, 0.322 mmol), and bidistilled H2O (211 µL, 11.7 mmol) in dry toluene (1.1 mL) was reacted. After extraction, the crude product was purified by silica FC (PE/Et2O 7:3), delivering 23 as a pale-yellow solid (36 mg, 44% yield). Rf 0.20 (PE/Et2O 7:3, UV, CAM). 1H NMR (400 MHz, CDCl3, 27 °C) δ 6.83 (s, 4H, 4×CHarom), 5.37 (s, 1H, NH), 3.96 (dt, J = 10.1, 5.2 Hz, 1H, OCHH), 3.88 (td, J = 9.2, 4.5 Hz, 1H, OCHH), 3.77 (s, 3H, OCH3), 2.50–2.37 (m, 1H, CH), 2.00 (ddt, J = 14.1, 9.4, 4.7 Hz, 1H, CHHCH), 1.84 (ddt, J = 14.2, 8.9, 5.4 Hz, 1H, CHHCH), 1.27 (s, 9H, C(CH3)3), 1.16 (d, J = 6.9 Hz, 3H, CHCH3). 13C NMR (101 MHz, CDCl3, 27 °C) δ 175.3 (C=O), 154.0 (Cq.), 153.1 (Cq.), 115.4 (2 2×CHarom), 114.8 (2×CHarom), 66.3 (OCH2), 55.8 (OCH3), 51.1 (C(CH3)3), 38.5 (CH), 34.0 (CH2CH), 28.9 (C(CH3)3), 18.0 (CHCH3). GC-MS: tR = 10.125 min (Method A); m/z (%): 39 (4), 41 (17), 42 (3), 44 (6), 55 (9), 56 (5), 57 (35), 58 (8), 73 (3), 77 (4), 81 (3), 83 (3), 100 (100), 101 (6), 109 (9), 124 (7), 137 (3), 156 (29), 157 (3). IR ῡ (cm−1): 3347, 2962, 2926, 2215, 1645, 1538, 1508, 1454, 1361, 1287, 1229, 1184, 1109, 1076, 1030, 991, 970, 912, 882, 819, 727, 694, 627. HRMS (ESI+): calcd. for C16H26NO3 [M+H]+ 280.1907, found 280.1910.
4-(4-Acetamidophenyl)-N-(tert-butyl)-2-methylbutanamide (24). Following the general procedure (condition B), the mixture of N-(4-(3-iodobutyl)phenyl)acetamide S17 (100 mg, 0.315 mmol), DMAP (2.3 mg, 0.0189mmol), Pd(PPh3)4 (21.9 mg, 0.0189 mmol), Et3N (75 µL, 0.536 mmol), t-butyl isocyanide (78 µL, 0.654 mmol), and bidistilled H2O (227 µL, 12.6 mmol) in dry toluene (1.1 mL) was reacted. After extraction, the crude product was purified by silica FC (PE/AcOEt 2:8), delivering 24 as a pale-yellow solid (67 mg, 72% yield). Rf 0.38 (PE/AcOEt 2:8, UV, CAM). 1H NMR (400 MHz, CDCl3, 27 °C) δ 7.42–7.34 (m, 2H, 2×3-CHarom), 7.19–7.00 (m, 3H, 2×2-CHarom + AcNH), 5.16 (bs, 1H, NH), 2.65–2.56 (m, 1H, CHHCH2CH), 2.55–2.47 (m, 1H, CHHCH2CH), 2.16 (s, 3H, CH3CO), 2.04–1.87 (m, 2H, CH + CHHCH), 1.68–1.55 (m, 1H, CHHCH), 1.34 (s, 9H, C(CH3)3), 1.10 (d, J = 6.6 Hz, 3H, CHCH3). 13C NMR (101 MHz, CDCl3, 27 °C) δ 175.5 (CHC=O), 168.2 (CH3C=O), 138.1 (C-Cq.), 135.8 (N-Cq.), 129.0 (2×2-CHarom), 120.2 (2×3-CHarom), 51.1 (C(CH3)3), 41.4 (CH), 35.8 (CH2CH), 33.0 (CH2CH2CH), 29.0 (C(CH3)3), 24.7 (CH3CO), 18.2 (CHCH3). GC-MS: tR = 11.845 min (Method A); m/z (%), 39 (3), 41 (12), 42 (4), 43 (16), 44 (6), 55 (3), 56 (5), 57 (17), 58 (10), 72 (6), 73 (90), 74 (18), 77 (5), 78 (6), 79 (3), 91 (3), 100 (4), 105 (4), 106 (34), 107 (4), 114 (3), 119 (6), 120 (4), 129 (100), 130 (8), 132 (3), 146 (3), 148 (3), 162 (6), 207 (3), 290 (3) [M]+. IR ῡ (cm−1): 3321, 3196, 3129, 3072, 2967, 2922, 2864, 1677, 1644, 1607, 1541, 1514, 1458, 1411, 1390, 1372, 1362, 1317, 1267, 1229, 1121, 1062, 1022, 962, 914, 882, 822, 763, 720, 672, 650, 609. HRMS (ESI+): calcd. for C17H27N2O2 [M+H]+ 291.2067, found 291.2069.
tert-Butyl (4-(4-(tert-butylamino)-3-methyl-4-oxobutyl)phenyl) carbamate (25). Following the general procedure (condition B), the mixture of tert-butyl (4-(3-iodobutyl)phenyl)carbamate S20 (100 mg, 0.266 mmol), DMAP (2 mg, 0.0160 mmol), Pd(PPh3)4 (18.5 mg, 0.0160 mmol), Et3N (63 µL, 0.453 mmol), t-butyl isocyanide (66 µL, 0.586 mmol), and bidistilled H2O (192 µL, 10.7mmol) in dry toluene (1.1 mL) was reacted. After extraction, the crude product was purified by silica FC (PE/Et2O 7:3), delivering 25 as a pale-yellow solid (66 mg, 74% yield). Rf 0.31 (PE/Et2O 7:3, UV, CAM). 1H NMR (400 MHz, CDCl3, 27 °C) δ 7.29–7.24 (m, 2H, 2×CHarom), 7.09 (d, J = 8.1 Hz, 2H, 2×CHarom), 6.47 (bs, 1H, OCONH), 5.18 (bs, 1H, CHCONH), 2.69–2.41 (m, 2H, CH2CH2CH), 2.05–1.86 (m, 2H, CHHCH + CH), 1.70–1.55 (m, 1H, CHHCH), 1.51 (s, 9H, OC(CH3)3), 1.35 (s, 9H, NC(CH3)3), 1.10 (d, J = 6.6 Hz, 3H, CHCH3). 13C NMR (101 MHz, CDCl3, 27 °C) δ 175.6 (CHC=O), 153.0 (OC=ONH), 136.7 (C-Cq), 136.3 (N-Cq), 129.0 (2×CHarom), 118.9 (2×CHarom), 80.5 (OC(CH3)3), 51.2 (NC(CH3)3), 41.3 (CH), 35.9 (CH2CH), 32.9 (CH2CH2CH), 29.0 (NC(CH3)3), 28.5 (OC(CH3)3), 18.2 (CHCH3). GC-MS: the compound degrades during the analysis. A big and broad peak (tR = 11.485 min) is obtained in the chromatograph alongside the product without the Boc- protecting group. tR = 11.485 min (Method C); m/z (%): 39 (13), 40 (5), 41 (25), 42 (8), 43 (8), 44 (51), 45 (5), 51 (3), 53 (3), 55 (6), 56 (11), 57 (13), 58 (9), 59 (3), 65 (3), 69 (3), 72 (5), 73 (100), 74 (17), 75 (4), 77 (7), 78 (5), 79 (4), 91 (4), 96 (4), 100 (4), 105 (3), 106 (33), 107 (5), 118 (3), 119 (11), 120 (4), 129 (52), 130 (5), 132 (3), 133 (4), 135 (8), 146 (4), 147 (8), 191 (4), 193 (9), 194 (3), 207 (39), 208 (6), 209 (5), 221 (5), 248 (10), 249 (4), 267 (3), 281 (11), 282 (4), 283 (3). IR ῡ (cm−1): 3348, 3272, 2965, 2931, 1699, 1651, 1603, 1536, 1478, 1451, 1413, 1391, 1363, 1318, 1246, 1229, 1158, 1089, 1054, 1021, 947, 904, 833, 805, 735, 719, 665, 635, 607. HRMS (ESI+): calcd. for C20H33N2O3 [M+H]+ 349.2486, found 349.2480.
N-(tert-Butyl)-2-methyl-4-(1-methyl-1H-indol-3-yl)butanamide (26). Following the general procedure (condition B), the mixture of 3-(3-iodobutyl)-1-methyl-1H-indole S22 (100 mg, 0.319 mmol), DMAP (2.3 mg, 0.0190 mmol), Pd(PPh3)4 (22.0 mg, 0.0190 mmol), Et3N (78 µL, 0.542 mmol), t-butyl isocyanide (79 µL, 0.703 mmol), and bidistilled H2O (230 µL, 12.8 mmol) in dry toluene (1.1 mL) was reacted. After extraction, the crude product was purified by silica FC (PE/Et2O 6:4), delivering 26 as a pale-yellow solid (48 mg, 53% yield). Rf 0.35 (PE/Et2O 6:4, UV, CAM). 1H NMR (400 MHz, CDCl3, 27 °C) δ 7.59 (d, J = 7.9 Hz, 1H, 4-CH indole), 7.28 (d, J = 8.2 Hz, 1H, 7-CH indole), 7.21 (t, J = 7.2 Hz, 1H, 6-CH indole), 7.09 (t, J = 7.4 Hz, 1H, 5-CH indole), 6.84 (s, 1H, 2-CH indole), 5.17 (s, 1H, NH), 3.74 (s, 3H, N-CH3), 2.82–2.67 (m, 2H, CH2CH2CH), 2.16–1.98 (m, 2H, CHHCH + CH), 1.80–1.69 (m, 1H, CHHCH), 1.34 (s, 9H, C(CH3)3), 1.14 (d, J = 6.6 Hz, 3H, CHCH3). 13C NMR (101 MHz, CDCl3, 27 °C) δ 175.9 (C=O), 137.2 (8-C indole), 128.0 (9-C indole), 126.3 (2-CH indole), 121.7 (6-CH indole), 119.2 (4-CH indole), 118.7 (5-CH indole), 114.7 (3-C indole), 109.3 (7-CH indole), 51.1 (C(CH3)3), 41.7 (CHCH3), 35.1 (CH2CH), 32.7 (NCH3), 29.0 (C(CH3)3), 22.9 (CH2CH2CH), 18.2 (CHCH3). GC-MS: tR = 11.090 min (Method A); m/z (%): 39 (5), 41 (20), 42 (14), 56 (8), 57 (23), 58 (13), 72 (7), 73 (100), 74 (22), 77 (11), 100 (6), 102 (7), 103 (6), 115 (12), 128 (10), 129 (89), 130 (11), 131 (6), 143 (14), 144 (79), 145 (12), 157 (41), 158 (28), 214 (5), 286 (32), 287 (6). IR ῡ (cm−1): 3279, 3082, 2969, 2932, 1642, 1616, 1554, 1470, 1451, 1425, 1361, 1325, 1292, 1265, 1234, 1208, 1177, 1156, 1119, 1090, 1075, 1058, 1045, 1026, 1011, 947, 932, 920, 893, 836, 811, 765, 734, 699, 651, 608. HRMS (ESI+): calcd. for C18H27N2O [M+H]+ 287.2118, found 287.2114.
N-Cyclohexyl-2-methyl-4-(1-methyl-1H-indol-3-yl)butanamide (27). Following the general procedure (condition B), the mixture of 3-(3-iodobutyl)-1-methyl-1H-indole S22 (100 mg, 0.319 mmol), DMAP (2.3 mg, 0.0190 mmol), Pd(PPh3)4 (22.0 mg, 0.0190 mmol), Et3N (78 µL, 0.542 mmol), c-Hex isocyanide (87 µL, 0.703 mmol), and bidistilled H2O (230 µL, 12.8 mmol) in dry toluene (1.1 mL) was reacted. After extraction, the crude product was purified by silica FC (PE/Et2O 6:4), delivering 27 as a pale-yellow solid (38 mg, 38% yield). Rf 0.28 (PE/Et2O 6:4, UV, CAM). 1H NMR (400 MHz, CDCl3, 27 °C) δ 7.58 (d, J = 7.9 Hz, 1H, 4-CH indole), 7.28 (d, J = 8.2 Hz, 1H, 7-CH indole), 7.21 (td, J = 7.0, 0.8 Hz, 1H, 6-CH indole), 7.09 (td, J = 7.4, 1.0 Hz, 1H, 5-CH indole), 6.82 (s, 1H, 2-CH indole), 5.25 (d, J = 7.7 Hz, 1H, NH), 3.85–3.75 (m, 1H, CH c-Hex), 3.73 (s, 3H, NCH3), 2.83–2.67 (m, 2H, CH2CH2CH), 2.24–2.12 (m, 1H, CHCH3), 2.12–2.00 (m, 1H, CHHCH), 1.97–1.85 (m, 2H, 2×CHH c-Hex), 1.82–1.74 (m, 1H, CHHCH), 1.74–1.65 (m, 2H, 2×CHH c-Hex), 1.64–1.54 (m, 1H, CHH c-Hex), 1.45–1.29 (m, 2H, 2×CHH c-Hex), 1.16 (d, J = 6.8 Hz, 3H, CHCH3), 1.14–1.02 (m, 3H, CHH c-Hex + 2×CHH c-Hex). 13C NMR (101 MHz, CDCl3, 27 °C) δ 175.5 (C=O), 137.2 (8-C indole), 128.0 (9-C indole), 126.3 (2-CH indole), 121.6 (6-CH indole), 119.1 (4-CH indole), 118.7 (5-CH indole), 114.6 (3-C indole), 109.3 (7-CH indole), 48.0 (CH c-Hex), 41.2 (CHCH3), 35.0 (CH2CH), 33.5 (2-CH2 c-Hex), 33.3 (6-CH2 c-Hex), 32.7 (NCH3), 25.7 (CH2 c-Hex), 25.0 (2×CH2 c-Hex), 22.9 (CH2CH2CH), 18.2 (CHCH3). GC-MS: tR = 13,240 min (Method A); m/z (%): 39 (4), 41 (16), 42 (9), 43 (7), 44 (10), 54 (5), 55 (18), 56 (11), 57 (4), 67 (8), 72 (3), 73 (7), 74 (100), 75 (4), 77 (8), 82 (6), 83 (4), 98 (3), 102 (4), 103 (4), 115 (7), 117 (3), 126 (10), 127 (3), 128 (6), 129 (3), 130 (3), 131 (4), 143 (9), 144 (50), 145 (7), 155 (38), 156 (6), 157 (21), 158 (19), 159 (3), 312 (13) [M]+. IR ῡ (cm−1): 3677, 3289, 3055, 2927, 2852, 1635, 1543, 1471, 1449, 1424, 1375, 1325, 1249, 1232, 1153, 1130, 1015, 942, 891, 736, 669. HRMS (ESI+): calcd. for C20H29N2O [M+H]+ 313.2274, found 313.2270.
N-(tert-Butyl)-4-((tert-butyldimethylsilyl)oxy)-2-methylbutanamide (28). Following the general procedure (condition B), the mixture of tert-butyl(3-iodobutoxy)dimethylsilane S26 (100 mg, 0.318 mmol), DMAP (2.3 mg, 0.0191 mmol), Pd(PPh3)4 (22 mg, 0.0191 mmol), Et3N (75 µL, 0.541 mmol), t-butyl isocyanide (79 µL, 0.700 mmol), and bidistilled H2O (229 µL, 12.7 mmol) in dry toluene (1.1 mL) was reacted. After extraction, the crude product was purified by silica FC (PE/Et2O 7:3), delivering 28 as a pale-yellow oil (65 mg, 71% yield). Rf 0.52 (PE/Et2O 7:3, basic KMnO4). 1H NMR (400 MHz, CDCl3, 27 °C) δ 5.43 (bs, 1H, NH), 3.69–3.56 (m, 2H, OCH2), 2.40–2.26 (m, H, CH), 1.76 (ddt, J = 13.8, 9.3, 4.8 Hz, 1H, CHHCH), 1.60–1.49 (m, 1H, CHHCH), 1.34 (s, 9H, NH-C(CH3)3), 1.10 (d, J = 6.9 Hz, 3H, CHCH3), 0.90 (s, 9H, Si-C(CH3)3), 0.05 (s, 6H Si(CH3)2). 13C NMR (101 MHz, CDCl3, 27 °C) δ 175.7 (C=O), 60.8 (OCH2), 51.1 (N-C(CH3)3), 38.0 (CH), 37.4 (CH2CH), 29.0 (N-C(CH3)3), 26.1 (Si-C(CH3)3), 18.4 (Si-C(CH3)3), 17.8 (CHCH3), -5.1 (Si-CH3), -5.2 (Si-CH3). GC-MS: Rt 8,035 min (Method A), m/z (%): 39 (6), 41 (27), 42 (5), 43 (4), 44 (4), 45 (6), 47 (5) 55 (10), 56 (11), 57 (33), 58 (16), 59 (12), 61 (4), 72 (3), 73 (52), 74 (89), 75 (42), 76 (6), 77 (3), 84 (3), 85 (4), 88 (4), 89 (5), 99 (3), 100 (12), 101 (6), 114 (7), 115 (9), 116 (4), 129 (19), 130 (3), 156 (5), 157 (4), 174 (100), 175 (13), 176 (4), 215 (9), 216 (5), 230 (33), 231 (7). IR ῡ (cm−1): 3300, 2961, 2930, 2858, 1643, 1552, 1462, 1389, 1361, 1256, 1227, 1089, 1024, 1005, 984, 938, 894, 834, 771, 684, 665. HRMS (ESI+): calcd. for C15H34NO2Si [M+H]+ 288.2353, found 288.2348.
4-(tert-Butylamino)-3-methyl-4-oxobutyl benzoate (30). Following the general procedure (condition B), the mixture of 3-iodobutyl benzoate S28 (100 mg, 0.329 mmol), DMAP (2.4 mg, 0.0197 mmol), Pd(PPh3)4 (23 mg, 0.0197 mmol), Et3N (78 µL, 0.559 mmol), t-butyl isocyanide (82 µL, 0.723 mmol), and bidistilled H2O (237 µL, 13.2 mmol) in dry toluene (1.2 mL) was reacted. After extraction, the crude product was purified by silica FC (PE/Et2O 6:4), delivering 30 as a pale-yellow solid (67 mg, 72% yield). Rf 0.27 (PE/Et2O 6:4, UV, CAM). 1H NMR (400 MHz, CDCl3, 27 °C) δ 8.04 (dd, J = 8.3, 1.2 Hz, 2H, 2× o-CHarom), 7.57 (tt, J = 7.4, 1.3 Hz, 1H, p-CHarom), 7.45 (t, J = 7.8 Hz, 2H, 2× m-CHarom), 5.46 (s, 1H, NH), 4.46–4.25 (m, 2H, OCH2), 2.34–2.21 (m, 1H, CHCH3), 2.10 (ddt, J = 14.4, 8.9, 5.6 Hz, 1H, CHHCH), 1.83 (ddt, J = 11.6, 8.0, 5.8 Hz, 1H, CHHCH), 1.34 (s, 9H, 3×CH3), 1.18 (d, J = 6.8 Hz, 3H, CHCH3). 13C NMR (101 MHz, CDCl3, 27 °C) δ 174.9 (OC=O), 166.8 (NC=O), 133.1 (p-CHarom), 129.7 (2×o-CHarom), 128.5 (2×m-CHarom), 63.2 (C(CH3)3), 51.3 (OCH2), 38.9 (CHCH3), 33.4 (CH2CH2CH), 28.9 (C(CH3)3), 18.3 (CHCH3). GC-MS: tR = 9.940 min (Method A); m/z (%): 39 (4), 41 (21), 42 (7), 43 (3), 44 (4), 51 (8), 55 (9), 56 (31), 57 (37), 58 (39), 72 (4), 73 (13), 74 (3), 77 (33), 78 (3), 79 (4), 99 (4), 100 (43), 101 (3), 105 (100), 106 (8), 122 (3), 123 (22), 129 (25), 140 (7), 155 (3), 178 (3), 262 (5). IR ῡ (cm−1): 3383, 3276, 2971, 2924, 1699, 1665, 1599, 1584, 1518, 1451, 1389, 1362, 1318, 1280, 1224, 1202, 1178, 1141, 1116, 1102, 1087, 1072, 1032, 998, 978, 947, 909, 857, 812, 801, 714, 688, 675. HRMS (ESI+): calcd. for C16H24NO3 [M+H]+ 278.1751, found 278.1746.


 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}