
Molecular Modeling Is Key to Understanding Supramolecular Resorcinarenyl Capsules, Inclusion Complex Formation and Organic Reactions in Nanoconfined Space


Abstract

1. Introduction
2. Discussion
2.1. Atwood Capsule
2.1.1. Capsule Assembly
- Early-stage formation of the hexameric capsule.
- Water molecules.
2.1.2. Formation of Inclusion Complexes
2.1.3. Study of Mechanisms Inside the Atwood Capsule
2.2. Pyrogallol[4]arene
2.2.1. Capsule Assembly
2.2.2. Formation of Inclusion Complexes
2.2.3. Study of Mechanisms Inside the Pyrogallolarenyl Capsule
2.3. Velcrand
2.3.1. Encapsulation of Guests
2.3.2. Co-Encapsulation of Guests
2.4. Octa Acid
2.4.1. Encapsulation of Guests
2.4.2. Studies of Excited States
3. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Filipe, H.A.L.; Loura, L.M.S. Molecular dynamics simulations: Advances and applications. Molecules 2022, 27, 2105. [Google Scholar] [CrossRef] [PubMed]
- Zois, K.P.; Tzeli, D. A critical look at density functional theory in chemistry: Untangling its strengths and weaknesses. Atoms 2024, 12, 65. [Google Scholar] [CrossRef]
- Wang, Y.; Ribeiro, J.M.L.; Tiwary, P. Machine learning approaches for analyzing and enhancing molecular dynamics simulations. Curr. Opin. Struct. Biol. 2020, 61, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Sulimov, V.B.; Kutov, D.C.; Sulimov, A.V. Advances in docking. Curr. Med. Chem. 2020, 26, 7555–7580. [Google Scholar] [CrossRef]
- Aldossary, A.; Campos-Gonzalez-Angulo, J.A.; Pablo-García, S.; Leong, S.X.; Rajaonson, E.M.; Thiede, L.; Tom, G.; Wang, A.; Avagliano, D.; Aspuru-Guzik, A. In silico chemical experiments in the age of AI: From quantum chemistry to machine learning and back. Adv. Mater. 2024, 36, 2402369. [Google Scholar] [CrossRef]
- Frederix, P.W.J.M.; Patmanidis, I.; Marrink, S.J. Molecular simulations of self-assembling bio-inspired supramolecular systems and their connection to experiments. Chem. Soc. Rev. 2018, 47, 3470–3489. [Google Scholar] [CrossRef]
- Calbo, J. Supramolecular polymer chemistry meets computational chemistry: Theoretical simulations on advanced self-assembling chiral materials. Supramol. Chem. 2018, 30, 876–890. [Google Scholar] [CrossRef]
- Moradi, S.; Nowroozi, A.; Shahlaei, M. Shedding light on the structural properties of lipid bilayers using molecular dynamics simulation: A review study. RSC Adv. 2019, 9, 4644–4658. [Google Scholar] [CrossRef]
- Abadie, M.J.M.; Pinteala, M.; Rotaru, A. (Eds.) New Trends in Macromolecular and Supramolecular Chemistry for Biological Applications; Springer: Cham, Switzerland, 2021. [Google Scholar]
- van Lommel, R.; Zhao, J.; de Borggraeve, W.M.; de Proft, F.; Alonso, M. Molecular dynamics based descriptors for predicting supramolecular gelation. Chem. Sci. 2020, 11, 4226–4238. [Google Scholar] [CrossRef]
- Wang, M.; Ma, A.; Wang, H.; Lou, X. Atomic molecular dynamics simulation advances of de novo-designed proteins. Q. Rev. Biophys. 2024, 57, e14. [Google Scholar] [CrossRef]
- Colaço, M.C.; Glitz, V.A.; Jacobs, A.K.; Port, V.C.; Caramori, G.F. Supramolecular chemistry: Exploring the use of electronic structure, molecular dynamics, and machine learning approaches. Eur. J. Org. Chem. 2024, 27, e202400367. [Google Scholar] [CrossRef]
- Cox, C.J.T.; Hale, J.; Molinska, P.; Lewis, J.E.M. Supramolecular and molecular capsules, cages and containers. Chem. Soc. Rev. 2024, 53, 10380–10408. [Google Scholar] [CrossRef] [PubMed]
- Rebek, J., Jr. Hydrogen-Bonded Capsules: Molecular Behavior in Small Spaces; World Scientific: Singapore, 2015. [Google Scholar]
- Kobayashi, K.; Yamanaka, M. Self-assembled capsules based on tetrafunctionalized calix[4]resorcinarene cavitands. Chem. Soc. Rev. 2015, 44, 449–466. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Liu, Q.; Zhou, L.; Sun, H.; Yao, X.; Hu, X.-Y. State-of-the-art and recent progress in resorcinarene-based cavitand. Chin. Chem. Lett. 2023, 34, 108559. [Google Scholar] [CrossRef]
- MacGillivray, L.R.; Atwood, J.L. A chiral spherical molecular assembly held together by 60 hydrogen bonds. Nature 1997, 389, 469–472. [Google Scholar] [CrossRef]
- Li, T.-R.; Huck, F.; Piccini, G.M.; Tiefenbacher, K. Mimicry of the proton wire mechanism of enzymes inside a supramolecular capsule enables β-selective O-glycosylations. Nat. Chem. 2022, 14, 985–994. [Google Scholar] [CrossRef]
- Osman, Z.; Sperni, L.; Fabris, F.; Scarso, A. Supramolecular catalysis in phosphite hydrolysis to H-phosphonate by the resorcinarene hexameric capsule. Tetrahedron Lett. 2024, 139, 154985. [Google Scholar] [CrossRef]
- Némethová, I.; Schmid, D.; Tiefenbacher, K. Supramolecular capsule catalysis enables the exploration of terpenoid chemical space untapped by nature. Angew. Chem. Int. Ed. 2023, 62, e202218625. [Google Scholar] [CrossRef]
- Bordignon, F.; Calmanti, R.; Perosa, A.; Fabris, F.; Scarso, A. Confinement effects in catalysis: Steering the product selectivity in cannabidiol isomerization by the resorcinarene supramolecular capsule. ChemCatChem 2024, 16, e202400278. [Google Scholar] [CrossRef]
- Gambaro, S.; Talotta, C.; Sala, P.D.; Sorienten, A.; De Rosa, M.; Gaeta, C.; Neri, P. Kinetic and thermodynamic modulation of dynamic imine libraries driven by the hexameric resorcinarene capsule. J. Am. Chem. Soc. 2020, 142, 4914–14923. [Google Scholar] [CrossRef]
- Atwood, J.L.; Barbour, L.J.; Jerga, A. Hydrogen-bonded molecular capsules are stable in polar media. Chem. Commun. 2001, 22, 2376–2377. [Google Scholar] [CrossRef] [PubMed]
- Avram, L.; Cohen, Y. The role of water molecules in a resorcinarene capsule as probed by NMR diffusion measurements. Org. Lett. 2002, 4, 4365–4368. [Google Scholar] [CrossRef] [PubMed]
- Avram, L.; Cohen, Y. Spontaneous formation of hexameric resorcinarene capsule in chloroform solution as detected by diffusion NMR. J. Am. Chem. Soc. 2002, 124, 15148–15149. [Google Scholar] [CrossRef] [PubMed]
- Shivanyuk, A.; Rebek, J., Jr. Reversible encapsulation by self-assembling resorcinarene subunits. Proc. Natl. Acad. Sci. USA 2001, 98, 7662–7665. [Google Scholar] [CrossRef]
- Shivanyuk, A.; Rebek, J., Jr. Reversible encapsulation of multiple, neutral guests in hexameric resorcinarene hosts. Chem. Commun. 2001, 2424–2425. [Google Scholar] [CrossRef]
- Avram, L.; Cohen, Y. Discrimination of guests encapsulation in large hexameric molecular capsules in solution: Pyrogallol[4]arene versus resorcin[4]arene capsules. J. Am. Chem. Soc. 2003, 125, 16180–16181. [Google Scholar] [CrossRef]
- Yi, S.; Mileo, E.; Kaifer, A.E. EPR and NMR investigation on the interactions of nitroxide probes with resorcin[4]arene molecular capsules. Org. Lett. 2009, 11, 5690–5693. [Google Scholar] [CrossRef]
- Mileo, E.; Yi, S.; Bhattacharya, P.; Kaifer, A.E. Probing the inner space of resorcinarene molecular capsules with nitroxide guests. Angew. Chem. Int. Ed. 2009, 48, 5337–5340. [Google Scholar] [CrossRef]
- Beyeh, N.K.; Kogej, M.; Åhman, A.; Rissanen, K.; Schalley, C.A. Flying capsules: Mass spectrometric detection of pyrogallarene and resorcinarene hexamers. Angew. Chem. Int. Ed. 2006, 45, 5214–5218. [Google Scholar] [CrossRef]
- Jans, A.C.H.; Gómez-Suárez, A.; Nolan, S.P.; Reek, J.N.H. A switchable gold catalyst by encapsulation in a self-assembled cage. Chem. Eur. J. 2016, 22, 14836–14839. [Google Scholar] [CrossRef]
- Philip, I.E.; Kaifer, A.E. Electrochemically driven formation of a molecular capsule around the ferrocenium ion. J. Am. Chem. Soc. 2002, 124, 12678–12679. [Google Scholar] [CrossRef] [PubMed]
- Horiuchi, S.; Yamaguchi, T.; Tessarolo, J.; Tanaka, H.; Sakuda, E.; Arikawa, Y.; Meggers, E.; Clever, G.H.; Umakoshi, K. Symmetry-breaking host-guest assembly in a hydrogen-bonded supramolecular system. Nat. Commun. 2023, 14, 155. [Google Scholar] [CrossRef] [PubMed]
- Philip, I.; Kaifer, A.E. Noncovalent encapsulation of cobaltocenium inside resorcinarene molecular capsules. J. Org. Chem. 2005, 70, 1558–1564. [Google Scholar] [CrossRef]
- Bianchini, G.; Scarso, A.; Sorella, G.L.; Strukul, G. Switching the activity of a photoredox catalyst through reversible encapsulation and release. Chem. Commun. 2012, 48, 12082. [Google Scholar] [CrossRef]
- Zhang, T.; Le Corre, L.; Reinaud, O.; Colasson, B. A promising approach for controlling the second coordination sphere of biomimetic metal complexes: Encapsulation in a dynamic hydrogen-bonded capsule. Chem. Eur. J. 2021, 27, 434–443. [Google Scholar] [CrossRef]
- Horiuchi, S.; Tanaka, H.; Sakuda, E.; Arikawa, Y.; Umakoshi, K. Encapsulation and enhanced luminescence properties of IrIII complexes within a hexameric self-assembled capsule. Chem. Eur. J. 2016, 22, 17533–17537. [Google Scholar] [CrossRef]
- Adriaenssens, L.; Escribano-Cuesta, A.; Homs, A.; Echavarren, A.M.; Ballester, P. Encapsulation studies of cationic gold complexes within a self-assembled hexameric resorcin[4]arene capsule. Eur. J. Org. Chem. 2013, 2013, 1494–1500. [Google Scholar] [CrossRef]
- Hkiri, S.; Steinmetz, M.; Schurhammer, R.; Sémeril, D. Encapsulated neutral ruthenium catalyst for substrate-selective oxidation of alcohols. Chem. Eur. J. 2022, 28, e202201887. [Google Scholar] [CrossRef]
- Jongkind, L.J.; Rahimi, M.; Poole, D., III; Ton, S.J.; Fogg, D.E.; Reek, J.N.H. Protection of ruthenium olefin metathesis catalysts by encapsulation in a self-assembled resorcinarene capsule. ChemCatChem 2020, 12, 4019–4023. [Google Scholar] [CrossRef]
- Cavarzan, A.; Scarso, A.; Sgarbossa, P.; Strukul, G.; Reek, J.N.H. Supramolecular control on chemo- and regioselectivity via encapsulation of (NHC)-Au catalyst within a hexameric self-assembled host. J. Am. Chem. Soc. 2011, 133, 2848–2851. [Google Scholar] [CrossRef]
- Cavarzan, A.; Reek, J.N.H.; Trentin, F.; Scarso, A.; Strukul, G. Substrate selectivity in the alkyne hydration mediated by NHC-Au(I) controlled by encapsulation of the catalyst within a hydrogen bonded hexameric host. Catal. Sci. Technol. 2013, 3, 2898–2901. [Google Scholar] [CrossRef]
- La Manna, P.; De Rosa, M.; Talotta, C.; Gaeta, C.; Soriente, A.; Floresta, G.; Rescifina, A.; Neri, P. The hexameric resorcinarene capsule as an artificial enzyme: Ruling the regio and stereochemistry of a 1,3-dipolar cycloaddition between nitrones and unsaturated aldehydes. Org. Chem. Front. 2018, 5, 827–837. [Google Scholar] [CrossRef]
- La Manna, P.; De Rosa, M.; Talotta, C.; Rescifina, A.; Floresta, G.; Soriente, A.; Gaeta, C.; Neri, P. Synergic Interplay between halogen bonding and hydrogen bonding in the activation of a neutral substrate in a nanoconfined space. Angew. Chem. Int. Ed. 2020, 59, 811–818. [Google Scholar] [CrossRef]
- Bräuer, M.; Zhang, Q.; Tiefenbacher, K. Iminium catalysis inside a self-assembled supramolecular capsule: Modulation of enantiomeric excess. Angew. Chem. Int. Ed. 2016, 55, 7698–7701. [Google Scholar] [CrossRef]
- Capelli, R.; Piccini, G. Atomistic insights into hydrogen-bonded supramolecular capsule self-assembly dynamics. J. Phys. Chem. C 2024, 128, 635–641. [Google Scholar] [CrossRef]
- Trugilho, L.F.; Rizzi, L.G. Microcanonical characterization of first-order phase transitions in a generalized model for aggregation. J. Stat. Phys. 2022, 186, 40. [Google Scholar] [CrossRef]
- Laio, A.; Parrinello, M. Escaping free-energy minima. Proc. Natl. Acad. Sci. USA 2002, 99, 12562–12566. [Google Scholar] [CrossRef]
- Barducci, A.; Bussi, G.; Parrinello, M. Well-tempered metadynamics: A smoothly converging and tunable free-energy method. Phys. Rev. Lett. 2008, 100, 020603. [Google Scholar] [CrossRef]
- Tribello, G.A.; Bonomi, M.; Branduardi, D.; Camilloni, C.; Bussi, G. PLUMED 2: New feathers for an old bird. Comput. Phys. Commun. 2014, 185, 604–613. [Google Scholar] [CrossRef]
- PLUMED Consortium. Promoting transparency and reproducibility in enhanced molecular simulations. Nat. Methods 2019, 16, 670–673. [Google Scholar] [CrossRef]
- Shivanyuk, A.; Rebek, J., Jr. Assembly of resorcinarene capsules in wet solvents. J. Am. Chem. Soc. 2003, 125, 3432–3433. [Google Scholar] [CrossRef] [PubMed]
- Evan-Salem, T.; Baruch, I.; Avram, L.; Cohen, Y.; Palmer, L.C.; Rebek, J., Jr. Resorcinarenes are hexameric capsules in solution. Proc. Natl. Acad. Sci. USA 2006, 103, 12296–12300. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, M.; Shivanyuk, A.; Rebek, J., Jr. Kinetics and thermodynamics of hexameric capsule formation. J. Am. Chem. Soc. 2004, 126, 2939–2943. [Google Scholar] [CrossRef] [PubMed]
- Merget, S.; Catti, L.; Piccini, G.; Tiefenbacher, K. Requirements for terpene cyclizations inside the supramolecular resorcinarene capsule: Bound water and its protonation determine the catalytic activity. J. Am. Chem. Soc. 2020, 142, 4400–4410. [Google Scholar] [CrossRef]
- Zhang, Q.; Tiefenbacher, K. Hexameric resorcinarene capsule is a Brønsted acid: Investigation and application to synthesis and catalysis. J. Am. Chem. Soc. 2013, 135, 16213–16219. [Google Scholar] [CrossRef]
- La Manna, P.; Talotta, C.; Floresta, G.; De Rosa, M.; Soriente, A.; Rescifina, A.; Gaeta, G.; Neri, P. Mild Friedel-Crafts reactions inside a hexameric resorcinarene capsule: C- Cl bond activation through hydrogen bonding to bridging water molecules. Angew. Chem. Int. Ed. 2018, 57, 5423–5428. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Stewart, J.J.P. Optimization of parameters for semiempirical methods V: Modification of NDDO approximations and application to 70 elements. J. Mol. Model. 2007, 13, 1173–1213. [Google Scholar] [CrossRef]
- Katiyar, A.; Sovierzoski, J.C.F.; Calio, P.B.; Vartia, A.A.; Thompson, W.H. Water plays a diverse role in a hydrogen-bonded, hexameric supramolecular assembly. Chem. Commun. 2019, 55, 6591–6594. [Google Scholar] [CrossRef]
- Katiyar, A.; Sovierzoski, J.C.F.; Calio, P.B.; Vartia, A.A.; Thompson, W.H. Water plays a dynamical role in a hydrogen-bonded, hexameric supramolecular assembly. Phys. Chem. Chem. Phys. 2020, 22, 6167–6175. [Google Scholar] [CrossRef]
- Plimpton, S. Fast parallel algorithms for short-range molecular dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef]
- Thompson, A.P.; Aktulga, H.M.; Berger, R.; Bolintineau, D.S.; Brown, W.M.; Crozier, P.S.; in’t Veld, P.J.; Kohlmeyer, A.; Moore, S.G.; Nguyen, T.D.; et al. LAMMPS—A flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales. Comput. Phys. Commun. 2022, 271, 109171. [Google Scholar] [CrossRef]
- Salomon-Ferrer, R.; Götz, A.W.; Poole, D.; Le Grand, S.; Walker, R.C. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 2. explicit solvent particle mesh Ewald. J. Chem. Theory Comput. 2013, 9, 3878–3888. [Google Scholar] [CrossRef] [PubMed]
- Poole, D.A., III; Mathew, S.; Reek, J.N.H. Just add water: Modulating the structure-derived acidity of catalytic hexameric resorcinarene capsules. J. Am. Chem. Soc. 2021, 143, 16419–16427. [Google Scholar] [CrossRef] [PubMed]
- Slovak, S.; Avram, L.; Cohen, Y. Encapsulated or not encapsulated? Mapping alcohol sites in hexameric capsules of resorcin[4]arenes in solution by diffusion NMR spectroscopy. Angew. Chem. Int. Ed. 2010, 49, 428–431. [Google Scholar] [CrossRef]
- Slovak, S.; Cohen, Y. The effect of alcohol structures on the interaction mode with the hexameric capsule of resorcin[4]arene. Chem. Eur. J. 2012, 18, 8515–8520. [Google Scholar] [CrossRef]
- Payne, R.M.; Oliver, C.L. A propanol-seamed C-methylcalix[4]resorcinarene hexamer accessible via solution crystallization, liquid-assisted grinding and vapour sorption. CrystEngComm 2018, 20, 1919–1922. [Google Scholar] [CrossRef]
- Ugono, O.; Holman, K.T. An achiral form of the hexameric resorcin[4]arene capsule sustained by hydrogen bonding with alcohols. Chem. Commun. 2006, 2144–2146. [Google Scholar] [CrossRef]
- Chwastek, M.; Cmoch, P.; Szumna, A. Anion-based self-assembly of resorcin[4]arenes and pyrogallol[4]arenes. J. Am. Chem. Soc. 2022, 144, 5350–5358. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A., Jr.; Vreven, T.; Kudin, K.N.; Burant, J.C.; et al. Gaussian 03, Revision A.1; Gaussian, Inc.: Pittsburgh, PA, USA, 2003. [Google Scholar]
- Curtiss, L.A.; McGrath, M.P.; Blaudeau, J.-P.; Davis, N.E.; Binning, R.C., Jr.; Radom, L. Extension of Gaussian-2 theory to molecules containing third-row atoms Ga–Kr. J. Chem. Phys. 1995, 103, 6104–6113. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Sierka, M.; Hogekamp, A.; Ahlrichs, R. Fast evaluation of the Coulomb potential for electron densities using multipole accelerated resolution of identity approximation. J. Chem. Phys. 2003, 118, 9136–9148. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Ahlrichs, R.; Bär, M.; Häser, M.; Horn, H.; Kölmel, C. Electronic structure calculations on workstation computers: The program system turbomole. Chem. Phys. Lett. 1989, 162, 165–169. [Google Scholar] [CrossRef]
- Zhang, Q.; Catti, L.; Kaila, V.R.I.; Tiefenbacher, K. To catalyze or not to catalyze: Elucidation of the subtle differences between the hexameric capsules of pyrogallolarene and resorcinarene. Chem. Sci. 2017, 8, 1653–1657. [Google Scholar] [CrossRef]
- WebLab ViewerPro Version 4.0, Molecular Simulation, Inc.: San Diego, CA, USA, 2000.
- Shivanyuk, A. Nanoencapsulation of calix[4]arene inclusion complexes. J. Am. Chem. Soc. 2007, 129, 14196–14199. [Google Scholar] [CrossRef]
- PCMODEL, version 7.50.00; Serena Software: Bloomington, IN, USA, 2000.
- Horiuchi, S.; Matsuo, C.; Sakuda, E.; Arikawa, Y.; Clever, G.H.; Umakoshi, K. Anion-mediated encapsulation-induced emission enhancement of an IrIII complex within a resorcin[4]arene hexameric capsule. Dalton Trans. 2020, 49, 8472–8477. [Google Scholar] [CrossRef]
- Uratani, H.; Horiuchi, S. Encapsulation-induced hypsochromic shift of emission properties from a cationic Ir(III) complex in a hydrogen-bonded organic cage: A theoretical study. J. Chem. Phys. 2024, 161, 201101. [Google Scholar] [CrossRef]
- Venkatachalam, C.M.; Jiang, X.; Oldfield, T.; Waldman, M. LigandFit: A novel method for the shape-directed rapid docking of ligands to protein active sites. J. Mol. Graph. Model. 2003, 21, 289–307. [Google Scholar] [CrossRef]
- Mecozzi, S.; Rebek, J., Jr. The 55 % solution: A formula for molecular recognition in the liquid state. Chem. Eur. J. 1998, 4, 1016–1022. [Google Scholar] [CrossRef]
- Diaz, D.E.; Quist, D.A.; Herzog, A.E.; Schaefer, E.W.; Kipouros, I.; Bhadra, M.; Solomon, E.I.; Karlin, K.D. Impact of intramolecular hydrogen bonding on the reactivity of cupric superoxide complexes with O-H and C-H substrates. Angew. Chem. Int. Ed. 2019, 58, 17572–17576. [Google Scholar] [CrossRef] [PubMed]
- Ehudin, M.A.; Schaefer, A.W.; Adam, S.M.; Quist, D.A.; Diaz, D.E.; Tang, J.A.; Solomon, E.I.; Karlin, K.D. Influence of intramolecular secondary sphere hydrogen-bonding interactions on cytochrome c oxidase inspired low-spin heme-peroxo-copper complexes. Chem. Sci. 2019, 10, 2893–2905. [Google Scholar] [CrossRef] [PubMed]
- Connolly, M.L. The molecular surface package. J. Mol. Graph. 1993, 11, 139–141. [Google Scholar] [CrossRef] [PubMed]
- Lech, L.M.; Freiser, B.S.; Gord, J.R. Heteronuclear diatomic transition-metal cluster ions in the gas phase: Reactions of ScFe+ with hydrocarbons. J. Am. Chem. Soc. 1989, 111, 8588–8592. [Google Scholar] [CrossRef]
- Fortman, G.C.; Poater, A.; Levell, J.W.; Gaillard, S.; Slawin, A.M.Z.; Samuel, I.D.W.; Cavallo, L.; Nolan, S.P. A versatile gold synthon for acetylene C-H bond activation. Dalton Trans. 2010, 39, 10382–10390. [Google Scholar] [CrossRef]
- Case, D.A.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E., III; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Ghoreishi, D.; Gilson, M.K.; et al. AMBER 18; University of California: San Francisco, CA, USA, 2018. [Google Scholar]
- Gambaro, S.; De Rosa, M.; Soriente, A.; Talotta, C.; Floresta, G.; Rescifina, A.; Gaeta, C.; Neri, P. A hexameric resorcinarene capsule as a hydrogen bonding catalyst in the conjugate addition of pyrroles and indoles to nitroalkenes. Org. Chem. Front. 2019, 6, 2339–2347. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. GaussView 5.0; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Pratihar, S.; Roy, S. Nucleophilicity and site selectivity of commonly used arenes and heteroarenes. J. Org. Chem. 2010, 75, 4957–4963. [Google Scholar] [CrossRef]
- Kędziorek, M.; Mayer, P.; Mayr, H. Nucleophilic reactivities of azulene and fulvenes. Eur. J. Org. Chem. 2009, 2009, 1202–1206. [Google Scholar] [CrossRef]
- Zhu, J.; Pérez, M.; Stephan, D.W. C-C Coupling of benzyl fluorides catalyzed by an electrophilic phosphonium cation. Angew. Chem. Int. Ed. 2016, 55, 8448–8451. [Google Scholar] [CrossRef]
- Schmuck, C.; Rupprecht, D. The synthesis of highly functionalized pyrroles: A challenge in regioselectivity and chemical reactivity. Synthesis 2007, 20, 3095–3110. [Google Scholar] [CrossRef]
- Iuliano, V.; Talotta, C.; De Rosa, M.; Soriente, A.; Neri, P.; Rescifina, A.; Floresta, G.; Gaeta, C. Insights into the Friedel-Crafts benzoylation of N-methylpyrrole inside the confined space of the self-assembled resorcinarene capsule. Org. Lett. 2023, 25, 6464–6468. [Google Scholar] [CrossRef] [PubMed]
- De Rosa, M.; Gambaro, S.; Soriente, A.; Della Sala, P.; Iuliano, V.; Talotta, C.; Gaeta, C.; Rescifina, A.; Neri, P. Carbocation catalysis in confined space: Activation of trityl chloride inside the hexameric resorcinarene capsule. Chem. Sci. 2022, 13, 8618–8625. [Google Scholar] [CrossRef] [PubMed]
- Ferrino, G.; De Rosa, M.; Della Sala, P.; Gaeta, C.; Talotta, C.; Soriente, A.; Cao, Z.; Maity, B.; Cavallo, L.; Neri, P. The resorcinarene hexameric capsule as a supramolecular photoacid to trigger olefin hydroarylation in confined space. Chem. Eur. J. 2024, 30, e202303678. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Te Velde, G.; Bickelhaupt, F.M.; Baerends, E.J.; Fonseca Guerra, C.; van Gisbergen, S.J.A.; Snijders, J.G.; Ziegler, T. Chemistry with ADF. J. Comput. Chem. 2001, 22, 931–967. [Google Scholar] [CrossRef]
- Bannwarth, C.; Grimme, S. A simplified time-dependent density functional theory approach for electronic ultraviolet and circular dichroism spectra of very large molecules. Comput. Theor. Chem. 2014, 1040–1041, 45–53. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Weinhold, F.; Landis, C. A natural Bond Orbital Donor-Acceptor Perspective; Cambridge University Press: Cambridge, UK, 2005. [Google Scholar]
- Gerkensmeier, T.; Iwanek, W.; Agena, C.; Fröhlich, R.; Kotila, S.; Näther, C.; Mattay, J. Self-assembly of 2,8,14,20-tetraisobutyl-5,11,17,23-tetrahydroxyresorc[4]arene. Eur. J. Org. Chem. 1999, 1999, 2257–2262. [Google Scholar] [CrossRef]
- Avram, L.; Cohen, Y. Hexameric capsules of lipophilic pyrogallolarene and resorcinarene in solutions as probed by diffusion NMR: one hydroxyl makes the difference. Org. Lett. 2003, 5, 3329–3332. [Google Scholar] [CrossRef]
- Avram, L.; Cohen, Y. Self-recognition, structure, stability, and guest affinity of pyrogallol[4]arene and resorcin[4]arene capsules in solution. J. Am. Chem. Soc. 2004, 126, 11556–11563. [Google Scholar] [CrossRef]
- Cave, G.W.V.; Antesberger, J.; Barbour, L.J.; McKinlay, R.M.; Atwood, J.L. Inner core structure responds to communication between nanocapsule walls. Angew. Chem. Int. Ed. 2004, 43, 5263–5266. [Google Scholar] [CrossRef]
- Avram, L.; Cohen, Y. Molecules at close range: Encapsulated solvent molecules in Pyrogallol[4]arene hexameric capsules. Org. Lett. 2006, 8, 219–222. [Google Scholar] [CrossRef] [PubMed]
- Guralnik, V.; Avram, L.; Cohen, Y. Unique organization of solvent molecules within the hexameric capsules of pyrogallol[4]arene in solution. Org. Lett. 2014, 16, 5592–5595. [Google Scholar] [CrossRef] [PubMed]
- Chapin, J.C.; Kvasnica, M.; Purse, B.W. Molecular encapsulation in pyrogallolarene hexamers under nonequilibrium conditions. J. Am. Chem. Soc. 2012, 134, 15000–15009. [Google Scholar] [CrossRef]
- Journey, S.N.; Teppang, K.L.; Garcia, C.A.; Brim, S.A.; Onofrei, D.; Addison, J.B.; Holland, G.P.; Purse, B.W. Mechanically induced pyrogallol[4]arene hexamer assembly in the solid state extends the scope of molecular encapsulation. Chem. Sci. 2017, 8, 7737–7745. [Google Scholar] [CrossRef]
- HyperChem Professional 8.0; Hypercube, Inc.: Gainesville, FL, USA, 2007.
- Dewar, M.J.S.; Zoebisch, E.G.; Healy, E.F.; Stewart, J.J.P. Development and use of quantum mechanical molecular models. 76. AM1: A new general purpose quantum mechanical molecular model. J. Am. Chem. Soc. 1985, 107, 3902–3909. [Google Scholar] [CrossRef]
- Fischer, E. Einfluss der Configuration auf die Wirkung der Enzyme. Berichte Dtsch. Chem. Ges. 1894, 27, 2985–2993. [Google Scholar] [CrossRef]
- Dalgarno, S.J.; Szabo, T.; Siavosh-Haghighi, A.; Deakyne, C.A.; Adams, J.E.; Atwood, J.L. Exploring the limits of encapsulation within hexameric pyrogallol[4]arene nano-capsules. Chem. Commun. 2009, 1339–1341. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.J.A.; Vreven, T.; Kudin, K.N.; Burant, J.C.; et al. Gaussian 03 Revision C.02; Gaussian, Inc.: Wallingford, CT, USA, 2004. [Google Scholar]
- Dalgarno, S.J.; Tucker, S.A.; Bassil, D.B.; Atwood, J.L. Fluorescent guest molecules report ordered inner phase of host capsules in solution. Science 2005, 309, 2037–2039. [Google Scholar] [CrossRef]
- Yariv-Shoushan, S.; Cohen, Y. Encapsulated or not encapsulated? Ammonium salts can be encapsulated in hexameric capsules of pyrogallol[4]arene. Org. Lett. 2016, 18, 936–939. [Google Scholar] [CrossRef]
- La Manna, P.; Talotta, C.; Gaeta, C.; Cohen, Y.; Slovak, S.; Rescifina, A.; Sala, P.D.; De Rosa, M.; Soriente, A.; Neri, P. Supramolecular catalysis in confined space: Making the pyrogallol[4]arene capsule catalytically active in non-competitive solvent. Org. Chem. Front. 2022, 9, 2453–2463. [Google Scholar] [CrossRef]
- Moran, J.R.; Ericson, J.L.; Dalcanale, E.; Bryant, J.A.; Knobler, C.B.; Cram, D.J. Vases and kites as cavitands. J. Am. Chem. Soc. 1991, 113, 5707–5714. [Google Scholar] [CrossRef]
- Cram, D.J.; Choi, H.J.; Bryant, J.A.; Knobler, C.B. Host-guest complexation. 62. Solvophobic and entropic driving forces for forming velcraplexes, which are 4-fold, lock-key dimers in organic media. J. Am. Chem. Soc. 1992, 114, 7748–7765. [Google Scholar] [CrossRef]
- Heinz, T.; Rudkevich, D.M.; Rebek, J., Jr. Pairwise selection of guests in a cylindrical molecular capsule of nanometre dimensions. Nature 1998, 394, 764–766. [Google Scholar] [CrossRef]
- Mohamadi, F.; Richards, N.G.J.; Guida, W.C.; Liskamp, R.; Lipton, M.; Caufield, C.; Chang, G.; Hendrickson, T.; Still, W.C. Macromodel—An integrated software system for modeling organic and bioorganic molecules using molecular mechanics. J. Comput. Chem. 1990, 11, 440–467. [Google Scholar] [CrossRef]
- Ebbing, M.H.K.; Villa, M.-J.; Valpuesta, J.-M.; Prados, P.; de Mendoza, J. Resorcinarenes with 2-benzimidazolone bridges: Self-aggregation, self-assembled dimeric capsules, and guest encapsulation. Proc. Natl. Acad. Sci. USA 2002, 99, 4962–4966. [Google Scholar] [CrossRef]
- Ajami, D.; Rebek, J., Jr. Expanded capsules with reversibly added spacers. J. Am. Chem. Soc. 2006, 128, 5314–5315. [Google Scholar] [CrossRef]
- Ajami, D.; Rebek, J., Jr. More chemistry in small spaces. Acc. Chem. Res. 2013, 46, 990–999. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. Density functionals with broad applicability in chemistry. Acc. Chem. Res. 2008, 41, 157–167. [Google Scholar] [CrossRef]
- Ajami, D.; Rebek, J., Jr. Gas behavior in self-assembled capsules. Angew. Chem. Int. Ed. 2008, 47, 6059–6061. [Google Scholar] [CrossRef]
- Nicholls, A.; Sharp, K.A.; Honig, B. Protein folding and association: Insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins Struct. Funct. Bioinf. 1991, 11, 281–296. [Google Scholar] [CrossRef] [PubMed]
- Kitaigorodskii, A.I. Molecular Crystals and Molecules, 1st ed.; Academic Press: San Diego, CA, USA, 1973. [Google Scholar]
- Scarso, A.; Trembleau, L.; Rebek, J., Jr. Encapsulation induces helical folding of alkanes. Angew. Chem. Int. Ed. 2003, 42, 5499–5502. [Google Scholar] [CrossRef] [PubMed]
- Scarso, A.; Trembleau, L.; Rebek, J., Jr. Helical folding of alkanes in a self-assembled, cylindrical capsule. J. Am. Chem. Soc. 2004, 126, 13512–13518. [Google Scholar] [CrossRef] [PubMed]
- Ajami, D.; Iwasawa, T.; Rebek, J., Jr. Experimental and computational probes of the space in a self-assembled capsule. Proc. Natl. Acad. Sci. USA 2006, 103, 8934–8936. [Google Scholar] [CrossRef]
- Gavette, J.V.; Zhang, K.-D.; Ajami, D.; Rebek, J., Jr. Folded alkyl chains in water-soluble capsules and cavitands. Org. Biomol. Chem. 2014, 12, 6561–6563. [Google Scholar] [CrossRef]
- Tiefenbacher, K.; Ajami, D.; Rebek, J., Jr. Self-assembled capsules of unprecedented shapes. Angew. Chem. Int. Ed. 2011, 50, 12003–12007. [Google Scholar] [CrossRef]
- Wang, J.; Shi, X.; Cao, W. Theoretical studies on co-encapsulation of two different guests inside a cylindrical capsular host. Russ. J. Appl. Chem. 2015, 88, 682–686. [Google Scholar] [CrossRef]
- Wesolowski, T.A.; Parisel, O.; Ellinger, Y.; Weber, J. Comparative study of benzene···X (X = O2, N2, CO) complexes using density functional theory: the importance of an accurate exchange-correlation energy density at high reduced density gradients. J. Phys. Chem. A 1997, 101, 7818–7825. [Google Scholar] [CrossRef]
- Tzeli, D.; Petsalakis, I.D.; Theodorakopoulos, G. Compression in encapsulated carboxylic acid homodimers. Chem. Phys. Lett. 2013, 573, 48–55. [Google Scholar] [CrossRef]
- Tzeli, D.; Theodorakopoulos, G.; Petsalakis, I.D.; Ajami, D.; Rebek, J., Jr. Theoretical study of hydrogen bonding in homodimers and heterodimers of amide, boronic acid, and carboxylic acid, free and in encapsulation complexes. J. Am. Chem. Soc. 2011, 133, 16977–16985. [Google Scholar] [CrossRef]
- Xantheas, S.S. On the importance of the fragment relaxation energy terms in the estimation of the basis set superposition error correction to the intermolecular interaction energy. J. Chem. Phys. 1996, 104, 8821–8824. [Google Scholar] [CrossRef]
- Ajami, D.; Tolstoy, P.M.; Dube, H.; Odermatt, S.; Koeppe, B.; Guo, J.; Limbach, H.; Rebek, J., Jr. Encapsulated carboxylic acid dimers with compressed hydrogen bonds. Angew. Chem. Int. Ed. 2011, 50, 528–531. [Google Scholar] [CrossRef] [PubMed]
- Tzeli, D.; Petsalakis, I.D.; Theodorakopoulos, G.; Ajami, D.; Jiang, W.; Rebek, J., Jr. Encapsulated hydrogen-bonded dimers of amide and carboxylic acid. Chem. Phys. Lett. 2012, 548, 55–59. [Google Scholar] [CrossRef]
- Tzeli, D.; Petsalakis, I.D.; Theodorakopoulos, G.; Ajami, D.; Rebek, J., Jr. Theoretical study of free and encapsulated carboxylic acid and amide dimers. Int. J. Quantum Chem. 2013, 113, 734–739. [Google Scholar] [CrossRef]
- Zhang, K.-D.; Ajami, D.; Rebek, J., Jr. Hydrogen-bonded capsules in water. J. Am. Chem. Soc. 2013, 135, 18064–18066. [Google Scholar] [CrossRef]
- Dapprich, S.; Komáromi, I.; Byun, K.S.; Morokuma, K.; Frisch, M.J. A new ONIOM implementation in Gaussian98. Part I. The calculation of energies, gradients, vibrational frequencies and electric field derivatives. J. Mol. Struct. Theochem 1999, 461–462, 1–21. [Google Scholar] [CrossRef]
- Vreven, T.; Morokuma, K.; Farkas, O.; Schlegel, H.B.; Frisch, M.J. Geometry optimization with QM/MM, ONIOM, and other combined methods. I. Microiterations and constraints. J. Comput. Chem. 2003, 24, 760–769. [Google Scholar] [CrossRef]
- Vreven, T.; Morokuma, K. Hybrid methods: ONIOM(QM:MM) and QM/MM. Annu. Rep. Comput. Chem. 2006, 2, 35–51. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. A new local density functional for main-group thermochemistry, transition metal bonding, thermochemical kinetics, and noncovalent interactions. J. Chem. Phys. 2006, 125, 194101. [Google Scholar] [CrossRef]
- Ajami, D.; Dube, H.; Rebek, J., Jr. Boronic acid hydrogen bonding in encapsulation complexes. J. Am. Chem. Soc. 2011, 133, 9689–9691. [Google Scholar] [CrossRef]
- Porel, M.; Jayaraj, N.; Kaanumalle, L.S.; Maddipatla, M.V.S.N.; Parthasarathy, A.; Ramamurthy, V. Cavitand octa acid forms a nonpolar capsuleplex dependent on the molecular size and hydrophobicity of the guest. Langmuir 2009, 25, 3473–3481. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Dwyer, T.; Bukannan, H.; Blackmon, O.; Delpo, C.; Barnett, J.W.; Gibb, B.C.; Ashbaugh, H.S. Pressure induced wetting and dewetting of the nonpolar pocket of deep-cavity cavitands in water. J. Phys. Chem. B 2020, 124, 4781–4792. [Google Scholar] [CrossRef] [PubMed]
- Ashbaugh, H.S.; Gibb, B.C.; Suating, P. Cavitand complexes in aqueous solution: Collaborative experimental and computational studies of the wetting, assembly, and function of nanoscopic bowls in water. J. Phys. Chem. B 2021, 125, 3253–3268. [Google Scholar] [CrossRef]
- Meyer, E.A.; Castellano, R.K.; Diederich, F. Interactions with aromatic rings in chemical and biological recognition. Angew. Chem. Int. Ed. 2003, 42, 1210–1250. [Google Scholar] [CrossRef]
- Jayaraj, N.; Zhao, Y.; Parthasarathy, A.; Porel, M.; Liu, R.S.H.; Ramamurthy, V. Nature of supramolecular complexes controlled by the structure of the guest molecules: Formation of octa acid based capsuleplex and cavitandplex. Langmuir 2009, 25, 10575–10586. [Google Scholar] [CrossRef]
- Caldararu, O.; Olsson, M.A.; Riplinger, C.; Neese, F.; Ryde, U. Binding free energies in the SAMPL5 octa-acid host-guest challenge calculated with DFT-D3 and CCSD (T). J. Comput. Aided Mol. Des. 2017, 31, 87–106. [Google Scholar] [CrossRef]
- Kulasekharan, R.; Jayaraj, N.; Porel, M.; Choudhury, R.; Sundaresan, A.K.; Parthasarathy, A.; Ottaviani, M.F.; Jockusch, S.; Turro, N.J.; Ramamurthy, V. Guest rotations within a capsuleplex probed by NMR and EPR techniques. Langmuir 2010, 26, 6943–6953. [Google Scholar] [CrossRef]
- Choudhury, R.; Barman, A.; Prabhakar, R.; Ramamurthy, V. Hydrocarbons depending on the chain length and head group adopt different conformations within a water-soluble nanocapsule: 1H NMR and molecular dynamics studies. J. Phys. Chem. B 2013, 117, 398–407. [Google Scholar] [CrossRef]
- Allinger, N.L. Conformational analysis. 130. MM2. A hydrocarbon force field utilizing V1 and V2 torsional terms. J. Am. Chem. Soc. 1977, 99, 8127–8134. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef] [PubMed]
- Kaminski, G.A.; Friesner, R.A.; Tirado-Rives, J.; Jorgensen, W.L. Evaluation and reparametrization of the OPLS-AA force field for proteins via comparison with accurate quantum chemical calculations on peptides. J. Phys. Chem. B 2001, 105, 6474–6487. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Tirado-Rives, J. The OPLS [optimized potentials for liquid simulations] potential functions for proteins, energy minimizations for crystals of cyclic peptides and crambin. J. Am. Chem. Soc. 1988, 110, 1657–1666. [Google Scholar] [CrossRef] [PubMed]
- Tzanov, A.T.; Cuendet, M.A.; Tuckerman, M.E. How accurately do current force fields predict experimental peptide conformations? An adiabatic free energy dynamics study. J. Phys. Chem. B 2014, 118, 6539–6552. [Google Scholar] [CrossRef]
- Cendejas, K.; Parker, H.E.; Molina, D.; Choudhury, R. Supramolecular self-assembly of water-soluble cavitands: Investigated by molecular dynamics simulation. J. Incl. Phenom. Macrocycl. Chem. 2017, 89, 199–205. [Google Scholar] [CrossRef]
- Mikulskis, P.; Cioloboc, D.; Andrejić, M.; Khare, S.; Brorsson, J.; Genheden, S.; Mata, R.A.; Söderhjelm, P.; Ryde, U. Free-energy perturbation and quantum mechanical study of SAMPL4 octa-acid host–guest binding energies. J. Comput. Aided Mol. Des. 2014, 28, 375–400. [Google Scholar] [CrossRef]
- Das, A.; Sharma, G.; Kamatham, N.; Prabhakar, R.; Sen, P.; Ramamurthy, V. Ultrafast solvation dynamics reveal that octa acid capsule’s interior dryness depends on the guest. J. Phys. Chem. A 2019, 123, 5928–5936. [Google Scholar] [CrossRef]
- Francl, M.M.; Pietro, W.J.; Hehre, W.J.; Binkley, J.S.; Gordon, M.S.; DeFrees, D.J.; Pople, J.A. Self-consistent molecular orbital methods. XXIII. A polarization-type basis set for second-row elements. J. Chem. Phys. 1982, 77, 3654–3665. [Google Scholar] [CrossRef]
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; van der Spoel, D.; et al. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef]
- Case, D.A.; Cheatham, T.E., III; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef]
- Samanta, S.R.; Choudhury, R.; Ramamurthy, V. Photoisomerization and photooxygenation of 1, 4-diaryl-1, 3-dienes in a confined space. J. Phys. Chem. A 2014, 118, 10554–10562. [Google Scholar] [CrossRef] [PubMed]
- Rio, G.; Berthelot, J. Photooxidation of acyclic dienes: Trans-trans-1,4-diphenyl-1,3-butadiene photo oxide. Bull. Soc. Chim. Fr. 1969, 5, 1664–1667. [Google Scholar]
- Blackburn, E.V.; Timmons, C.J. The photocyclisation of stilbene analogues. Q. Rev. Chem. Soc. 1969, 23, 482–503. [Google Scholar] [CrossRef]
- Lindahl, E.; Hess, B.; Van Der Spoel, D. GROMACS 3.0: A package for molecular simulation and trajectory analysis. J. Mol. Model. 2001, 7, 306–317. [Google Scholar] [CrossRef]
- Shrake, A.; Rupley, J.A. Environment and exposure to solvent of protein atoms. Lysozyme and insulin. J. Mol. Biol. 1973, 79, 351–371. [Google Scholar] [CrossRef]
- Otolski, C.J.; Mohan Raj, A.; Sharma, G.; Prabhakar, R.; Ramamurthy, V.; Elles, C.G. Ultrafast trans → cis photoisomerization dynamics of alkyl-substituted stilbenes in a supramolecular capsule. J. Phys. Chem. A 2019, 123, 5061–5071. [Google Scholar] [CrossRef]
- Parthasarathy, A.; Ramamurthy, V. Role of free space and weak interactions on geometric isomerization of stilbenes held in a molecular container. Photochem. Photobiol. Sci. 2011, 10, 1455–1462. [Google Scholar] [CrossRef]
- Samanta, S.R.; Parthasarathy, A.; Ramamurthy, V. Supramolecular control during triplet sensitized geometric isomerization of stilbenes encapsulated in a water soluble organic capsule. Photochem. Photobiol. Sci. 2012, 11, 1652–1660. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R. g_mmpbsa—A GROMACS Tool for High-Throughput MM-PBSA Calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
- Myers, A.B.; Mathies, R.A. Excited-state torsional dynamics of cis-stilbene from resonance Raman intensities. J. Chem. Phys. 1984, 81, 1552–1558. [Google Scholar] [CrossRef]
- Kaanumalle, L.S.; Gibb, C.L.D.; Gibb, B.C.; Ramamurthy, V. A hydrophobic nanocapsule controls the photophysics of aromatic molecules by suppressing their favored solution pathways. J. Am. Chem. Soc. 2005, 127, 3674–3675. [Google Scholar] [CrossRef]
- Bouas-Laurent, H.; Desvergne, J.-P.; Castellan, A.; Lapouyade, R. Photodimerization of anthracenes in fluid solution: Structural aspects. Chem. Soc. Rev. 2000, 29, 43–55. [Google Scholar] [CrossRef]
- Bouas-Laurent, H.; Desvergne, J.-P.; Castellan, A.; Lapouyade, R. Photodimerization of anthracenes in fluid solutions: (part 2) mechanistic aspects of the photocycloaddition and of the photochemical and thermal cleavage. Chem. Soc. Rev. 2001, 30, 248–263. [Google Scholar] [CrossRef]
- Das, A.; Danao, A.; Banerjee, S.; Raj, A.M.; Sharma, G.; Prabhakar, R.; Srinivasan, V.; Ramamurthy, V.; Sen, P. Dynamics of anthracene excimer formation within a water-soluble nanocavity at room temperature. J. Am. Chem. Soc. 2021, 143, 2025–2036. [Google Scholar] [CrossRef]
- Warshel, A.; Levitt, M. Theoretical studies of enzymic reactions: Dielectric, electrostatic and steric stabilization of the carbonium ion in the reaction of lysozyme. J. Mol. Biol. 1976, 103, 227–249. [Google Scholar] [CrossRef]
- Valiev, M.; Bylaska, E.J.; Govind, N.; Kowalski, K.; Straatsma, T.P.; Van Dam, H.J.J.; Wang, D.; Nieplocha, J.; Apra, E.; Windus, T.L.; et al. NWChem: A comprehensive and scalable open-source solution for large scale molecular simulations. Comput. Phys. Commun. 2010, 181, 1477–1489. [Google Scholar] [CrossRef]
- Gao, Y.; Liu, H.; Zhang, S.; Gu, Q.; Shen, Y.; Ge, Y.; Yang, B. Excimer formation and evolution of excited state properties in discrete dimeric stacking of an anthracene derivative: A computational investigation. Phys. Chem. Chem. Phys. 2018, 20, 12129–12137. [Google Scholar] [CrossRef]
- Gupta, S.; Adhikari, A.; Mandal, A.K.; Bhattacharyya, K. Ramamurthy, V. Ultrafast singlet singlet energy transfer between an acceptor electrostatically attached to the walls of an organic capsule and the enclosed donor. J. Phys. Chem. C 2011, 115, 9593–9600. [Google Scholar] [CrossRef]
- Santos, F.d.S.; Ramasamy, E.; da Luz, L.C.; Ramamurthy, V.; Rodembusch, F.S. Spectroscopic insights of an emissive complex between 4′-N,N-diethylaminoflavonol in octa-acid deep-cavity cavitand and rhodamine 6G. Molecules 2023, 28, 4260. [Google Scholar] [CrossRef]
- Raj, A.M.; Porel, M.; Mukherjee, P.; Ma, X.; Choudhury, R.; Galoppini, E.; Sen, P.; Ramamurthy, V. Ultrafast electron transfer from upper excited state of encapsulated azulenes to acceptors across an organic molecular wall. J. Phys. Chem. C 2017, 121, 20205–20216. [Google Scholar] [CrossRef]
- Chuang, C.H.; Porel, M.; Choudhury, R.; Burda, C.; Ramamurthy, V. Ultrafast electron transfer across a nanocapsular wall: Coumarins as donors, viologen as acceptor, and octa ocid capsule as the mediator. J. Phys. Chem. B 2018, 122, 328–337. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Kamatham, N.; Mohan Raj, A.; Sen, P.; Ramamurthy, V. Marcus Relationship maintained during ultrafast electron transfer across a supramolecular capsular wall. J. Phys. Chem. A 2020, 124, 5297–5305. [Google Scholar] [CrossRef] [PubMed]
- Ramamurthy, V. Photochemistry in a capsule: Controlling excited state dynamics via confinement. Chem. Commun. 2022, 58, 6571–6585. [Google Scholar] [CrossRef]
- Jayaraj, N.; Jockusch, S.; Kaanumalle, L.S.; Turro, N.J.; Ramamurthy, V. Dynamics of capsuleplex formed between octaacid and organic guest molecules—Photophysical techniques reveal the opening and closing of capsuleplex. Can. J. Chem. 2011, 89, 203–213. [Google Scholar] [CrossRef]
- Tang, H.; De Oliveira, C.S.; Sonntag, G.; Gibb, C.L.D.; Gibb, B.C.; Bohne, C. Dynamics of a supramolecular capsule assembly with pyrene. J. Am. Chem. Soc. 2012, 134, 5544–5547. [Google Scholar] [CrossRef]
- Bhandari, S.; Zheng, Z.; Maiti, B.; Chuang, C.H.; Porel, M.; You, Z.Q.; Ramamurthy, V.; Burda, C.; Herbert, J.M.; Dunietz, B.D. What Is the Optoelectronic Effect of the Capsule on the Guest Molecule in Aqueous Host/Guest Complexes? A Combined Computational and Spectroscopic Perspective. J. Phys. Chem. C 2017, 121, 15481–15488. [Google Scholar] [CrossRef]




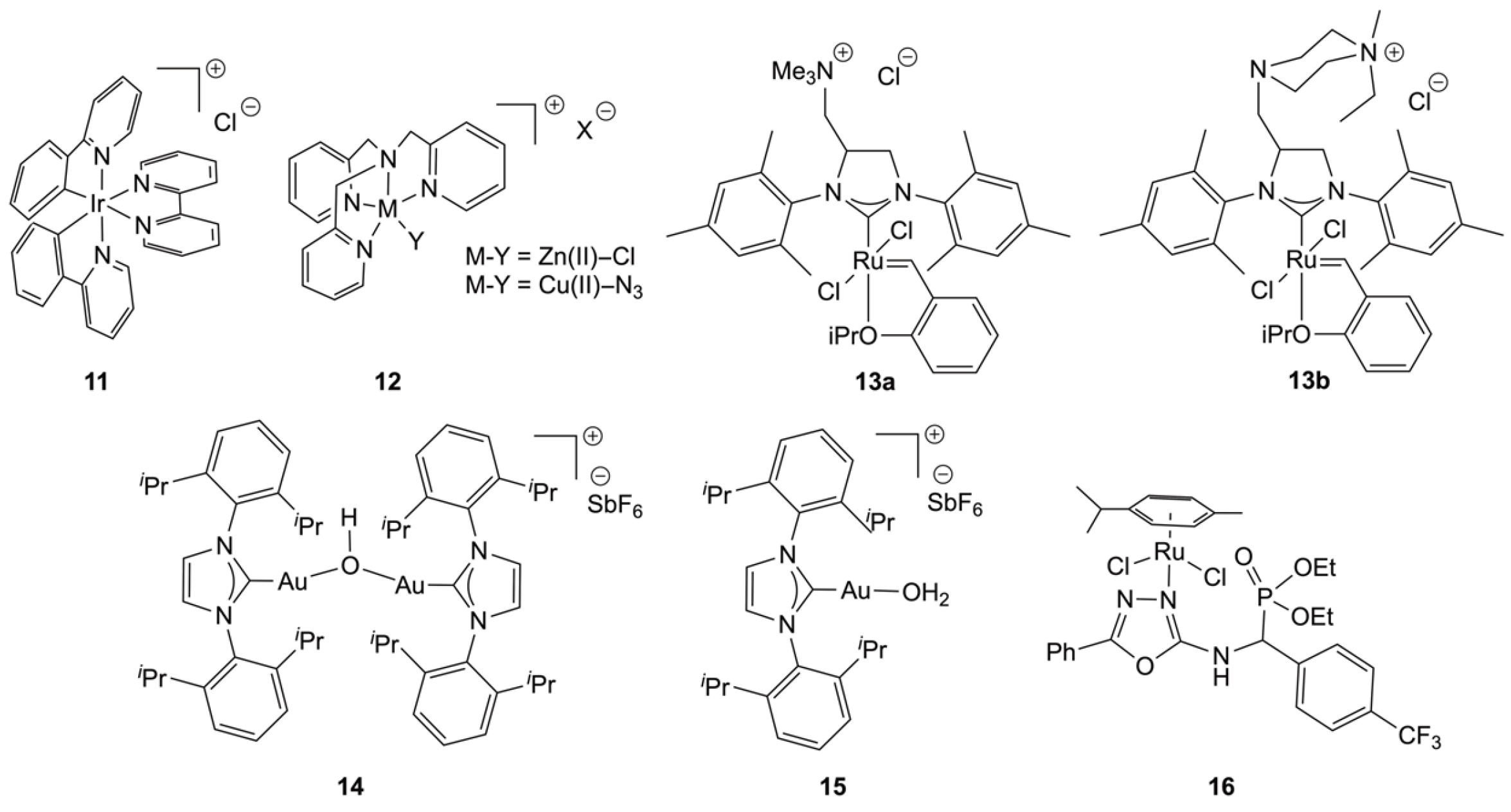
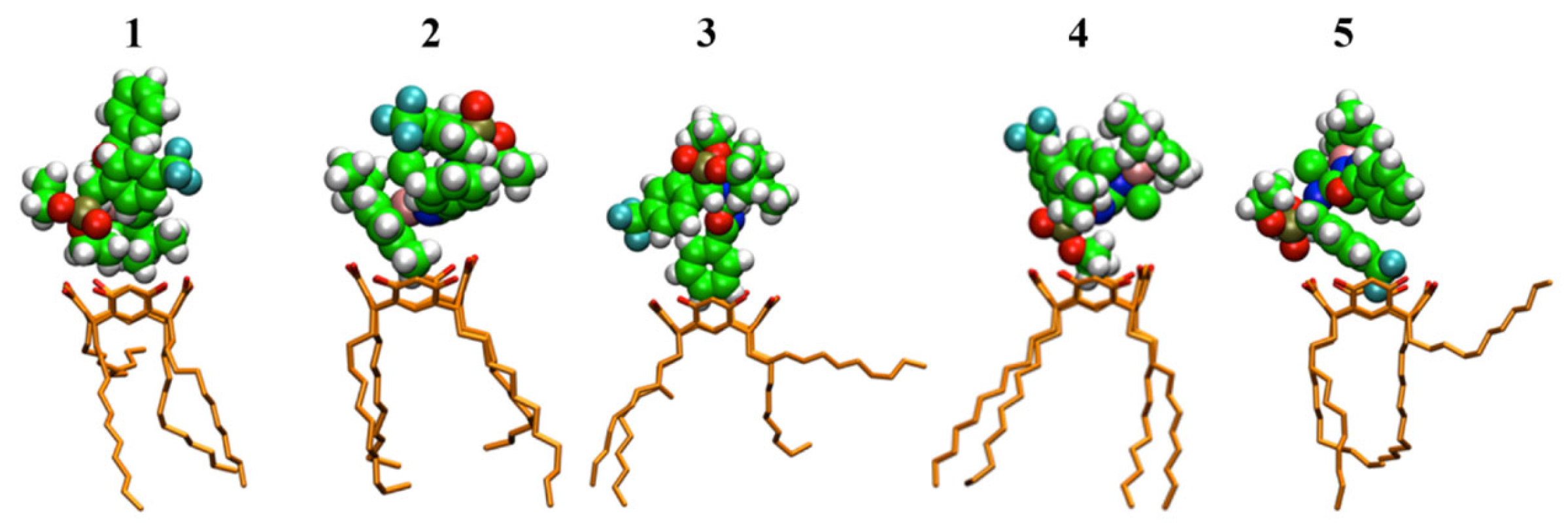









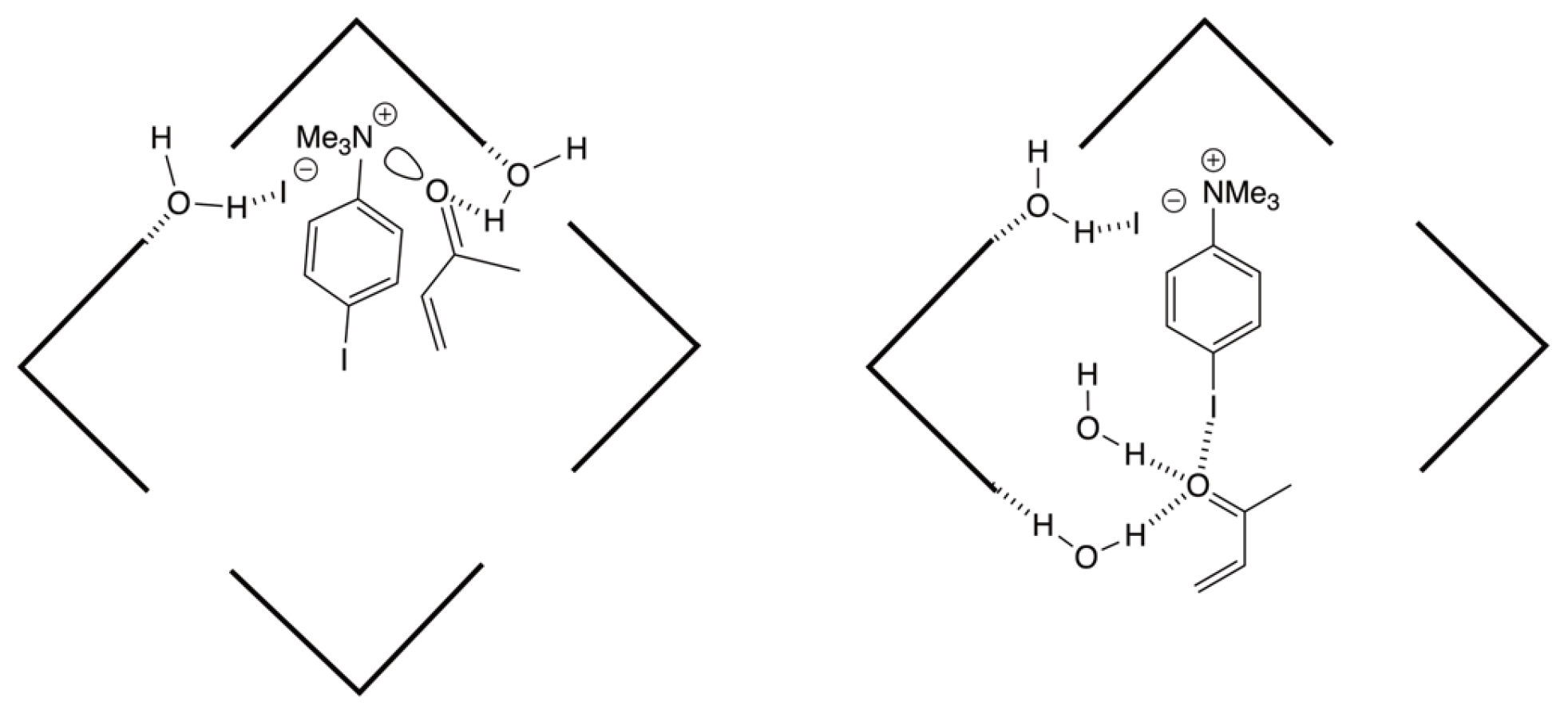





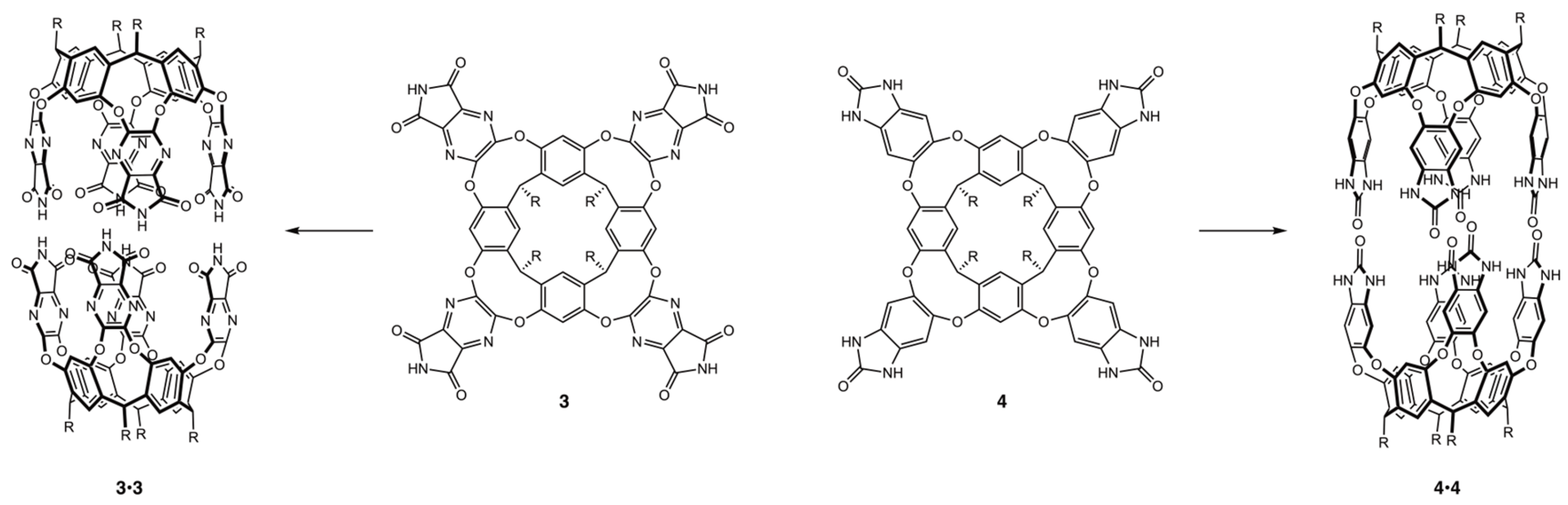










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Guest | Packing Coefficients Regarding Number of Guests | |||||||
|---|---|---|---|---|---|---|---|---|
| Volume (Å3) | in 3•3 Capsule (425 Å3) | in 3•564•3 Capsule (620 Å3) | ||||||
| 1 | 2 | 3 | 1 | 2 | 3 | 4 | ||
| Ethane | 46 | 11 | 22 | 33 | 7 | 15 | 22 | 30 |
| Cyclopropane | 56 | 13 | 26 | 39 | 9 | 18 | 27 | 36 |
| n-Butane | 80 | 19 | 38 | 57 | 13 | 26 | 39 | 52 |
| n-Hexane | 113 | 27 | 53 | 80 | 18 | 36 | 54 | 72 |
| Guest | Supramolecular Assembly | |||||
|---|---|---|---|---|---|---|
| 3•574•3 | 3•572•57′2•3 | 3•57′2•574•3 | 3•574•57′4•3 | |||
| Calculated Length (Å) | Accessible Cavity Length (Å) | |||||
| 19 | 20 | 24 | 28 | |||
| Extended | Coiled | Packing Coefficient (%) | ||||
| n-C14H30 | 19.2 | 14.4 | 50 | 47 | ||
| n-C15H32 | 20.5 | 15.3 | 53 | 50 | ||
| n-C16H34 | 21.7 | 16.2 | 55 | 53 | ||
| n-C17H36 | 23.0 | 17.2 | 53 | |||
| n-C18H38 | 24.3 | 18.1 | 54 | 52 | ||
| n-C19H40 | 25.5 | 19.1 | 53 | |||
| n-C20H42 | 26.8 | 20.1 | 53 | |||
| n-C21H44 | 28.0 | 21.0 | 55 | 54 | ||
| n-C22H46 | 29.3 | 21.9 | 55 | |||
| n-C23H48 | 30.6 | 22.7 | 56 | |||
| Dimer | Dimerization Energy (kcal/mol) | Interaction Energy with the Capsule (kcal/mol) | Total Interaction Energy with the Capsule (kcal/mol) |
|---|---|---|---|
| 79•79-trans | 15.4 | 38.7 | 56.3 |
| 78•79-trans | 13.8 | 40.8 | 56.7 |
| 78•78-trans | 11.5 | 41.7 | 55.3 |
| 78•77-trans | 9.4 | 44.7 | 56.3 |
| 77•79-trans | 7.6 | 44.3 | 57.2 |
| 77•77-trans | 7.6 | 47.7 | 58.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Steinmetz, M.; Sémeril, D. Molecular Modeling Is Key to Understanding Supramolecular Resorcinarenyl Capsules, Inclusion Complex Formation and Organic Reactions in Nanoconfined Space. Molecules 2025, 30, 2549. https://doi.org/10.3390/molecules30122549
Steinmetz M, Sémeril D. Molecular Modeling Is Key to Understanding Supramolecular Resorcinarenyl Capsules, Inclusion Complex Formation and Organic Reactions in Nanoconfined Space. Molecules. 2025; 30(12):2549. https://doi.org/10.3390/molecules30122549
Chicago/Turabian StyleSteinmetz, Maxime, and David Sémeril. 2025. "Molecular Modeling Is Key to Understanding Supramolecular Resorcinarenyl Capsules, Inclusion Complex Formation and Organic Reactions in Nanoconfined Space" Molecules 30, no. 12: 2549. https://doi.org/10.3390/molecules30122549
APA StyleSteinmetz, M., & Sémeril, D. (2025). Molecular Modeling Is Key to Understanding Supramolecular Resorcinarenyl Capsules, Inclusion Complex Formation and Organic Reactions in Nanoconfined Space. Molecules, 30(12), 2549. https://doi.org/10.3390/molecules30122549