
A Cost-Effective Liquid Chromatography Method with Ultraviolet Detection for Identity Screening and Assay of Injectable Antibiotics


Abstract
1. Introduction
2. Results and Discussion
2.1. Method Development and Optimization
2.2. Method Validation
2.2.1. Specificity
2.2.2. Robustness
2.2.3. Linearity
2.2.4. Accuracy
2.2.5. Precision
2.2.6. Sensitivity
2.3. Analysis of Commercial Samples
3. Materials and Methods
3.1. Reagents and Materials
3.2. Chromatographic Conditions
3.3. Screening Method
3.3.1. Preparation of Standard Solutions
3.3.2. Preparation of Sample Solutions
3.4. Quantification Method
3.4.1. Preparation of Standards
3.4.2. Preparation of Sample Solutions
3.5. Validation of the Screening and Quantification Method
3.6. Application of the Method
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zabala, G.A.; Bellingham, K.; Vidhamaly, V.; Boupha, P.; Boutsamay, K.; Newton, P.N.; Caillet, C. Substandard and falsified antibiotics: Neglected drivers of antimicrobial resistance? BMJ Glob. Health 2022, 7, e008587. [Google Scholar] [CrossRef] [PubMed]
- Wolie, Z.T.; Roberts, J.A.; Gilchrist, M.; McCarthy, K.; Sime, F.B. Current practices and challenges of outpatient parenteral antimicrobial therapy: A narrative review. J. Antimicrob. Chemother. 2024, 7, 2083–2102. [Google Scholar] [CrossRef] [PubMed]
- Carlier, M.; Stove, V.; Wallis, S.C.; De Waele, J.J.; Verstraete, A.G.; Lipman, J.; Roberts, J.A. Assays for therapeutic drug monitoring of β-lactam antibiotics: A structured review. Int. J. Antimicrob. Agents 2015, 46, 367–375. [Google Scholar] [CrossRef]
- Bottoni, P.; Caroli, S. Fake pharmaceuticals: A review of current analytical approaches. Microchem. J. 2019, 149, 104053. [Google Scholar] [CrossRef]
- Li, H.K.; Agweyu, A.; English, M.; Bejon, P. An unsupported preference for intravenous antibiotics. PLoS Med. 2015, 12, e1001825. [Google Scholar] [CrossRef]
- Wada, Y.H.; Abdulrahman, A.; Muhammad, M.I.; Owanta, V.C.; Chimelumeze, P.U.; Khalid, G.M. Falsified and substandard medicines trafficking: A wakeup call for the African continent. Public Health Pract. 2022, 3, 100240. [Google Scholar] [CrossRef]
- Ozawa, S.; Evans, D.R.; Bessias, S.; Haynie, D.G.; Yemeke, T.T.; Laing, S.K.; Herrington, J.E. Prevalence and estimated economic burden of substandard and falsified medicines in low-and middle-income countries: A systematic review and meta-analysis. JAMA Netw. Open 2018, 1, e181662. [Google Scholar] [CrossRef]
- Höllein, L.; Kaale, E.; Mwalwisi, Y.H.; Schulze, M.H.; Holzgrabe, U. Routine quality control of medicines in developing countries: Analytical challenges, regulatory infrastructures and the prevalence of counterfeit medicines in Tanzania. TrAC Trends Anal. Chem. 2016, 76, 60–70. [Google Scholar] [CrossRef]
- Opuni, K.F.; Sunkwa-Mills, G.; Antwi, M.; Squire, A.; Afful, G.Y.; de Wit, T.F.R.; Kretchy, I. Quality assessment of medicines in selected resource-limited primary healthcare facilities using low-to medium-cost field testing digital technologies. Digit. Health 2024, 10, 20552076241299064. [Google Scholar] [CrossRef]
- Bakker, I.M.; Ohana, D.; Venhuis, B.J. Current challenges in the detection and analysis of falsified medicines. J. Pharm. Biomed. Anal. 2021, 197, 113948. [Google Scholar]
- Sorato, M.M.; Davari, M.; Kebriaeezadeh, A. Improving access to medicines to reduce marketing and use of substandard and falsified medicines in Africa: Scoping review. J. Med. Access 2024, 8, 27550834241236598. [Google Scholar] [CrossRef] [PubMed]
- Fahad Abdulaziz Alghannam, A.; Aslanpour, Z.; Evans, S.; Schifano, F. A systematic review of counterfeit and substandard medicines in field quality surveys. Integr. Pharm. Res. Pract. 2014, 3, 71–88. [Google Scholar]
- Rebiere, H.; Guinot, P.; Chauvey, D.; Brenier, C. Fighting falsified medicines: The analytical approach. J. Pharm. Biomed. Anal. 2017, 142, 286–306. [Google Scholar] [CrossRef]
- Johnston, A.; Holt, D.W. Substandard drugs: A potential crisis for public health. Br. J. Clin. Pharmacol. 2014, 78, 218–243. [Google Scholar] [CrossRef] [PubMed]
- Kelesidis, T.; Falagas, M.E. Substandard/counterfeit antimicrobial drugs. Clin. Microbiol. Rev. 2015, 28, 443–464. [Google Scholar] [CrossRef]
- Roncone, A.; Kelly, S.D.; Giannioti, Z.; Hauk, C.; Caillet, C.; Newton, P.N.; Perez-Mon, C.; Bontempo, L. Stable isotope ratio analysis: An emerging tool to trace the origin of falsified medicines. TrAC Trends Anal. Chem. 2024, 174, 117666. [Google Scholar] [CrossRef]
- Janvier, S.; De Spiegeleer, B.; Vanhee, C.; Deconinck, E. Falsification of biotechnology drugs: Current dangers and/or future disasters? J. Pharm. Biomed. Anal. 2018, 161, 175–191. [Google Scholar] [CrossRef]
- Bliese, S.L.; Maina, M.; Were, P.; Lieberman, M. Detection of degraded, adulterated, and falsified ceftriaxone using paper analytical devices. Anal. Methods 2019, 11, 4727–4732. [Google Scholar] [CrossRef]
- Akpobolokemi, T.; Martinez-Nunez, R.T.; Raimi-Abraham, B.T. Tackling the global impact of substandard and falsified and unregistered/unlicensed anti-tuberculosis medicines. J. Med. Access 2022, 6, 23992026211070406. [Google Scholar] [CrossRef]
- Tie, Y.; Vanhee, C.; Deconinck, E.; Adams, E. Development and validation of chromatographic methods for screening and subsequent quantification of suspected illegal antimicrobial drugs encountered on the Belgian market. Talanta 2019, 194, 876–887. [Google Scholar] [CrossRef]
- Roth, L.; Nalim, A.; Turesson, B.; Krech, L. Global landscape assessment of screening technologies for medicine quality assurance: Stakeholder perceptions and practices from ten countries. Glob. Health 2018, 14, 43. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Ethiopian essential medicine list, Food. In Medicine and Healthcare Administration and Control Authority of Ethiopia, 15th ed.; World Health Organization: Geneva, Switzerland, 2015. [Google Scholar]
- EDQM Council of Europe. European Pharmacopoeia, 11th ed.; EDQM Council of Europe: Strasbourg, France, 2023. [Google Scholar]
- Mbinze, J.; Dispas, A.; Lebrun, P.; Mbay, J.M.T.; Habyalimana, V.; Kalenda, N.; Rozet, E.; Hubert, P.; Marini, R. Application of an innovative design space optimization strategy to the development of LC methods for the simultaneous screening of antibiotics to combat poor quality medicines. J. Pharm. Biomed. Anal. 2013, 85, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Kassahun, H.; Van Schepdael, A.; Ketema, G.; Adams, E. Development and validation of a simple and affordable LC-UV method for identification and assay of selected antimicrobial medicines. J. Pharm. Biomed. Anal. 2024, 244, 116127. [Google Scholar] [CrossRef]
- US Pharmacopeial Convention. United States Pharmacopeia, USP 41/The National Formulary; US Pharmacopeial Convention: Rockville, MD, USA, 2018. [Google Scholar]
- ICH Guidelines. Validation of analytical Procedures: Text and methodology. Q2 (R1) 2005, 1, 5. [Google Scholar]
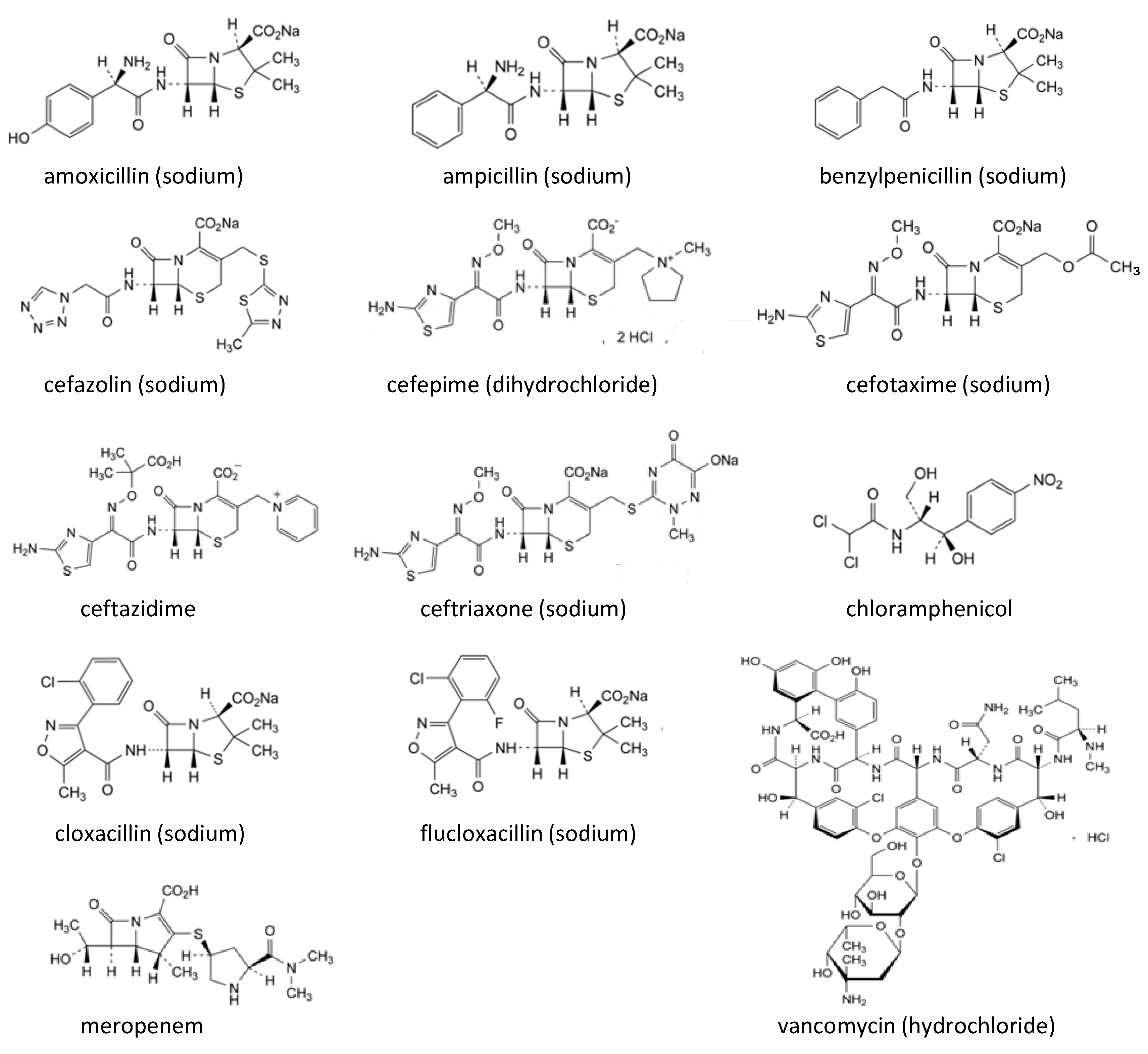
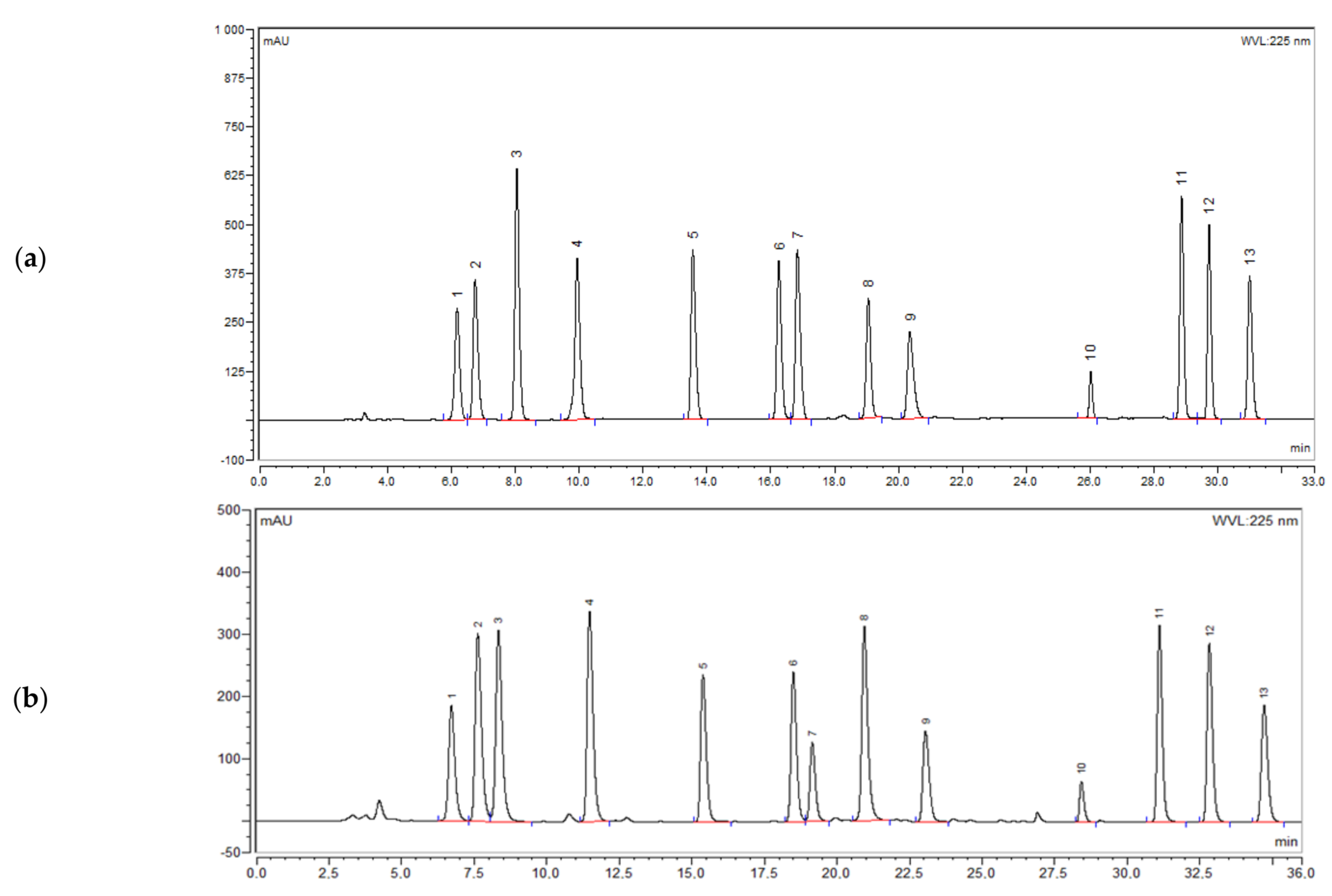
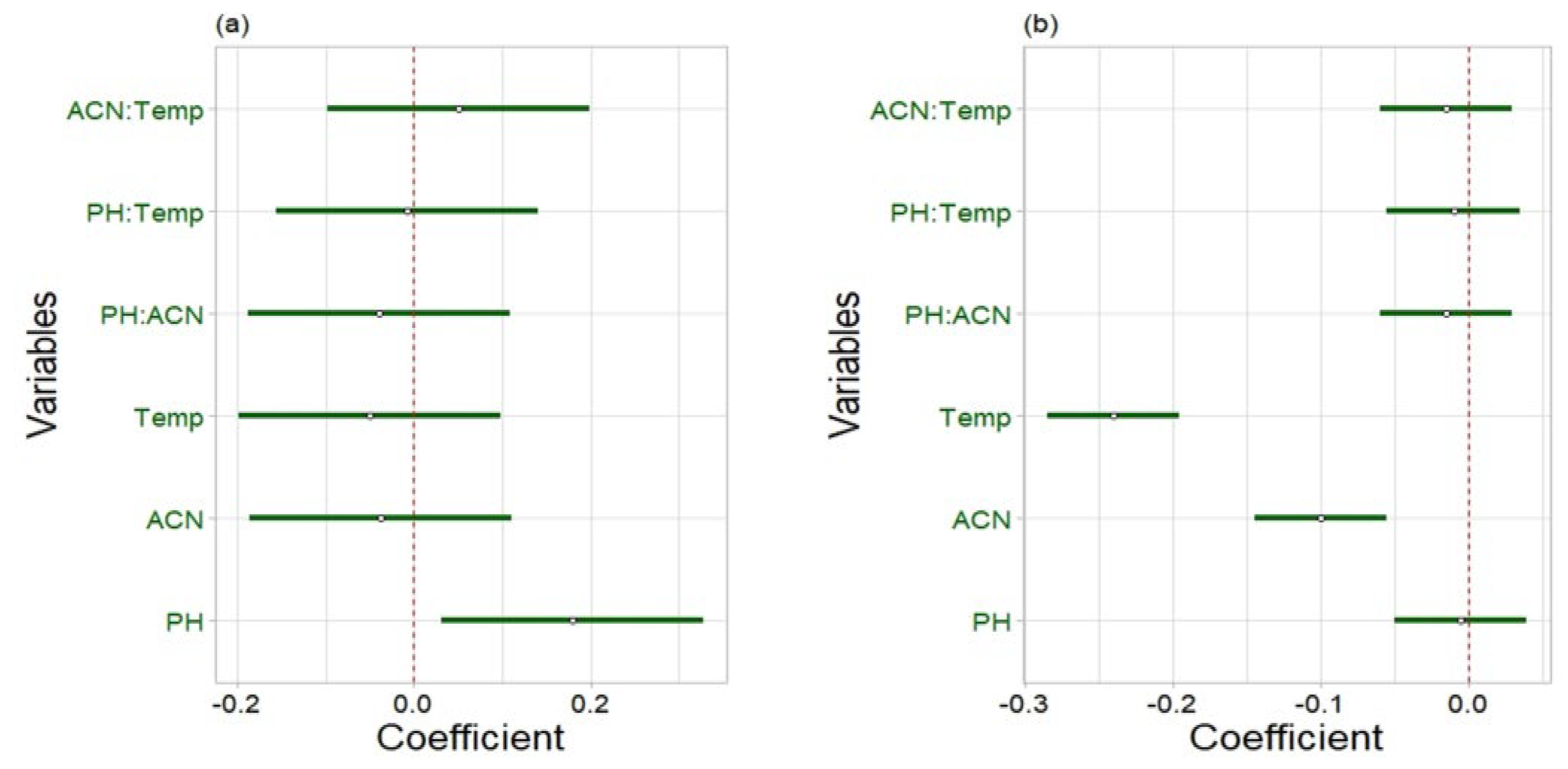


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analytes | Intraday Precision (% RSD), n = 6 | Interday Precision (% RSD), n = 18 | % Recovery (% RSD), n = 3 | ||
|---|---|---|---|---|---|
| 80% | 100% | 120% | |||
| Ceftazidime | 0.3 | 0.3 | 100.3 (0.2) | 99.4 (0.1) | 98.9 (0.2) |
| Ceftriaxone | 0.1 | 0.1 | 101.6 (0.4) | 99.9 (0.1) | 100.8 (0.3) |
| Cefepime | 0.5 | 0.5 | 99.4 (0.4) | 98.2 (0.2) | 98.3 (0.3) |
| Amoxicillin | 0.1 | 0.3 | 101.3 (0.1) | 100.8 (0.1) | 100.0 (0.1) |
| Meropenem | 0.4 | 0.9 | 101.5 (0.3) | 98.8 (0.2) | 99.6 (0.2) |
| Cefotaxime | 0.4 | 0.7 | 101.5 (0.2) | 99.0 (0.2) | 99.9 (0.2) |
| Cefazolin | 0.1 | 0.2 | 99.9 (0.4) | 99.6 (0.5) | 99.6 (0.1) |
| Vancomycin | 0.2 | 0.5 | 99.8 (0.3) | 99.1 (0.1) | 100.2 (0.1) |
| Ampicillin | 0.3 | 0.3 | 101.2 (0.4) | 99.4 (0.5) | 99.9 (0.1) |
| Benzylpenicillin | 0.2 | 0.6 | 100.0 (0.4) | 100.2 (0.1) | 99.8 (0.1) |
| Chloramphenicol | 0.1 | 0.8 | 100.1 (0.2) | 100.1 (0.1) | 100.5 (0.1) |
| Flucloxacillin | 0.2 | 0.7 | 99.5 (0.3) | 99.7 (0.1) | 99.5 (0.1) |
| Cloxacillin | 0.4 | 0.8 | 99.4 (0.3) | 99.5 (0.1) | 99.7 (0.2) |
| Analytes | LOQ (µg/mL) | LOD (µg/mL) |
|---|---|---|
| Ceftazidime | 0.018 | 0.005 |
| Ceftriaxone | 0.018 | 0.005 |
| Cefepime | 0.039 | 0.012 |
| Amoxicillin | 0.053 | 0.016 |
| Meropenem | 0.174 | 0.052 |
| Cefotaxime | 0.059 | 0.018 |
| Cefazolin | 0.242 | 0.072 |
| Vancomycin | 0.216 | 0.065 |
| Ampicillin | 0.592 | 0.178 |
| Benzylpenicillin | 0.735 | 0.221 |
| Chloramphenicol | 0.391 | 0.117 |
| Flucloxacillin | 0.068 | 0.020 |
| Cloxacillin | 0.144 | 0.043 |
| Analytes | Sample Code | % Content (% RSD) |
|---|---|---|
| Vancomycin 1 g | Sample 1 | 106.9 (0.9) |
| Sample 2 | 93.7 (0.9) | |
| Sample 3 | 104.1 (0.8) | |
| Ceftazidime 1 g | Sample 4 | 99.2 (0.9) |
| Sample 5 | 100.7 (0.7) | |
| Ceftriaxone 1 g | Sample 6 | 98.5 (0.1) |
| Sample 7 | 99.1 (0.2) | |
| Sample 8 | 97.9 (0.7) | |
| Sample 9 | 95.9 (0.8) | |
| Cefepime 1 g | Sample 10 | 90.7 (0.3) |
| Sample 11 | 92.5 (0.7) | |
| Sample 12 | 91.5 (0.8) | |
| Sample 13 | 90.4 (0.4) | |
| Meropenem 1 g | Sample 14 | 91.8 (0.2) |
| Cloxacillin 500 mg | Sample 15 | 101.3 (0.3) |
| Ampicillin 1 g | Sample 16 | 94.6 (0.4) |
| Cefotaxime 1 g | Sample 17 | 92.8 (0.8) |
| Analytes | Sample Code | Country of Origin |
|---|---|---|
| Vancomycin 1 g | Sample 1 | India |
| Sample 2 | India | |
| Sample 3 | India | |
| Ceftazidime 1 g | Sample 4 | India |
| Sample 5 | India | |
| Ceftriaxone 1 g | Sample 6 | India |
| Sample 7 | India | |
| Sample 8 | India | |
| Sample 9 | China | |
| Cefepime 1 g | Sample 10 | India |
| Sample 11 | India | |
| Sample 12 | India | |
| Sample 13 | Ethiopia | |
| Meropenem 1 g | Sample 14 | China |
| Cloxacillin 500 mg | Sample 15 | China |
| Ampicillin 1 g | Sample 16 | China |
| Cefotaxime 1 g | Sample 17 | India |
| Parameter | Lower Level (−) | Central Level (0) | Higher Level (+) |
|---|---|---|---|
| Buffer pH | 7.8 | 8 | 8.2 |
| % ACN in mobile phase B | 28 | 30 | 32 |
| Column temperature (°C) | 28 | 30 | 32 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Desta, H.K.; Ketema, G.; Van Schepdael, A.; Adams, E. A Cost-Effective Liquid Chromatography Method with Ultraviolet Detection for Identity Screening and Assay of Injectable Antibiotics. Molecules 2025, 30, 2151. https://doi.org/10.3390/molecules30102151
Desta HK, Ketema G, Van Schepdael A, Adams E. A Cost-Effective Liquid Chromatography Method with Ultraviolet Detection for Identity Screening and Assay of Injectable Antibiotics. Molecules. 2025; 30(10):2151. https://doi.org/10.3390/molecules30102151
Chicago/Turabian StyleDesta, Haile Kassahun, Gebremariam Ketema, Ann Van Schepdael, and Erwin Adams. 2025. "A Cost-Effective Liquid Chromatography Method with Ultraviolet Detection for Identity Screening and Assay of Injectable Antibiotics" Molecules 30, no. 10: 2151. https://doi.org/10.3390/molecules30102151
APA StyleDesta, H. K., Ketema, G., Van Schepdael, A., & Adams, E. (2025). A Cost-Effective Liquid Chromatography Method with Ultraviolet Detection for Identity Screening and Assay of Injectable Antibiotics. Molecules, 30(10), 2151. https://doi.org/10.3390/molecules30102151