
Novel Antibacterial 4-Piperazinylquinoline Hybrid Derivatives Against Staphylococcus aureus: Design, Synthesis, and In Vitro and In Silico Insights

 , , , , ,
, , , , ,  and
and
Abstract

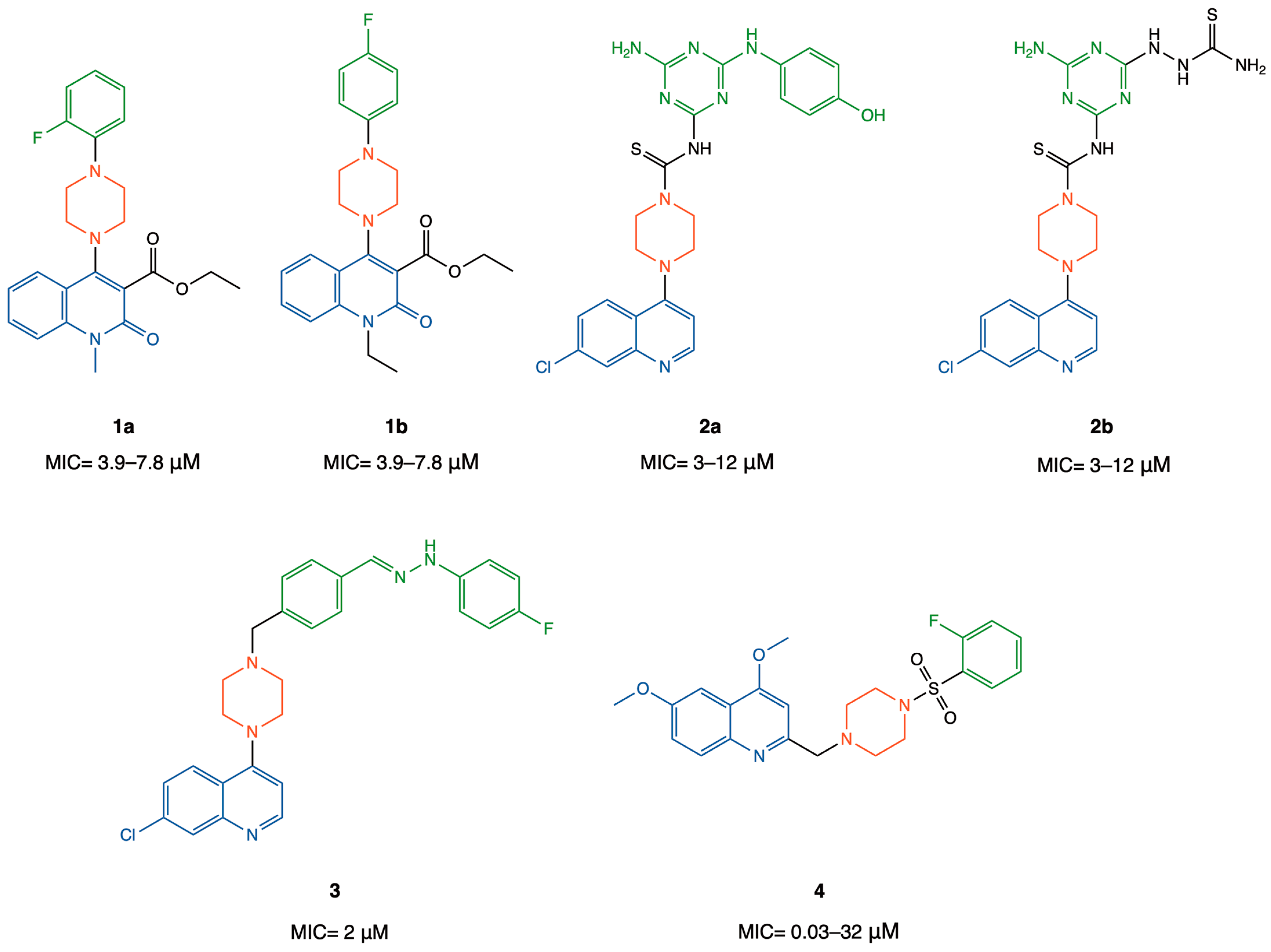
1. Introduction
2. Results and Discussion
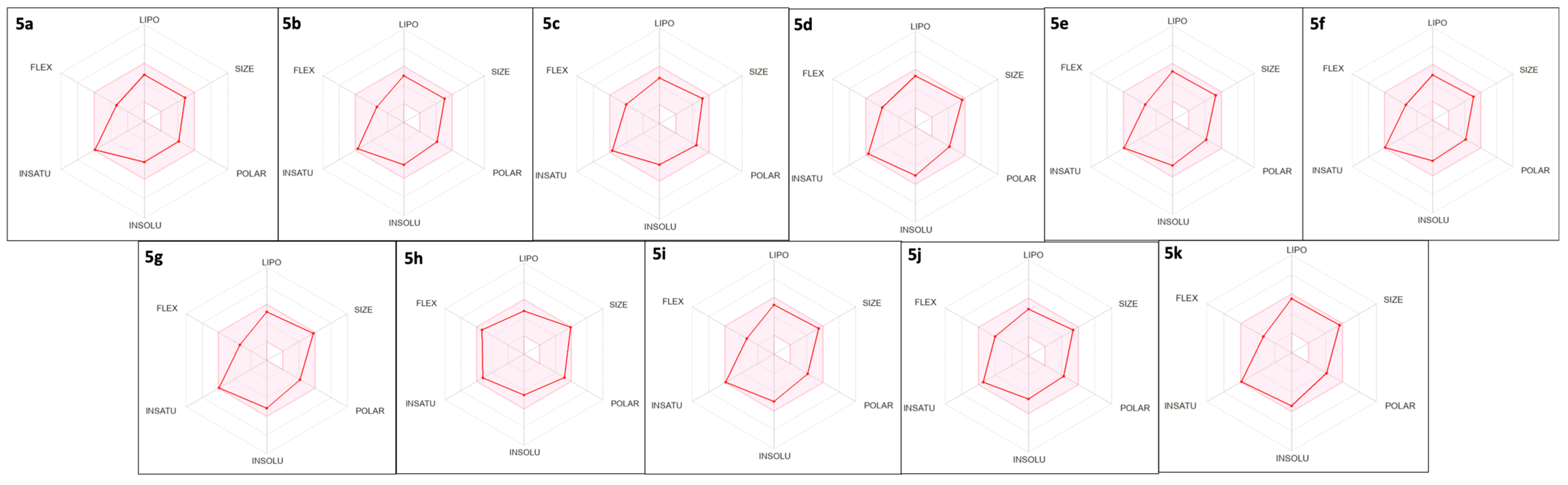
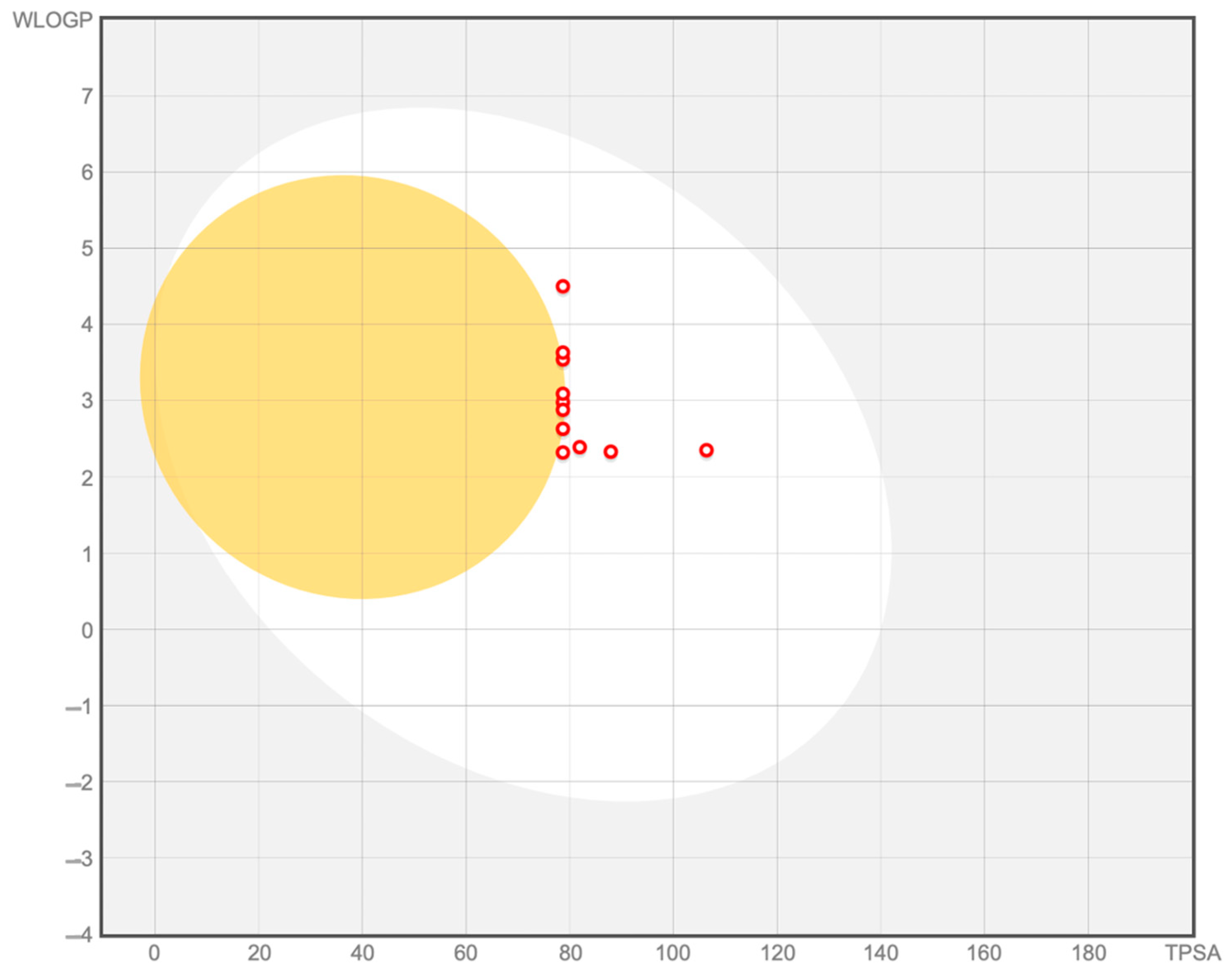
2.1. In-House Database Design and Physicochemical, Drug-Likeness, and ADME Properties Prediction
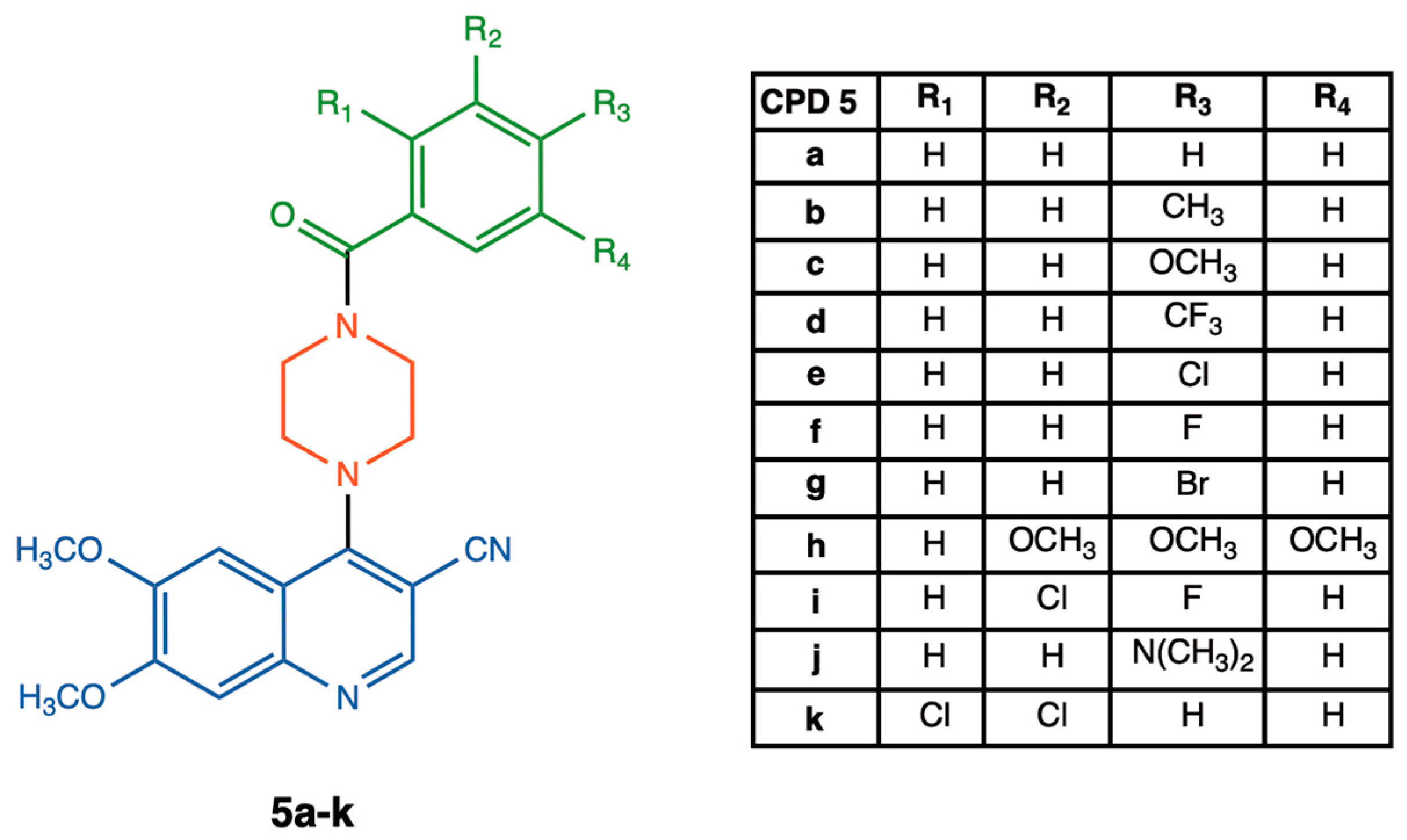
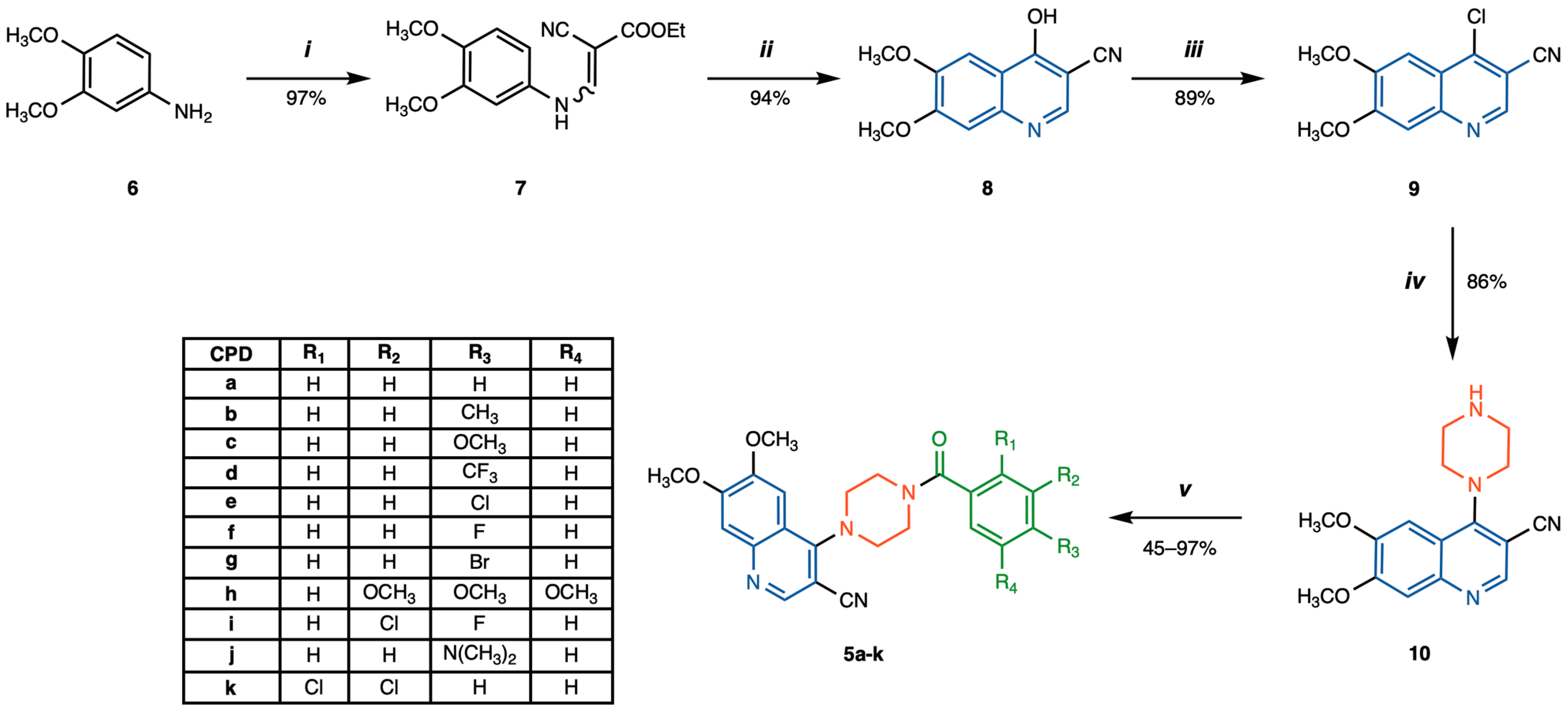
2.2. Synthesis of 4-(4-Benzoylpiperazin-1-yl)-6,7-dimethoxyquinoline-3-carbonitrile Derivatives 5a–k
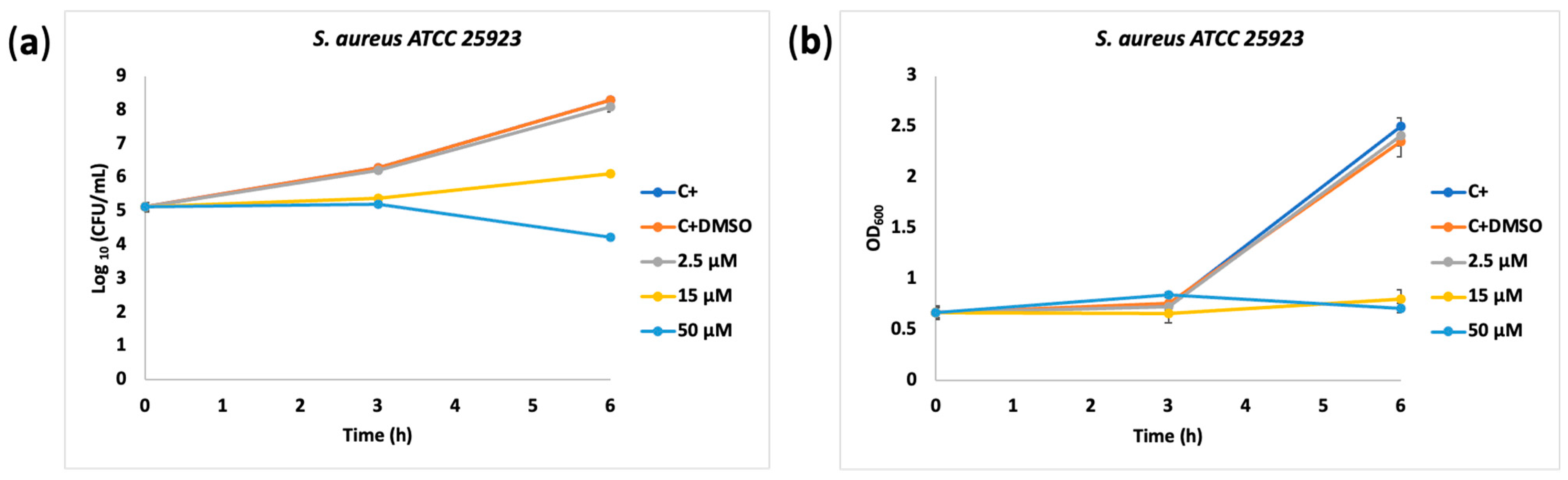
2.3. Biological Activity
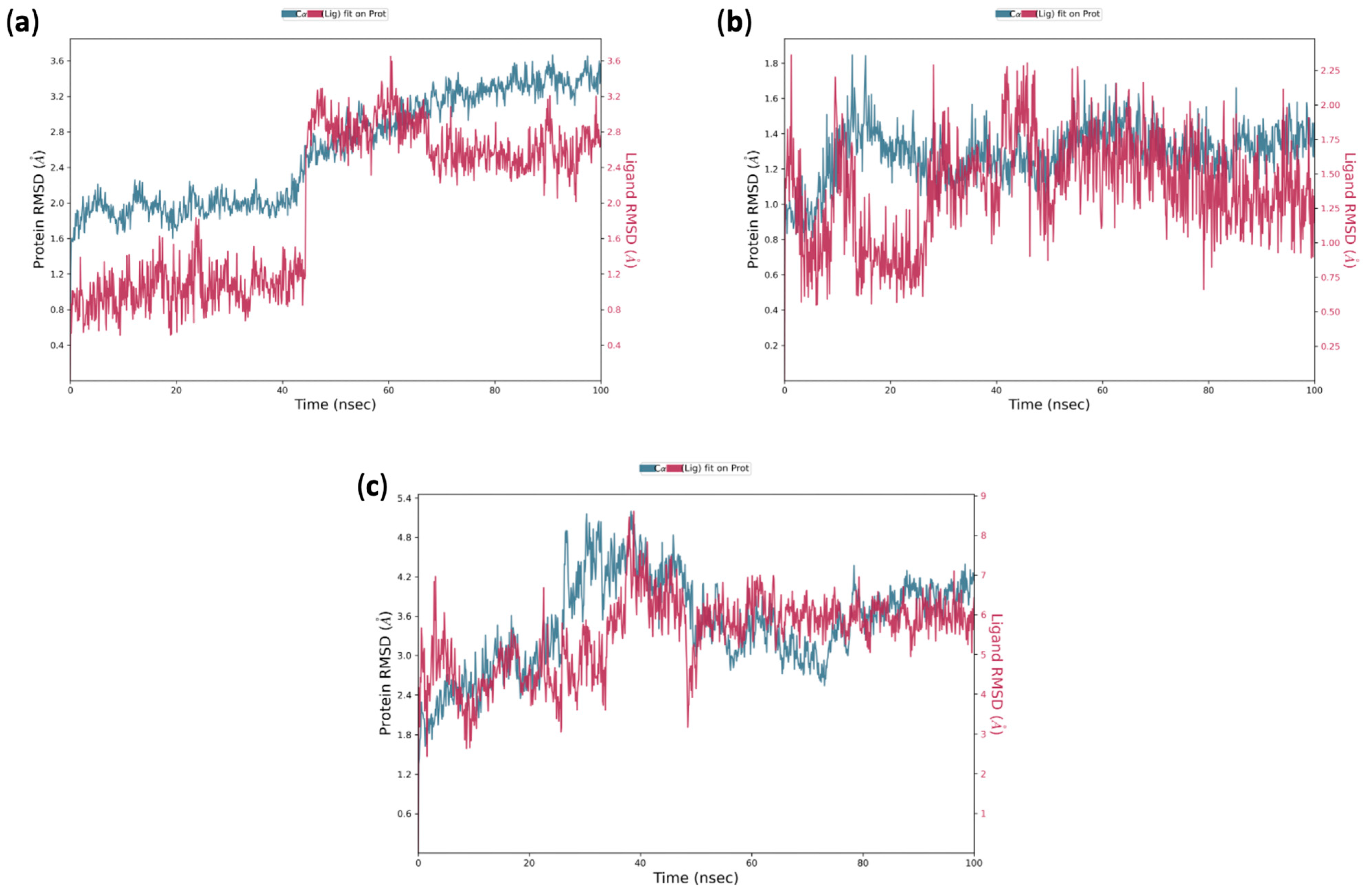
2.4. In Silico Insights into the Mechanism of Action of 5k on S. aureus: Induced Fit Docking and Molecular Dynamic Simulation Studies
3. Materials and Methods
3.1. Chemistry
3.1.1. General Information
3.1.2. Experimental Procedures and Characterization of 6,7-Dimethoxy-4-piperazinylquinoline Derivatives
- 6,7-dimethoxy-4-(piperazin-1-yl)quinoline-3-carbonitrile (10)
- General procedure for the synthesis of 4-(4-benzoylpiperazin-1-yl)-6,7-dimethoxyquinoline-3-carbonitrile 5a–k
- 4-(4-benzoylpiperazin-1-yl)-6,7-dimethoxyquinoline-3-carbonitrile (5a)
- 6,7-dimethoxy-4-(4-(4-methylbenzoyl)piperazin-1-yl)quinoline-3-carbonitrile (5b)
- 6,7-dimethoxy-4-(4-(4-methoxybenzoyl)piperazin-1-yl)quinoline-3-carbonitrile (5c)
- 6,7-dimethoxy-4-(4-(4-(trifluoromethyl)benzoyl)piperazin-1-yl)quinoline-3-carbonitrile (5d)
- 4-(4-(4-chlorobenzoyl)piperazin-1-yl)-6,7-dimethoxyquinoline-3-carbonitrile (5e)
- 4-(4-(4-fluorobenzoyl)piperazin-1-yl)-6,7-dimethoxyquinoline-3-carbonitrile (5f)
- 4-(4-(4-bromobenzoyl)piperazin-1-yl)-6,7-dimethoxyquinoline-3-carbonitrile (5g)
- 6,7-dimethoxy-4-(4-(3,4,5-trimethoxybenzoyl)piperazin-1-yl)quinoline-3-carbonitrile (5h)
- 4-(4-(3-chloro-4-fluorobenzoyl)piperazin-1-yl)-6,7-dimethoxyquinoline-3-carbonitrile (5i)
- 4-(4-(4-(dimethylamino)benzoyl)piperazin-1-yl)-6,7-dimethoxyquinoline-3-carbonitrile (5j)
- 4-(4-(2,3-dichlorobenzoyl)piperazin-1-yl)-6,7-dimethoxyquinoline-3-carbonitrile (5k)
3.2. Biology
3.2.1. Evaluation of Bacteriostatic and Growth-Inhibitory Properties of Novel 6,7-Dimethoxy-4-piperazinyl Quinoline Derivatives
3.2.2. Minimum Inhibitory Concentration (MIC) Determination
3.2.3. Cell Viability Assay
3.3. In Silico Studies
3.3.1. ADMET, Drug-Likeness, and Medicinal Chemistry Friendliness Parameters Prediction
3.3.2. Ligand Preparation
3.3.3. Protein Retrieval and Preparation
3.3.4. Structure-Based Studies: Induced Fit Docking (IFD) Simulations
3.3.5. Structure-Based Studies: Molecular Dynamics Simulation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Aatif, M.; Raza, M.A.; Javed, K.; Nashre-Ul-Islam, S.M.; Farhan, M.; Alam, M.W. Potential Nitrogen-Based Heterocyclic Compounds for Treating Infectious Diseases: A Literature Review. Antibiotics 2022, 11, 1750. [Google Scholar] [CrossRef] [PubMed]
- Heravi, M.M.; Zadsirjan, V. Prescribed drugs containing nitrogen heterocycles: An overview. RSC Adv. 2020, 10, 44247–44311. [Google Scholar] [CrossRef] [PubMed]
- Dube, P.S.; Legoabe, L.J.; Beteck, R.M. Quinolone: A versatile therapeutic compound class. Mol. Divers. 2023, 27, 1501–1526. [Google Scholar] [CrossRef]
- Senerovic, L.; Opsenica, D.; Moric, I.; Aleksic, I.; Spasić, M.; Vasiljevic, B. Quinolines and Quinolones as Antibacterial, Antifungal, Anti-virulence, Antiviral and Anti-parasitic Agents. Adv. Exp. Med. Biol. 2020, 1282, 37–69. [Google Scholar] [CrossRef]
- Kumar, P. A review on quinoline derivatives as anti-methicillin resistant Staphylococcus aureus (MRSA) agents. BMC Chem. 2020, 14, 17. [Google Scholar] [CrossRef] [PubMed]
- Tahir, S.; Mahmood, T.; Dastgir, F.; Haq, I.U.; Waseem, A.; Rashid, U. Design, synthesis and anti-bacterial studies of piperazine derivatives against drug resistant bacteria. Eur. J. Med. Chem. 2019, 166, 224–231. [Google Scholar] [CrossRef]
- Faizan, M.; Kumar, R.; Mazumder, A.; Salahuddin; Kukreti, N.; Kumar, A.; Chaitanya, M.V.N.L. The medicinal chemistry of piperazines: A review. Chem. Biol. Drug Des. 2024, 103, e14537. [Google Scholar] [CrossRef] [PubMed]
- Patil, M.; Noonikara Poyil, A.; Joshi, S.D.; Patil, S.A.; Bugarin, A. Design, synthesis, and molecular docking study of new piperazine derivative as potential antimicrobial agents. Bioorg. Chem. 2019, 92, 103217. [Google Scholar] [CrossRef] [PubMed]
- Bérubé, G. An overview of molecular hybrids in drug discovery. Expert. Opin. Drug Discov. 2016, 11, 281–305. [Google Scholar] [CrossRef] [PubMed]
- Ivasiv, V.; Albertini, C.; Gonçalves, A.E.; Rossi, M.; Bolognesi, M.L. Molecular Hybridization as a Tool for Designing Multitarget Drug Candidates for Complex Diseases. Curr. Top. Med. Chem. 2019, 19, 1694–1711. [Google Scholar] [CrossRef] [PubMed]
- Elebiju, O.F.; Ajani, O.O.; Oduselu, G.O.; Ogunnupebi, T.A.; Adebiyi, E. Recent advances in functionalized quinoline scaffolds and hybrids-Exceptional pharmacophore in therapeutic medicine. Front. Chem. 2022, 10, 1074331. [Google Scholar] [CrossRef]
- Patel, K.B.; Kumari, P. A review: Structure-activity relationship and antibacterial activities of Quinoline based hybrids. J. Mol. Struct. 2022, 1268, 133634. [Google Scholar] [CrossRef]
- Bala, I.A.; Al Sharif, O.F.; Asiri, A.M.; El-Shishtawy, R.M. Quinoline: A versatile bioactive scaffold and its molecular hybridization. Results Chem. 2024, 7, 101529. [Google Scholar] [CrossRef]
- Insuasty, D.; Vidal, O.; Bernal, A.; Marquez, E.; Guzman, J.; Insuasty, B.; Quiroga, J.; Svetaz, L.; Zacchino, S.; Puerto, G.; et al. Antimicrobial Activity of Quinoline-Based Hydroxyimidazolium Hybrids. Antibiotics 2019, 8, 239. [Google Scholar] [CrossRef]
- Verma, S.; Lal, S.; Narang, R.; Sudhakar, K. Quinoline Hydrazide/Hydrazone Derivatives: Recent Insights on Antibacterial Activity and Mechanism of Action. ChemMedChem 2023, 18, e202200571. [Google Scholar] [CrossRef]
- El-Azzouny, A.M.A.E.; Aboul-Enein, M.N.; Hamissa, M.F. Structural and biological survey of 7-chloro-4-(piperazin-1-yl)quinoline and its derivatives. Drug Dev. Res. 2020, 81, 786–802. [Google Scholar] [CrossRef]
- Banu, S.; Bollu, R.; Naseema, M.; Gomedhika, P.M.; Nagarapu, L.; Sirisha, K.; Kumar, C.G.; Gundasw, S.K. A novel templates of piperazinyl-1,2-dihydroquinoline-3-carboxylates: Synthesis, anti-microbial evaluation and molecular docking studies. Bioorg. Med. Chem. Lett. 2018, 28, 1166–1170. [Google Scholar] [CrossRef]
- Pathak, P.; Thakur, A.; Bhat, H.R.; Singh, U.P. Hybrid 4-Aminoquinoline-1,3,5-triazine Derivatives: Design, Synthesis, Characterization, and Antibacterial Evaluation. J. Heterocycl. Chem. 2015, 52, 1108–1113. [Google Scholar] [CrossRef]
- Subramanian, S.; Eswaran, S. Design, synthesis and study of antibacterial and antitubercular activity of quinoline hydrazone hybrids. Heterocycl. Comm. 2020, 26, 137–147. [Google Scholar] [CrossRef]
- Gnanavelu, K.; Vinay Kumar, K.S.; Eswaran, S.; Sivashanmugam, K. Novel quinoline-piperazine hybrids: The design, synthesis and evaluation of antibacterial and antituberculosis properties. RSC Med. Chem. 2023, 14, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Lauria, A.; La Monica, G.; Bono, A.; Martorana, A. Quinoline anticancer agents active on DNA and DNA-interacting proteins: From classical to emerging therapeutic targets. Eur. J. Med. Chem. 2021, 220, 113555. [Google Scholar] [CrossRef]
- Martorana, A.; La Monica, G.; Lauria, A. Quinoline-Based Molecules Targeting c-Met, EGF, and VEGF Receptors and the Proteins Involved in Related Carcinogenic Pathways. Molecules 2020, 25, 4279. [Google Scholar] [CrossRef]
- Mingoia, F.; Di Sano, C.; D’Anna, C.; Fazzari, M.; Minafra, L.; Bono, A.; La Monica, G.; Martorana, A.; Almerico, A.M.; Lauria, A. Synthesis of new antiproliferative 1,3,4-substituted-pyrrolo [3,2-c]quinoline derivatives, biological and in silico insights. Eur. J. Med. Chem. 2023, 258, 115537. [Google Scholar] [CrossRef]
- La Monica, G.; Pizzolanti, G.; Baiamonte, C.; Bono, A.; Alamia, F.; Mingoia, F.; Lauria, A.; Martorana, A. Design and Synthesis of Novel Thieno[3,2-c]quinoline Compounds with Antiproliferative Activity on RET-Dependent Medullary Thyroid Cancer Cells. ACS Omega 2023, 8, 34640–34649. [Google Scholar] [CrossRef] [PubMed]
- Dulsat, J.; López-Nieto, B.; Estrada-Tejedor, R.; Borrell, J.I. Evaluation of Free Online ADMET Tools for Academic or Small Biotech Environments. Molecules 2023, 28, 776. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Urban, L.; Bojanic, D. Maximising use of in vitro ADMET tools to predict in vivo bioavailability and safety. Expert. Opin. Drug Metab. Toxicol. 2007, 3, 641–665. [Google Scholar] [CrossRef]
- Vrbanac, J.; Slauter, R. ADME in Drug Discovery. In A Comprehensive Guide to Toxicology in Nonclinical Drug Development; Academic Press: Cambridge, MA, USA, 2016; pp. 39–67. [Google Scholar]
- Stegemann, S.; Leveiller, F.; Franchi, D.; de Jong, H.; Lindén, H. When poor solubility becomes an issue: From early stage to proof of concept. Eur. J. Pharm. Sci. 2007, 31, 249–261. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2017-1: QikProp; Schrödinger, LLC: New York, NY, USA, 2017.
- Daina, A.; Zoete, V. A BOILED-Egg To Predict Gastrointestinal Absorption and Brain Penetration of Small Molecules. ChemMedChem 2016, 11, 1117–1121. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. J. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Egan, W.J.; Merz, K.M.; Baldwin, J.J. Prediction of drug absorption using multivariate statistics. J. Med. Chem. 2000, 43, 3867–3877. [Google Scholar] [CrossRef] [PubMed]
- Muegge, I.; Heald, S.L.; Brittelli, D. Simple selection criteria for drug-like chemical matter. J. Med. Chem. 2001, 44, 1841–1846. [Google Scholar] [CrossRef]
- Baell, J.B.; Holloway, G.A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [PubMed]
- Brenk, R.; Schipani, A.; James, D.; Krasowski, A.; Gilbert, I.H.; Frearson, J.; Wyatt, P.G. Lessons learnt from assembling screening libraries for drug discovery for neglected diseases. ChemMedChem 2008, 3, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Weyesa, A.; Mulugeta, E. Recent advances in the synthesis of biologically and pharmaceutically active quinoline and its analogues: A review. RSC Adv. 2020, 10, 20784–20793. [Google Scholar] [CrossRef] [PubMed]
- Wissner, A.; Berger, D.M.; Boschelli, D.H.; Floyd, M.B., Jr.; Greenberger, L.M.; Gruber, B.C.; Johnson, B.D.; Mamuya, N.; Nilakantan, R.; Reich, M.F.; et al. 4-Anilino-6,7-dialkoxyquinoline-3-carbonitrile inhibitors of epidermal growth factor receptor kinase and their bioisosteric relationship to the 4-anilino-6,7-dialkoxyquinazoline inhibitors. J. Med. Chem. 2000, 43, 3244–3256. [Google Scholar] [CrossRef] [PubMed]
- Capri, F.C.; Di Leto, Y.; Presentato, A.; Mancuso, I.; Scatassa, M.L.; Alduina, R. Characterization of Staphylococcus Species Isolates from Sheep Milk with Subclinical Mastitis: Antibiotic Resistance, Enterotoxins, and Biofilm Production. Foodborne Pathog. Dis. 2024, 21, 10–18. [Google Scholar] [CrossRef]
- Secretary, E. OPINION 36: Designation of Strain ATCC 10145 as the Neotype Strain of Pseudomonas aeruginosa (Schroeter) Migula. Int. J. Syst. Bacteriol. 1970, 20, 15–16. [Google Scholar] [CrossRef]
- Rubino, S.; Pibiri, I.; Minacori, C.; Alduina, R.; Di Stefano, V.; Orecchio, S.; Buscemi, S.; Girasolo, M.A.; Tesoriere, L.; Attanzio, A. Synthesis, structural characterization, anti-proliferative and antimicrobial activity of binuclear and mononuclear Pt(II) complexes with perfluoroalkyl-heterocyclic ligands. Inorganica Chim. Acta 2018, 483, 180–190. [Google Scholar] [CrossRef]
- Saladino, M.L.; Markowska, M.; Carmone, C.; Cancemi, P.; Alduina, R.; Presentato, A.; Scaffaro, R.; Biały, D.; Hasiak, M.; Hreniak, D.; et al. Graphene Oxide Carboxymethylcellulose Nanocomposite for Dressing Materials. Materials 2020, 13, 1980. [Google Scholar] [CrossRef]
- Ronkin, S.M.; Badia, M.; Bellon, S.; Grillot, A.L.; Gross, C.H.; Grossman, T.H.; Mani, N.; Parsons, J.D.; Stamos, D.; Trudeau, M.; et al. Discovery of pyrazolthiazoles as novel and potent inhibitors of bacterial gyrase. Bioorg. Med. Chem. Lett. 2010, 20, 2828–2831. [Google Scholar] [CrossRef]
- Martínez-Botella, G.; Loch, J.T.; Green, O.M.; Kawatkar, S.P.; Olivier, N.B.; Boriack-Sjodin, P.A.; Keating, T.A. Sulfonylpiperidines as novel, antibacterial inhibitors of Gram-positive thymidylate kinase (TMK). Bioorg. Med. Chem. Lett. 2013, 23, 169–173. [Google Scholar] [CrossRef] [PubMed]
- Oefner, C.; Parisi, S.; Schulz, H.; Lociuro, S.; Dale, G.E. Inhibitory properties and X-ray crystallographic study of the binding of AR-101, AR-102 and iclaprim in ternary complexes with NADPH and dihydrofolate reductase from Staphylococcus aureus. Acta Crystallogr. D Biol. Crystallogr. 2009, 65, 751–757. [Google Scholar] [CrossRef]
- Qiu, X.; Janson, C.A.; Smith, W.W.; Green, S.M.; McDevitt, P.; Johanson, K.; Carter, P.; Hibbs, M.; Lewis, C.; Chalker, A.; et al. Crystal structure of Staphylococcus aureus tyrosyl-tRNA synthetase in complex with a class of potent and specific inhibitors. Protein Sci. 2001, 10, 2008–2016. [Google Scholar] [CrossRef]
- Axerio-Cilies, P.; See, R.H.; Zoraghi, R.; Worral, L.; Lian, T.; Stoynov, N.; Jiang, J.; Kaur, S.; Jackson, L.; Gong, H.; et al. Cheminformatics-driven discovery of selective, nanomolar inhibitors for staphylococcal pyruvate kinase. ACS Chem. Biol. 2012, 7, 350–359. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.I.; Liu, G.Y.; Song, Y.; Yin, F.; Hensler, M.E.; Jeng, W.Y.; Nizet, V.; Wang, A.H.; Oldfield, E. A cholesterol biosynthesis inhibitor blocks Staphylococcus aureus virulence. Science 2008, 319, 1391–1394. [Google Scholar] [CrossRef]
- Tan, C.M.; Therien, A.G.; Lu, J.; Lee, S.H.; Caron, A.; Gill, C.J.; Lebeau-Jacob, C.; Benton-Perdomo, L.; Monteiro, J.M.; Pereira, P.M.; et al. Restoring methicillin-resistant Staphylococcus aureus susceptibility to β-lactam antibiotics. Sci. Transl. Med. 2012, 4, 126ra35. [Google Scholar] [CrossRef]
- Vincetti, P.; Caporuscio, F.; Kaptein, S.; Gioiello, A.; Mancino, V.; Suzuki, Y.; Yamamoto, N.; Crespan, E.; Lossani, A.; Maga, G.; et al. Discovery of Multitarget Antivirals Acting on Both the Dengue Virus NS5-NS3 Interaction and the Host Src/Fyn Kinases. J. Med. Chem. 2015, 58, 4964–4975. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2017–2: LigPrep; Schrödinger, LLC: New York, NY, USA, 2017.
- Banks, J.L.; Beard, H.S.; Cao, Y.; Cho, A.E.; Damm, W.; Farid, R.; Felts, A.K.; Halgren, T.A.; Mainz, D.T.; Maple, J.R.; et al. Integrated Modeling Program, Applied Chemical Theory (IMPACT). J. Comput. Chem. 2005, 26, 1752–1780. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- RCSB PDB. Available online: www.rcsb.org (accessed on 10 December 2024).
- Schrödinger Release 2017-2: Protein Preparation Wizard; Epik, Schrödinger, LLC: New York, NY, USA, 2017.
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.-J.; Wang, J.-Z.; Deng, W.-Q.; Zou, K. Induced-fit docking and binding free energy calculation on furostanol saponins from Tupistra chinensis as epidermal growth factor receptor inhibitors. Med. Chem. Res. 2013, 22, 4970–4979. [Google Scholar] [CrossRef]
- Zhong, H.; Tran, L.M.; Stang, J.L. Induced-fit docking studies of the active and inactive states of protein tyrosine kinases. J. Mol. Graph. Model. 2009, 28, 336–346. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Aslanian, R.; Madison, V.S. Induced-fit docking of mometasone furoate and further evidence for glucocorticoid receptor 17alpha pocket flexibility. J. Mol. Graph. Model. 2008, 27, 512–521. [Google Scholar] [CrossRef] [PubMed]
- Sherman, W.; Beard, H.S.; Farid, R. Use of an induced fit receptor structure in virtual screening. Chem. Biol. Drug Des. 2006, 67, 83–84. [Google Scholar] [CrossRef] [PubMed]
- Sherman, W.; Day, T.; Jacobson, M.P.; Friesner, R.A.; Farid, R. Novel procedure for modeling ligand/receptor induced fit effects. J. Med. Chem. 2006, 49, 534–553. [Google Scholar] [CrossRef]
- Schrödinger Release 2023-4: Desmond Molecular Dynamics System; D.E. Shaw Research; Maestro-Desmond Interoperability Tools, Schrödinger: New York, NY, USA, 2023.
- Hooper, D.C. Mechanisms of action of antimicrobials: Focus on fluoroquinolones. Clin. Infect. Dis. 2001, 32 (Suppl. S1), S9–S15. [Google Scholar] [CrossRef] [PubMed]
- Murínová, S.; Dercová, K. Response mechanisms of bacterial degraders to environmental contaminants on the level of cell walls and cytoplasmic membrane. Int. J. Microbiol. 2014, 2014, 873081. [Google Scholar] [CrossRef]
- Sikkema, J.; de Bont, J.A.; Poolman, B. Mechanisms of membrane toxicity of hydrocarbons. Microbiol. Rev. 1995, 59, 201–222. [Google Scholar] [CrossRef] [PubMed]
- Treangen, T.J.; Maybank, R.A.; Enke, S.; Friss, M.B.; Diviak, L.F.; Karaolis, D.K.; Koren, S.; Ondov, B.; Phillippy, A.M.; Bergman, N.H.; et al. Complete Genome Sequence of the Quality Control Strain Staphylococcus aureus subsp. aureus ATCC 25923. Genome Announc. 2014, 2, e01110-14. [Google Scholar] [CrossRef]
- Walton, I.M.; Cox, J.M.; Benson, C.A.; Patel, D.G.; Chen, Y.S.; Benedict, J.B. The role of atropisomers on the photo-reactivity and fatigue of diarylethene-based metal-organic frameworks. New J. Chem. 2016, 40, 101–106. [Google Scholar] [CrossRef]
- Richter, M.F.; Hergenrother, P.J. The challenge of converting Gram-positive-only compounds into broad-spectrum antibiotics. Ann. N. Y. Acad. Sci. 2019, 1435, 18–38. [Google Scholar] [CrossRef] [PubMed]
- Saxena, D.; Maitra, R.; Bormon, R.; Czekanska, M.; Meiers, J.; Titz, A.; Verma, S.; Chopra, S. Tackling the outer membrane: Facilitating compound entry into Gram-negative bacterial pathogens. Npj Antimicrob. Resist. 2023, 1, 17. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound * | MW | C LogPo/w | TPSA | Csp3 | RB | ESOL LogS | QPPCaco |
|---|---|---|---|---|---|---|---|
| 5a | 402.45 | 2.56 | 78.69 | 0.26 | 5 | −4.23 | 860.5 |
| 5b | 416.47 | 2.92 | 78.69 | 0.29 | 5 | −4.53 | 860.3 |
| 5c | 432.47 | 2.54 | 87.92 | 0.29 | 6 | −4.30 | 858.4 |
| 5d | 470.44 | 3.60 | 78.69 | 0.29 | 6 | −5.10 | 917.9 |
| 5e | 436.89 | 3.10 | 78.69 | 0.26 | 5 | −4.82 | 859.3 |
| 5f | 420.44 | 2.84 | 78.69 | 0.26 | 5 | −4.39 | 859.2 |
| 5g | 481.34 | 3.18 | 78.69 | 0.26 | 5 | −5.14 | 789.5 |
| 5h | 492.52 | 2.55 | 106.38 | 0.35 | 8 | −4.47 | 938.4 |
| 5i | 454.88 | 3.32 | 78.69 | 0.26 | 5 | −4.99 | 710.91 |
| 5j | 445.51 | 2.57 | 81.93 | 0.32 | 6 | −4.47 | 828.9 |
| 5k | 471.34 | 3.55 | 78.69 | 0.26 | 5 | −5.42 | 1013.8 |
| Compound | QP Stars * | LRoF | GV | VV | EV | MV | PAINS | Brenk Alerts |
|---|---|---|---|---|---|---|---|---|
| 5a | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 5b | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 5c | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 5d | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 5e | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 5f | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 5g | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 0 |
| 5h | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 0 |
| 5i | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 5j | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 |
| 5k | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 0 |
| Target | PDB | Compound | IFD Score | Glide Score | Prime Energy |
|---|---|---|---|---|---|
| Tyrosyl–tRNA synthetase (TyrRS) * | 1JIJ [48] | Ref. inhibitors # | −676.10 ÷ −681.22 | −9.552 ÷ −11.473 | −13,327.4 ÷ −13,390.2 |
| 5k | −678.81 | −9.268 | −13,390.3 | ||
| Pyruvate Kinase (PK) * | 3T0T [49] | Ref. inhibitors | −2473.28 ÷ −2476.14 | −10.038 ÷ −11.023 | −49,244.4 ÷ −49,320.6 |
| 5k | −2473.46 | −9.572 | −49,277.2 | ||
| Gyrase B * | 3G7B [45] | Ref. inhibitors | −442.53 ÷ −445.73 | −6.890 ÷ −7.911 | −8704.1 ÷ −8755.1 |
| 5k | −442.92 | −8.751 | −8682.8 | ||
| FtsZ | 4DXD [51] | Ref. inhibitors | −696.44 ÷ −696.62 | −11.082 ÷ −11.814 | −13,696.0 ÷ −13,707.3 |
| 5k | −693.60 | −9.507 | 13,681.3 | ||
| CrtM | 2ZCQ [50] | Ref. inhibitors | −686.06 ÷ −688.91 | −10.927 ÷ −13.583 | −13,429.6 ÷ −13,506.6 |
| 5k | −682.43 | −10.927 | −13,429.6 | ||
| Thymidylate Kinase (TMK) | 4HLC [46] | Ref. inhibitors | −453.73 ÷ −455.97 | −8.513 ÷ −10.406 | −8823.6 ÷ −8910.8 |
| 5k | −449.39 | −8.185 | |||
| Dihydrofolate reductase (DHFR) | 3FYW [47] | Ref. inhibitors | −373.07 ÷ −375.62 | −8.021 ÷ −9.797 | −7291.2 ÷ −7306.7 |
| 5k | −364.46 | −7.644 | −7135.7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
La Monica, G.; Gallo, A.; Bono, A.; Alamia, F.; Lauria, A.; Alduina, R.; Martorana, A. Novel Antibacterial 4-Piperazinylquinoline Hybrid Derivatives Against Staphylococcus aureus: Design, Synthesis, and In Vitro and In Silico Insights. Molecules 2025, 30, 28. https://doi.org/10.3390/molecules30010028
La Monica G, Gallo A, Bono A, Alamia F, Lauria A, Alduina R, Martorana A. Novel Antibacterial 4-Piperazinylquinoline Hybrid Derivatives Against Staphylococcus aureus: Design, Synthesis, and In Vitro and In Silico Insights. Molecules. 2025; 30(1):28. https://doi.org/10.3390/molecules30010028
Chicago/Turabian StyleLa Monica, Gabriele, Annamaria Gallo, Alessia Bono, Federica Alamia, Antonino Lauria, Rosa Alduina, and Annamaria Martorana. 2025. "Novel Antibacterial 4-Piperazinylquinoline Hybrid Derivatives Against Staphylococcus aureus: Design, Synthesis, and In Vitro and In Silico Insights" Molecules 30, no. 1: 28. https://doi.org/10.3390/molecules30010028
APA StyleLa Monica, G., Gallo, A., Bono, A., Alamia, F., Lauria, A., Alduina, R., & Martorana, A. (2025). Novel Antibacterial 4-Piperazinylquinoline Hybrid Derivatives Against Staphylococcus aureus: Design, Synthesis, and In Vitro and In Silico Insights. Molecules, 30(1), 28. https://doi.org/10.3390/molecules30010028