
New Triterpenoids from the Leaves of Heritiera littoralis and Their Anti-Inflammatory Activity

 ,
,
Abstract

1. Introduction
2. Results and Discussion
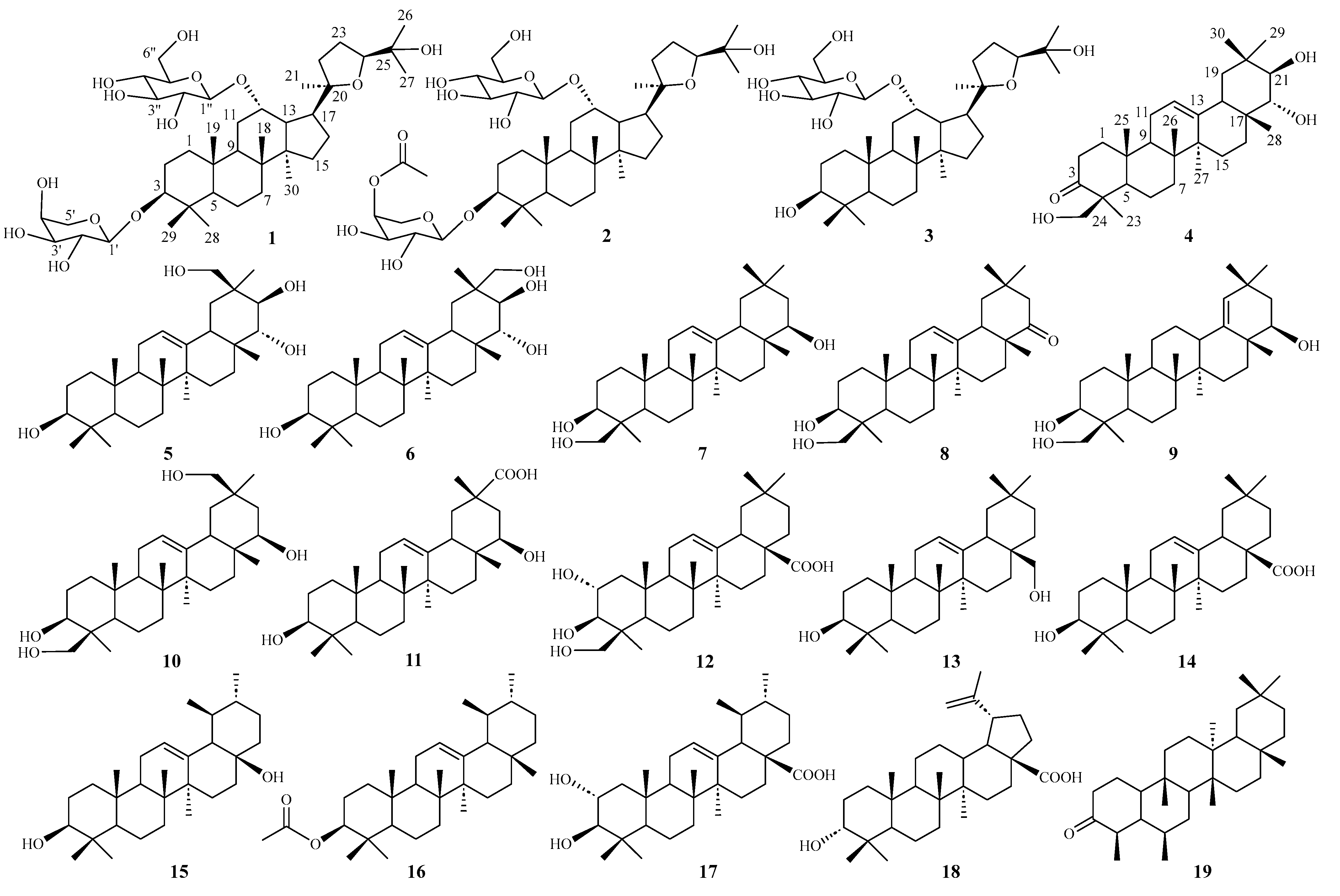
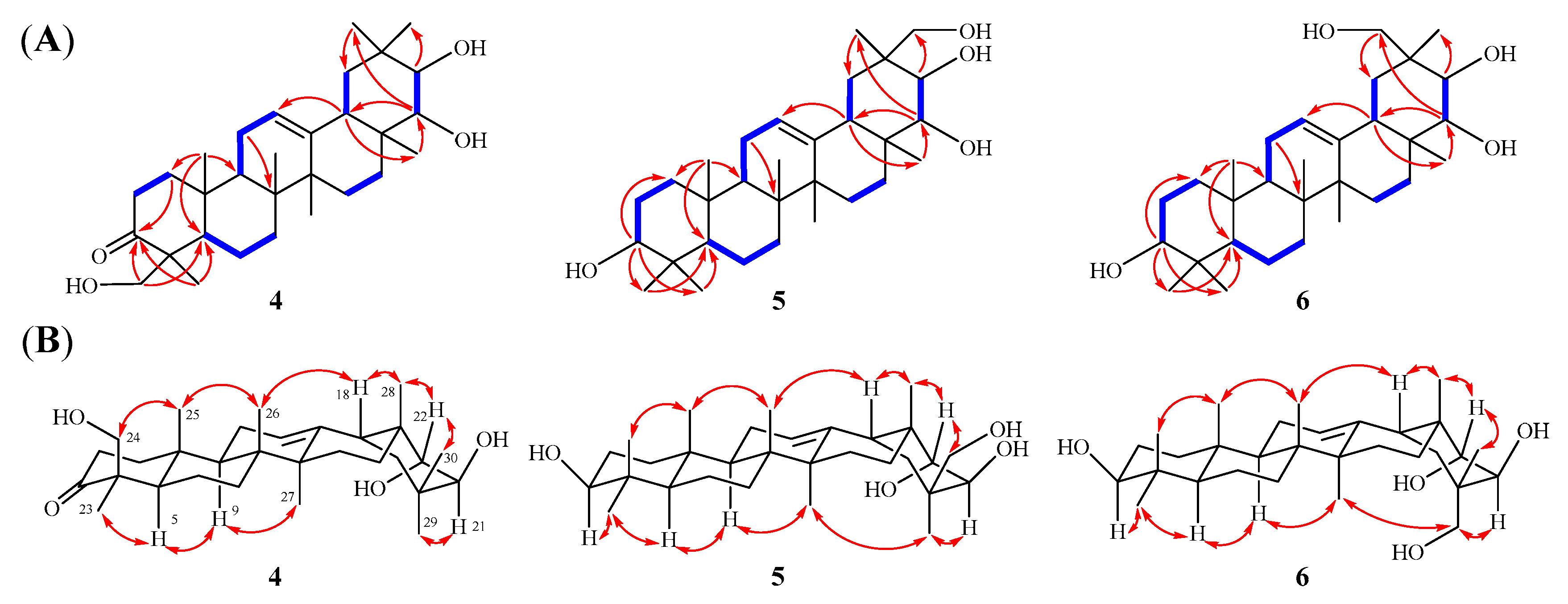
2.1. Elucidation of the Chemical Structures of Heritieras C–H (1–6)
2.2. Anti-Inflammatory Assay of the Isolates
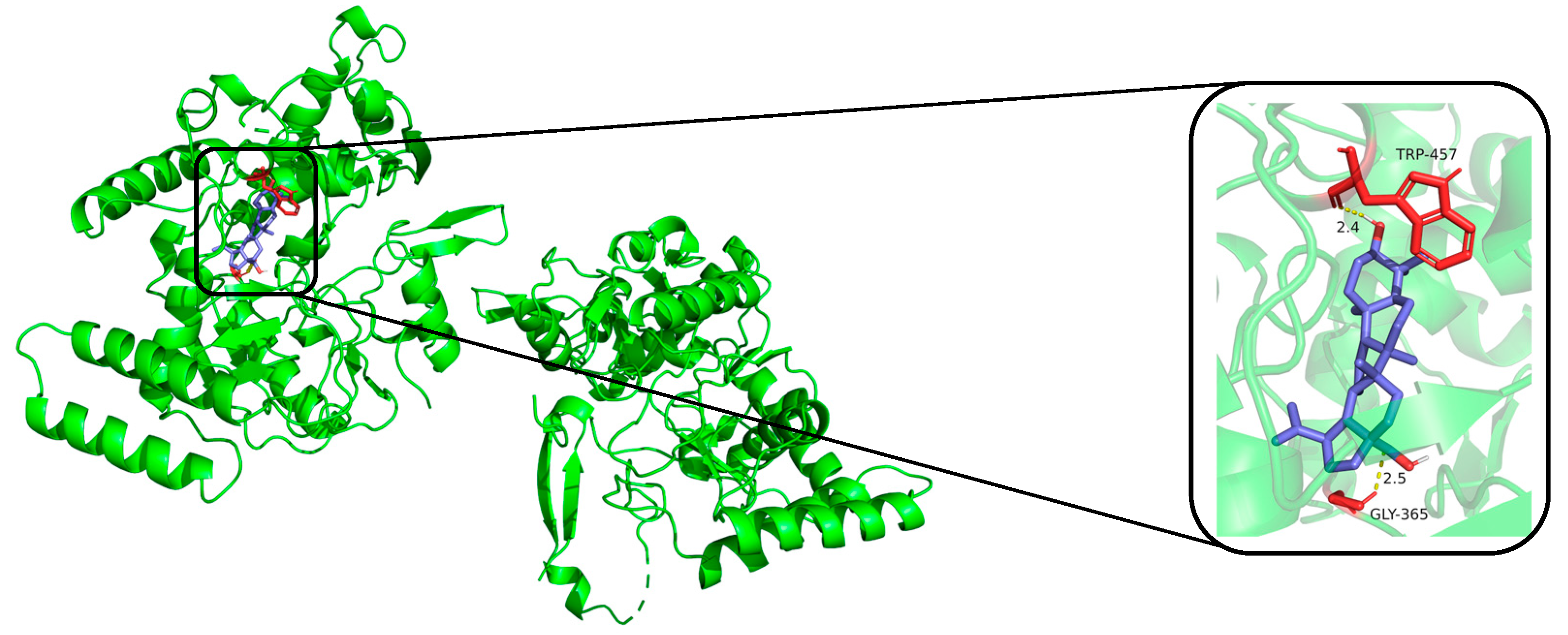
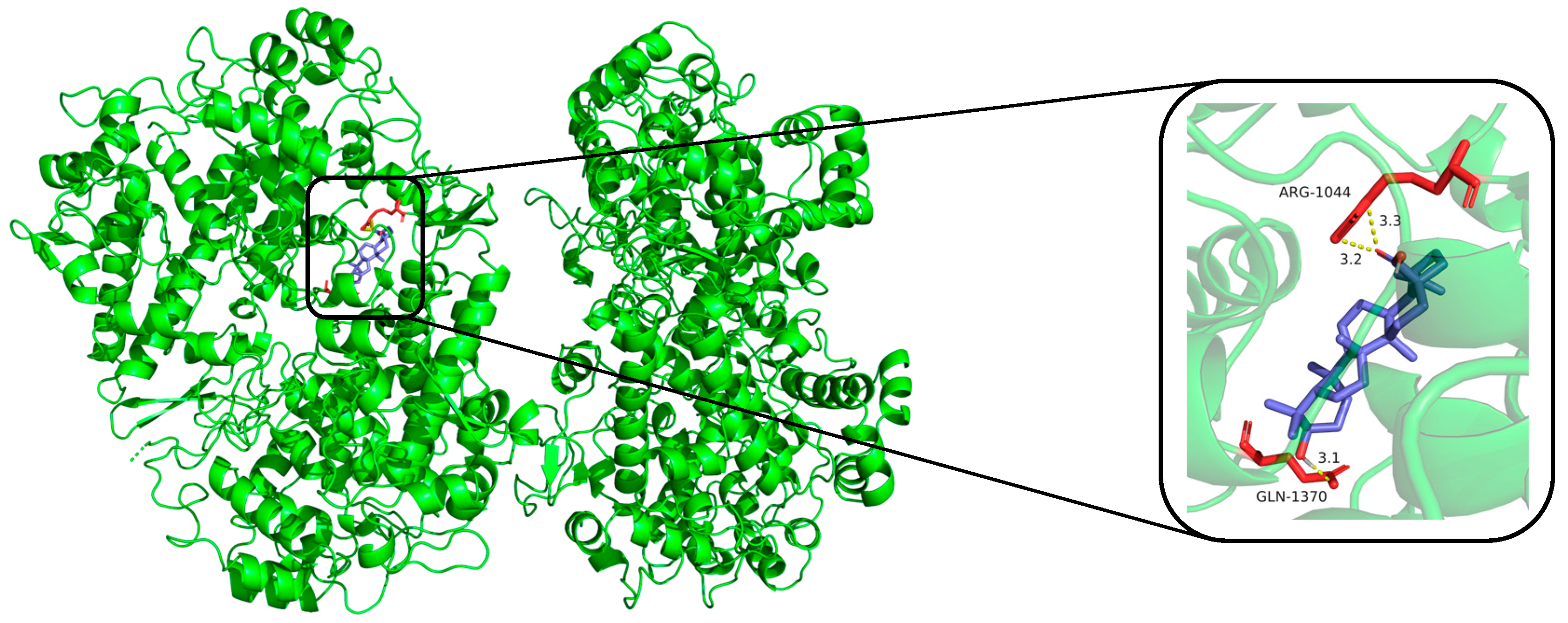
2.3. Predicted Binding Modes of Compounds and iNOS, COX-2 Using Molecular Docking Analysis
3. Experimental
3.1. General Experimental Procedures
3.2. Plant Material
3.3. Extraction and Isolation
3.4. Characterization of the Isolates
3.5. Enzymatic Hydrolysis of Compounds 1–3
3.6. Anti-Inflammatory Assay
3.7. Molecular Docking Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- The Biodiversity Committee of Chinese Academy of Sciences. Catalogue of Life China: 2024 Annual Checklist; The Biodiversity Committee of Chinese Academy of Sciences: Beijing, China, 2024. [Google Scholar]
- Calixto-Júnior, J.T.; Morais, S.M.; Colares, A.V.; Coutinho, H.D.M. The Genus Luehea (Malvaceae-Tiliaceae): Review about chemical and pharmacological aspects. J. Pharm. 2016, 2016, 1368971. [Google Scholar] [CrossRef]
- Editorial Committee of Flora of China. Flora of China. Science Press: Beijing, China, 1984; Volume 49, pp. 39–40. [Google Scholar]
- Lin, P. Medicinal plants of mangrove in China. J. Mar. Drugs 1984, 12, 45–51. [Google Scholar]
- Shao, C.L.; Fu, X.M.; Wang, C.Y.; Han, L.; Fang, Y.C.; Li, G.Q.; Zeng, X.Q.; Liu, G.X.; Guan, H.S. Investigation on the status of mangrove resources and medicinal research in China III. status of folk medicinal usage and medicinal research. J. Ocean Univ. Chin. 2009, 39, 712–718. [Google Scholar]
- Du, Q.; Wei, W.M.; Mi, D.Q. Knowledge and existing status of medicinal ethnobotany of mangrove among Jing People in Guangxi. Guihaia 2016, 36, 405–412. [Google Scholar]
- Ning, X.Q.; Li, J.F.; Huang, Y.; Tan, Y.F.; Deng, J.G. Study on the species of medicinal mangroves and their folk medicinal efficacy in Guangxi. Guide Chin. Med. 2013, 11, 73–75. [Google Scholar]
- Fan, H.Q.; Liang, S.C. (Eds.) Research and Management on China Mangroves; Science Press: Beijing, China, 1995; pp. 164–172. [Google Scholar]
- Cui, J.G.; Lu, Y.; Huang, Y.M. Review on bioactive substances from mangrove. Nat. Prod. Res. Dev. 2017, 29, 1626–1633. [Google Scholar]
- Ge, L.; Li, Y.J.; Yang, K.D. Chemical constituents of the leaves of Heritiera littoralis. Chem. Nat. Compd. 2016, 52, 603–604. [Google Scholar] [CrossRef]
- Miles, D.H.; Vallapa, C.W. Toxicants from mangrove plants, VII. Vallapin and vallapianin, novel sesquiterpene lactones from the mangrove plant Heritiera littoralis. J. Nat. Prod. 1991, 54, 286–289. [Google Scholar] [CrossRef]
- Takeda, Y.; Miyazaki, K.; Shimizu, H.; Masuda, T.; Otsuka, H. A new phenylpropanoid-glycerol conjugate from Heritiera littoralis Dryand. Nat. Med. 2000, 54, 22–25. [Google Scholar]
- Tian, Y.; Wu, J.; Xi, S.H.; Zhang, D.J.; Xu, L.R.; Zhang, S. Studies on the triterpenoid components of Heritiera littoralis. Chin. Trad. Herb. Drugs 2007, 3, 35–36. [Google Scholar]
- Tian, Y.; Wu, J.; Zhang, S. Flavonoids from leaves of Heritiera littoralis. J. Chin. Pharm. Sci. 2004, 13, 214–216. [Google Scholar]
- Christopher, R.; Nyandoro, S.S.; Chacha, M.; De Koning, C.B. A new cinnamoylgly-coflavonid antimycobacterial and antioxidant constituents from Heritiera littoralis leaf extracts. Nat. Prod. Res. 2014, 28, 351–358. [Google Scholar] [CrossRef]
- Liang, X.Q.; Niu, P.; Li, J.; Guan, X.L.; Zhang, Y.J.; Li, J. Discovery of anti-inflammatory triterpenoid glucosides from the Heritiera littoralis Dryand. Molecules 2023, 28, 1658. [Google Scholar] [CrossRef] [PubMed]
- Tewtrakul, S.; Tansakul, P.; Daengrot, C.; Ponglimanont, C.; Karalai, C. Anti-inflammatory principles from Heritiera littoralis bark. Phytomedicine 2010, 17, 851–855. [Google Scholar] [CrossRef] [PubMed]
- Nagai, N.; Tanaka, N.; Ichikawa, S.; Tanaka, O. Triterpenes of betula platyphylla sukatchev var. Japonica hara and the configuration at C-24 of ocotillol-II and its related compounds. Tetrahedron Lett. 1968, 9, 4239–4242. [Google Scholar]
- Liang, X.Q.; Deng, S.P.; Huang, Y.; Pan, L.W.; Chang, Y.L.; Hou, P.; Ren, C.Y.; Xu, F.W.; Yang, R.Y.; Li, K.Y.; et al. Triterpenoids from the leaves of Cyclocarya paliurus and their glucose uptake activity in 3T3-L1 adipocytes. Molecules 2023, 28, 3294. [Google Scholar] [CrossRef] [PubMed]
- Fei, Y.H.; Lou, H.X. Natural Medicinal Chemistry, 7th ed.; People’s Publishing Agence: Beijing, China, 2016. [Google Scholar]
- Seger, C.; Pointinger, S.; Greger, H.; Hofer, O. Isoeichlerianic acid from Aglaia silvestris and revision of the stereochemistry of foveolin B. Tetrahedron Lett. 2008, 49, 4313–4315. [Google Scholar] [CrossRef]
- Cui, B.S.; Li, S. New triterpenoid saponins from the leaves of Cyclocarya paliurus. Chin. Chem. Lett. 2015, 26, 585–589. [Google Scholar] [CrossRef]
- Arao, T.; Kinjo, J.; Nohara, T.; Isobe, R. Oleanene-type triterpene glycosides from Puerariae Radix. II. Isolation of saponins and the application of tandem mass spectrometry to their structure determination. Chem. Pharm. Bull. 1995, 43, 1176–1179. [Google Scholar] [CrossRef] [PubMed]
- Miyao, H.; Sakai, Y.; Takeshita, T.; Hinjo, J.; Nohara, T. Triterpene saponins from Abrus cantoniensis (Leguminosae). I. Isolatioin and characterization of four new saponins and a new sapogenol. Chem. Pharm. Bull. 1996, 44, 1222–1227. [Google Scholar] [CrossRef]
- Ding, P.L.; Hou, A.J.; Chen, D.F. Three new isoprenylated flavonoids from the roots of Sophora flavescens. J. Asian Nat. Prod. Res. 2005, 7, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Macías, F.A.; Simonet, A.M.; Galindo, J.C.G.; Pacheco, P.C.; Sánchez, J.A. Bioactive polar triterpenoids from Melilotus messanensis. Phytochemistry 1998, 49, 709–717. [Google Scholar] [CrossRef]
- Tava, A.; Biazzi, E.; Mella, M.; Quadrelli, P.; Avato, P. Artefact formation during acid hydrolysis of saponins from Medicago spp. Phytochemistry 2017, 138, 116–127. [Google Scholar] [CrossRef] [PubMed]
- Kinjo, J.; Fujishima, Y.; Saino, K.; Tian, R.H.; Nohara, T. Five new triterpene glycosides from Wisteria branchybotrys (Leguminosae). Chem. Pharm. Bull. 1995, 43, 636–640. [Google Scholar] [CrossRef]
- Chen, Y. Triterpenoids from Tripterygium willfordii. J. Hubei Univ. Technol. 2000, 04, 42–44. [Google Scholar]
- Lee, I.K.; Kim, D.H.; Lee, S.Y.; Kim, K.R.; Kang, R.L. Triterpenoic acids of Prunella vulgarisvar. lilacina and their cytotoxic activities in vitro. Arch. Pharm. Res. 2008, 31, 1578–1583. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; Iuchi, M.; Fujita, Y.; Minami, H.; Fukuyama, Y. Coumaroyl triterpenes from Casuarina equisetifolia. Phytochemistry 1999, 51, 543–550. [Google Scholar] [CrossRef]
- Zhu, L.L.; Zhong, L.J.; Xu, M.J.; Chen, J.K.; Liang, L.F. Chemical Constituents of Triterpenes from Sabia discolor Dunn. Chem. Ind. Forest Prod. 2022, 42, 95–100. [Google Scholar]
- Siddiqui, S.; Hafeez, F.; Begum, S.; Siddiqui, B.S. Kaneric Acid, a new triterpene from the leaves of Nerium oleander. J. Nat. Prod. 1986, 49, 1086–1090. [Google Scholar] [CrossRef]
- Shiojima, K.; Suzuki, H.; Kodera, N.; Ageta, H.; Chang, H.C.; Chen, Y.P. Composite constituents: Thirty-nine triterpenoids including two novel compounds from Ixeris chinensis. Chem. Pharm. Bull. 1996, 44, 509–514. [Google Scholar] [CrossRef]
- Cui, Y.; Zhang, X.M.; Chen, J.J.; Zhang, Y.; Lin, X.K.; Zhou, L. Chemical constituents from root of Actinidia chinensis. China J. Chin. Mater. Med. 2007, 32, 1663–1665. [Google Scholar]
- Xu, J.; Gao, H.Y.; Ma, S.L.; Li, H.H.; Bai, L.M. Chemical constituents and bioactivity of Kalimeris indica. Chin. Trad. Herb. Drugs 2014, 45, 3246–3250. [Google Scholar]
- Lee, S.R.; Lee, S.; Moon, E.; Park, H.J.; Park, H.B.; Kim, K.H. Bioactivity-guided isolation of anti-inflammatory triterpenoids from the sclerotia of Poria cocos using LPS-stimulated RAW 264. 7 cells. Bioorg. Chem. 2017, 70, 94–99. [Google Scholar] [CrossRef]
- Kim, A.T.; Kim, D.O. Anti-inflammatory effects of vanadium-binding protein from Halocynthia roretzi in LPS-stimulated RAW 264. 7 macrophages through NF-κB and MAPK pathways. Int. J. Biol. Macromol. 2019, 133, 732–738. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.S.; Kim, Y.J.; Kim, B.Y.; Park, G.; Jeong, S.J. The anti-neuroinflammatory activity of tectorigenin pretreatment via downregulated NF-κB and ERK/JNK pathways in BV-2 microglial and microglia inactivation in mice with lipopolysaccharide. Front. Pharmacol. 2018, 9, 462–475. [Google Scholar] [CrossRef] [PubMed]
- Stefani, H.A.; Gueogjan, K.; Manarin, F.; Farsky Sandra, H.P.; Zukerman-Schpector, J.; Caracelli, I.; Pizano Rodrigues, S.R.; Muscará, M.N.; Teixeira, S.A.; Santin, J.R.; et al. Synthesis, biological evaluation and molecular docking studies of 3-(triazolyl)-coumarin derivatives: Effect on inducible nitric oxide synthase. Eur. J. Med. Chem. 2012, 58, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Rupa, E.J.; Zheng, S.; Nahar, J.; Yang, D.C.; Kang, S.C.; Wang, Y. Panos-fermented extract-mediated nanoemulsion: Preparation, characterization, and in vitro anti-inflammatory effects on RAW 264.7 cells. Molecules 2022, 27, 218. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.J.; Huang, X.S.; Chen, H.C.; Zhou, D.X.; Yang, Z.M.; Wang, K.; Liu, W.; Deng, S.P.; Yang, R.Y.; Li, J.; et al. Discovery of anti-inflammatory terpenoids from Mallotus conspurcatus Croizat. J. Ethnopharmacol. 2019, 231, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Yang, X.; Ma, J.; Yang, Y.; Xie, C.; Tuerhong, M.; Jin, D.Q.; Xu, J.; Lee, D.; Ohizumi, Y.; et al. Nitric oxide inhibitory daphnane diterpenoids as potential anti-neuroinflammatory agents for AD from the twigs of Trigonostemon thyrsoideus. Bioorg. Chem. 2017, 75, 149–156. [Google Scholar] [CrossRef]
- Garcin, E.D.; Arvai, A.S.; Rosenfeld, R.J.; Kroeger, M.D.; Crane, B.R.; Andersson, G.; Andrews, G.; Hamley, P.J.; Nicholls, P.R.; Mallinder, D.J.; et al. Anchored plasticity opens doors for selective inhibitor design in nitric oxide synthase. Nat. Chem. Biol. 2008, 4, 700–707. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 | 2 | 3 | ||||
|---|---|---|---|---|---|---|
| NO. | δH (J in Hz) | δC (DEPT) | δH (J in Hz) | δC (DEPT) | δH (J in Hz) | δC (DEPT) |
| 1 | 3.16, dt (10.3, 3.1); 2.08, m | 36.1, CH2 | 3.20, dt (10.3, 3.1); 2.13, m | 35.6, CH2 | 3.12, dt (10.3, 3.1); 2.16, m | 36.0, CH2 |
| 2 | 2.07, m; 1.87, m | 22.3, CH2 | 2.13, m; 1.86, m | 22.0, CH2 | 2.08, m; 1.75, m | 27.0, CH2 |
| 3 | 3.61, brs | 81.8, CH | 3.58, brs | 81.5, CH | 3.59, brs | 75.7, CH |
| 4 | 38.5, C | 38.0, C | 38.9, C | |||
| 5 | 1.60, m | 51.4, CH | 1.64, m | 50.9, CH | 1.79, m | 50.3, CH |
| 6 | 1.57, m; 1.45, m | 18.8, CH2 | 1.57, m; 1.45, m | 18.3, CH2 | 1.54, m; 1.46, m | 18.8, CH2 |
| 7 | 1.50, m; 1.16, m | 36.8, CH2 | 1.52, m; 1.16, m | 36.4, CH2 | 1.63, m; 1.21, m | 36.8, CH2 |
| 8 | 41.7, C | 41.5, C | 41.8, C | |||
| 9 | 1.91, m | 54.4, CH | 1.92, m | 53.9, CH | 2.05, m | 54.7, CH |
| 10 | 41.9, C | 39.9, C | 40.5, C | |||
| 11 | 2.93, dt (8.5, 3.9); 1.53, m | 35.0, CH2 | 2.93, dt (8.5, 3.9); 1.52, m | 34.5, CH2 | 2.96, dt (12.2, 4.1); 1.54, m | 34.8, CH2 |
| 12 | 4.47, m | 78.0, CH | 4.47, m | 77.8, CH | 4.54, m | 77.2, CH |
| 13 | 1.79, m | 41.6, CH | 1.79, m | 41.2, CH | 1.84, m | 41.5, CH |
| 14 | 50.5, C | 50.0, C | 50.5, C | |||
| 15 | 1.37, m; 0.99, m | 32.0, CH2 | 1.37, m; 0.99, m | 31.5, CH2 | 1.44, m; 1.03, m | 31.9, CH2 |
| 16 | 1.84, m; 1.76, m | 26.7, CH2 | 1.86, m; 1.75, m | 26.4, CH2 | 1.87, m; 1.64, m | 26.8, CH2 |
| 17 | 1.88, m | 50.0, CH | 1.88, m | 49.2, CH | 1.90, m | 49.7, CH |
| 18 | 1.03, s | 17.4, CH3 | 1.04, s | 17.0, CH3 | 1.07, s | 17.5, CH3 |
| 19 | 1.41, s | 17.4, CH3 | 1.42, s | 16.8, CH3 | 1.43, s | 17.3, CH3 |
| 20 | 86.9, C | 86.4, C | 86.9, C | |||
| 21 | 1.18, s | 25.0, CH3 | 1.18, s | 24.5, CH3 | 1.20, s | 24.8, CH3 |
| 22 | 1.77, m; 1.57, m | 34.6, CH2 | 1.75, m; 1.57, m | 34.2, CH2 | 1.77, m; 1.60, m | 34.7, CH2 |
| 23 | 2.05, m; 1.95, m | 27.1, CH2 | 2.06, m; 1.95, m | 26.7, CH2 | 2.05, m; 1.97, m | 26.7, CH2 |
| 24 | 3.95, t (7.2) | 84.7, CH | 3.96, t (7.2) | 84.2, CH | 3.98, t (7.2) | 84.6, CH |
| 25 | 71.7, C | 71.2, C | 71.7, C | |||
| 26 | 1.41, s | 26.7, CH3 | 1.42, s | 26.2, CH3 | 1.44, s | 26.5, CH3 |
| 27 | 1.48, s | 28.1, CH3 | 1.48, s | 27.7, CH3 | 1.49, s | 28.0, CH3 |
| 28 | 0.95, s | 23.6, CH3 | 0.95, s | 23.1, CH3 | 0.95, s | 23.4, CH3 |
| 29 | 1.26, s | 30.5, CH3 | 1.27, s | 29.8, CH3 | 1.26, s | 30.4, CH3 |
| 30 | 0.63, s | 17.2, CH3 | 0.61, s | 16.7, CH3 | 0.81, s | 17.1, CH3 |
| 1′ | 4.72, d (6.4) | 102.3, CH | 4.73, d (6.5) | 102.2, CH | ||
| 2′ | 4.43, dd (8.2, 6.4) | 72.9, CH | 4.31, m | 75.1, CH | ||
| 3′ | 4.23, dd (8.2, 3.4) | 75.2, CH | 4.23, m | 75.1, CH | ||
| 4′ | 4.37, m | 69.6, CH | 5.41, m | 73.3, CH | ||
| 5′ | 4.33, m; 3.76, m | 66.8, CH2 | 4.34, m; 3.53, m | 63.5, CH2 | ||
| 1″ | 5.14, d (8.0) | 102.7, CH | 5.14, d (8.0) | 102.3, CH | 5.17, d (7.8) | 102.0, CH |
| 2″ | 4.01, m | 75.8, CH | 4.02, m | 75.3, CH | 4.04, m | 75.5, CH |
| 3″ | 4.30, m | 79.1, CH | 4.29, m | 78.7, CH | 4.30, m | 78.9, CH |
| 4″ | 4.16, m | 72.9, CH | 4.15, m | 72.3, CH | 4.14, m | 72.7, CH |
| 5″ | 4.03, m | 78.3, CH | 4.04, m | 77.8, CH | 4.04, m | 78.2, CH |
| 6″ | 4.54, dd (11.3, 3.0); 4.37, dd (11.3, 5.5) | 63.9, CH2 | 4.54, dd (11.3, 3.0); 4.37, dd (11.3, 5.5) | 63.3, CH2 | 4.54, dd (11.3, 3.0); 4.37, dd (11.3, 5.5) | 63.9, CH2 |
| -OOCCH3 | 170.7, C | |||||
| -OOCCH3 | 1.98, s | 20.9, CH3 | ||||
| 4 a | 5 a | 6 b | ||||
|---|---|---|---|---|---|---|
| NO. | δH (J in Hz) | δC (DEPT) | δH (J in Hz) | δC (DEPT) | δH (J in Hz) | δC (DEPT) |
| 1 | 1.33, m; 1.78, m | 40.8, CH2 | 1.00, m; 1.58, m | 39.5, CH2 | 1.01, m; 1.60, m | 39.5, CH2 |
| 2 | 2.37, m; 2.83, m | 36.0, CH2 | 1.86, m; 1.86, m | 28.5, CH2 | 1.86, m; 1.86, m | 28.5, CH2 |
| 3 | 214.9, C | 3.46, dd (11.2, 5.0) | 78.4, CH | 3.46, dd (11.2, 5.0) | 78.4, CH | |
| 4 | 55.6, C | 42.5, C | 39.5, C | |||
| 5 | 1.39, m | 58.2, CH | 0.88, m | 56.1, CH | 0.86, m | 56.1, CH |
| 6 | 1.18, m; 1.64, m | 20.4, CH2 | 1.44, m; 1.63, m | 19.2, CH2 | 1.45, m; 1.61, m | 19.2, CH2 |
| 7 | 1.30, m; 1.49, m | 33.3, CH2 | 1.35, m; 1.56, m | 33.2, CH2 | 1.33, m; 1.55, m | 33.2, CH2 |
| 8 | 40.6, C | 39.8, C | 39.8, C | |||
| 9 | 1.68, m | 47.9, CH | 1.68, m | 48.4, CH | 1.69, m | 48.4, CH |
| 10 | 37.4, C | 37.6, C | 37.6, C | |||
| 11 | 1.68, m; 1.91, m | 24.6, CH2 | 1.90, m; 1.90, m | 24.3, CH2 | 1.92, m; 1.94, m | 24.3, CH2 |
| 12 | 5.35, t (3.6) | 122.6, CH | 5.36, t (3.4) | 123.4, CH | 5.41, t (3.6) | 123.0, CH |
| 13 | 145.0, C | 144.8, C | 145.1, C | |||
| 14 | 42.4, C | 41.1, C | 42.5, C | |||
| 15 | 0.97, m; 1.84, m | 26.9, CH2 | 1.00, m; 1.90, m | 26.9, CH2 | 1.01, m; 1.90, m | 27.0, CH2 |
| 16 | 1.01, m; 1.96, m | 27.8, CH2 | 1.10, m; 2.06, m | 28.0, CH2 | 1.05, m; 2.14, m | 27.8, CH2 |
| 17 | 39.6, C | 39.8, C | 41.5, C | |||
| 18 | 2.61, dd (13.8, 4.3) | 44.3, CH | 2.89, dd (13.8, 4.3) | 44.6, CH | 2.75, dd (13.8, 4.3) | 43.7, CH |
| 19 | 1.34, m; 2.09, m | 47.6, CH2 | 1.81, m; 2.06, m | 43.9, CH2 | 1.40, m; 2.58, m | 41.4, CH2 |
| 20 | 37.0, C | 40.6, C | 40.7, C | |||
| 21 | 3.87, s | 75.0, CH | 4.15, d (3.4) | 76.6, CH | 4.51, d (3.5) | 70.8, CH |
| 22 | 3.78, t (3.0) | 80.0, CH | 3.84, s | 79.8, CH | 3.87, s | 80.2, CH |
| 23 | 1.52, s | 21.2, CH3 | 1.26, s | 29.1, CH3 | 1.25, s | 29.1, CH3 |
| 24 | 3.89, d (11.1) 4.37, d (11.1) | 65.6, CH2 | 1.08, s | 16.9, CH3 | 1.08, s | 17.0, CH3 |
| 25 | 1.17, s | 16.1, CH3 | 0.96, s | 16.1, CH3 | 0.98, s | 16.1, CH3 |
| 26 | 1.06, s | 17.4, CH3 | 1.31, s | 17.4, CH3 | 1.08, s | 17.5, CH3 |
| 27 | 1.22, s | 27.0, CH3 | 1.06, s | 27.1, CH3 | 1.33, s | 27.1, CH3 |
| 28 | 1.29, s | 22.7, CH3 | 1.34, s | 22.8, CH3 | 1.35, s | 22.7, CH3 |
| 29 | 1.46, s | 21.7, CH3 | 1.44, s | 27.4, CH3 | 3.66, d (10.1) 4.07, d (10.1) | 72.0, CH3 |
| 30 | 1.25, s | 31.9, CH3 | 4.33, d (10.3) 4.42, d (10.3) | 67.4, CH2 | 1.54, s | 17.9, CH2 |
| Compounds | IC50 (μM) |
|---|---|
| 1 | 47.12 ± 0.61 |
| 12 | 25.23 ± 0.76 |
| 16 | 45.31 ± 0.45 |
| 17 | 39.98 ± 0.42 |
| 18 | 18.13 ± 0.32 |
| Dexamethasone b | 16.37 ± 0.82 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ren, C.; Liang, X.; Pi, R.; Xin, J.; Yang, B.; Zheng, Q.; Li, Y.; Li, J.; Zhang, Y. New Triterpenoids from the Leaves of Heritiera littoralis and Their Anti-Inflammatory Activity. Molecules 2025, 30, 131. https://doi.org/10.3390/molecules30010131
Ren C, Liang X, Pi R, Xin J, Yang B, Zheng Q, Li Y, Li J, Zhang Y. New Triterpenoids from the Leaves of Heritiera littoralis and Their Anti-Inflammatory Activity. Molecules. 2025; 30(1):131. https://doi.org/10.3390/molecules30010131
Chicago/Turabian StyleRen, Chenyang, Xiaoqin Liang, Rui Pi, Jiwen Xin, Bo Yang, Qingfang Zheng, Yanqin Li, Jun Li, and Yanjun Zhang. 2025. "New Triterpenoids from the Leaves of Heritiera littoralis and Their Anti-Inflammatory Activity" Molecules 30, no. 1: 131. https://doi.org/10.3390/molecules30010131
APA StyleRen, C., Liang, X., Pi, R., Xin, J., Yang, B., Zheng, Q., Li, Y., Li, J., & Zhang, Y. (2025). New Triterpenoids from the Leaves of Heritiera littoralis and Their Anti-Inflammatory Activity. Molecules, 30(1), 131. https://doi.org/10.3390/molecules30010131