
Targeting MAO-B with Small-Molecule Inhibitors: A Decade of Advances in Anticancer Research (2012–2024)
 , and
, and
Abstract
1. Introduction
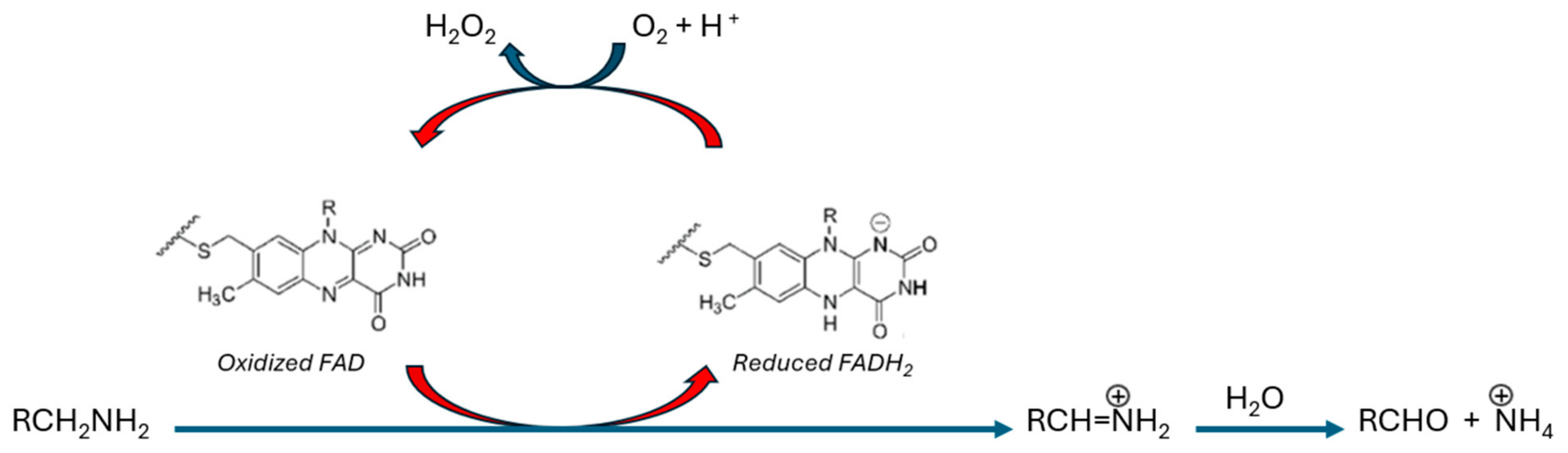
MAO-B Catalysis
2. MAO-B and Cancer
2.1. Bladder Cancer
2.2. Glioma
2.3. Colorectal Cancer
2.4. Liver Cancer
2.5. Lung Cancer
2.6. Kidney Cancer
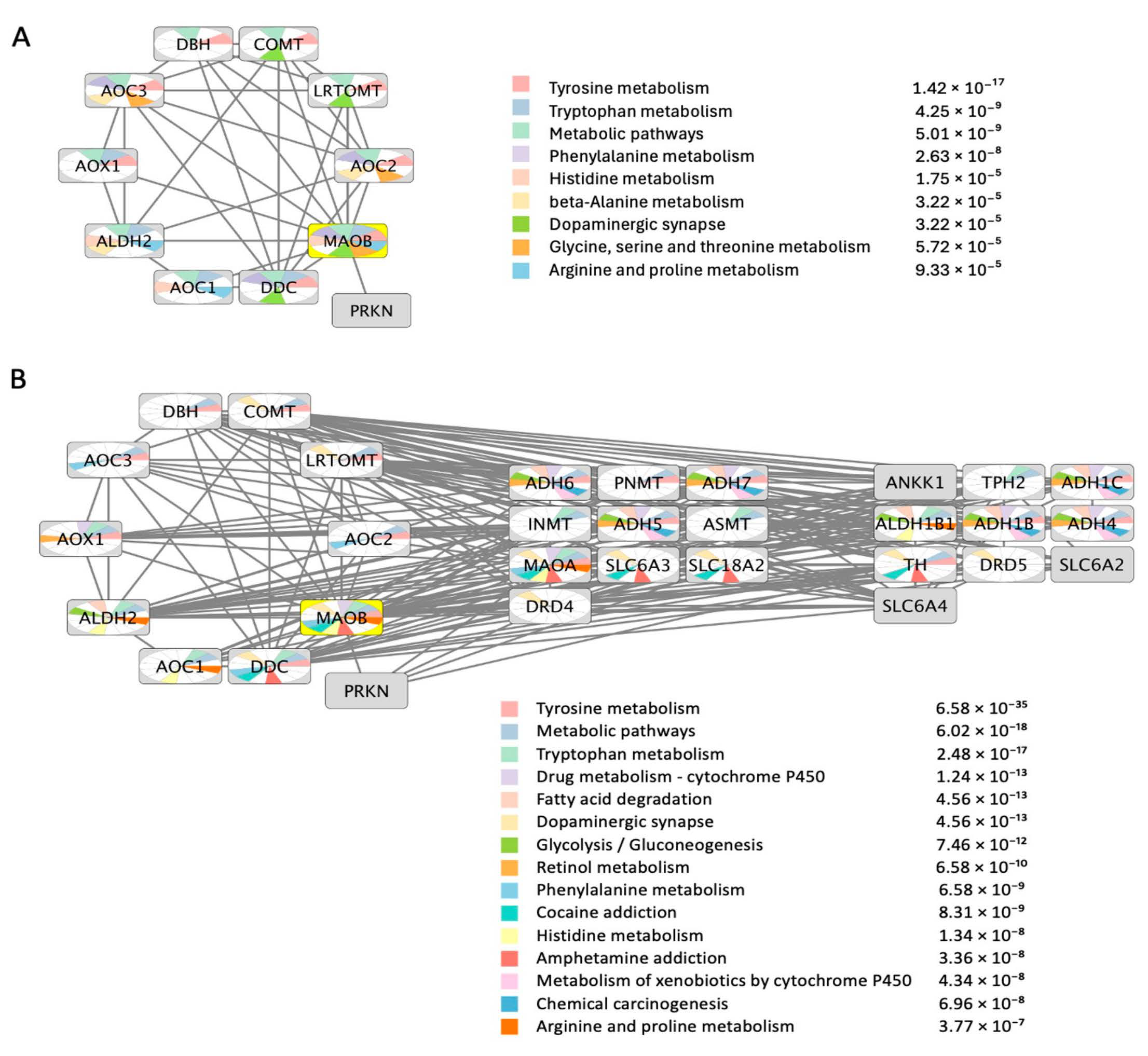
3. MAO-B Protein-Protein Functional Interactions Network
4. The Structural Basis of MAO-B Ligand Interactions in the Binding Pocket
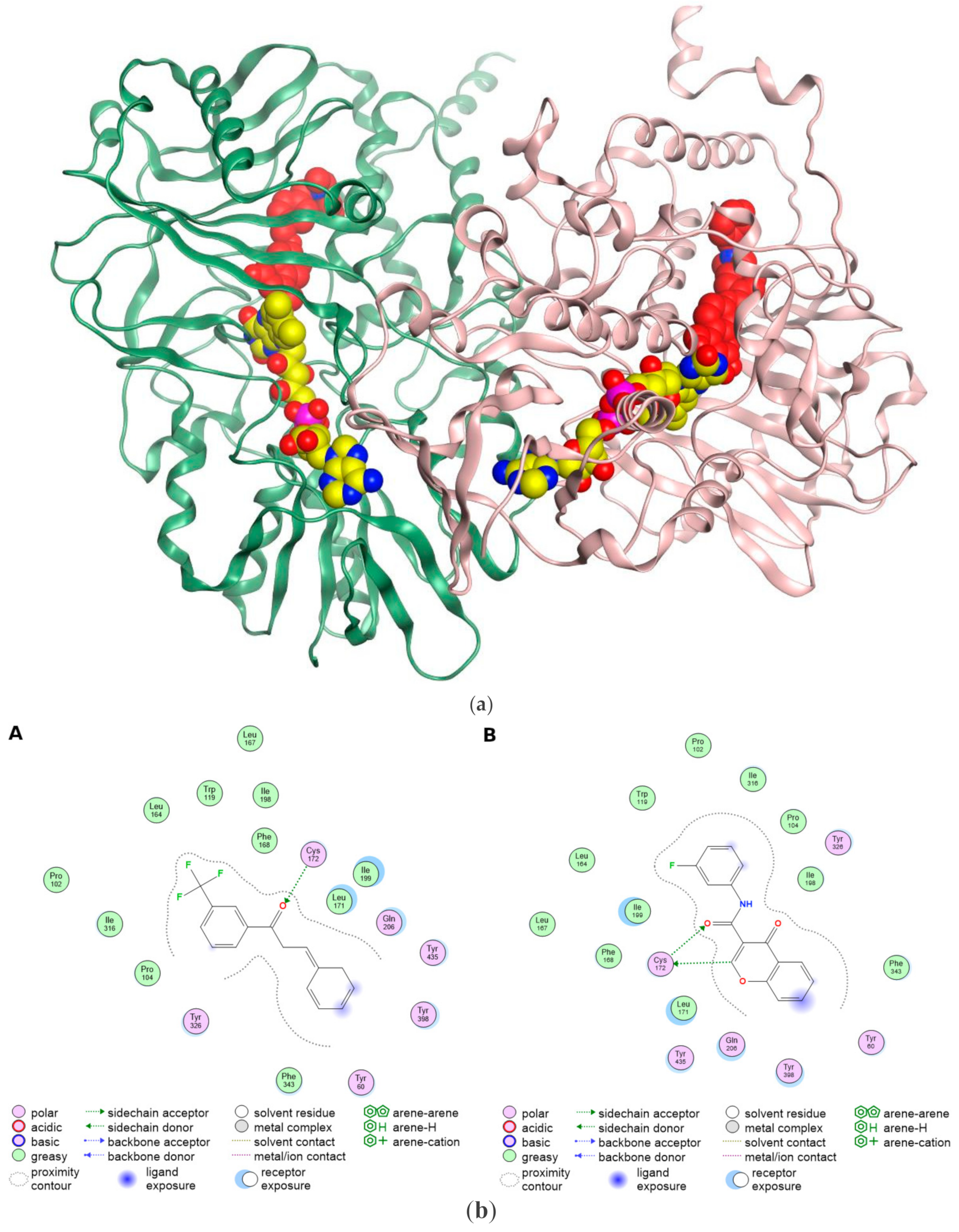
5. MAO-B Inhibitors as Anticancer Agents
5.1. Phenyl Alkyl Amine Derivatives

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Scaffold | Mode of Action | Compound | Preclinical and Clinical Trial/s |
|---|---|---|---|
| Synthetic propargyl phenyl analog of alkyl amine | Irreversible selective MAOB inhibitor | 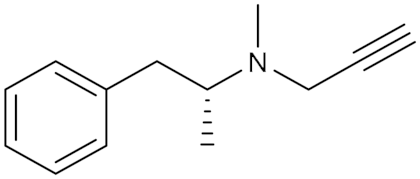 Selegiline (1) IC50 = 20 nM Ki = 0.5 nM | In vitro and in vivo studies [15] Phase II clinical trial ID: NCT04586543 [24] |
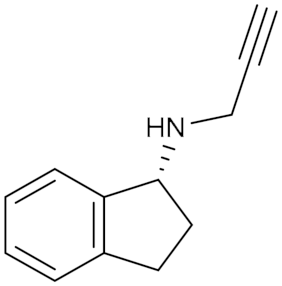 Rasagiline (2) IC50 = 4.4 nM Ki = 0.2 nM | In vitro and in vivo studies [15] | ||
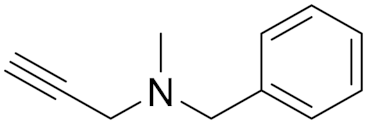 Pargyline (3) IC50 = 8.2 nM Ki = 5 nM | In vitro studies [13,68] | ||
| Synthetic cyclopropylamine derivative | Irreversible Non-selective MAO A/B inhibitor + LSD1 inhibitor |  Tranylcypromine TCP (4) IC50 = 0.95 µM Ki = 10 nM | Phase I clinical trial ID: NCT02273102 [22] Phase I/II clinical trial ID: NCT02261779 [23] |
| Synthetic hydrazine derivative | Irreversible Non-selective MAO A/B Inhibitor | 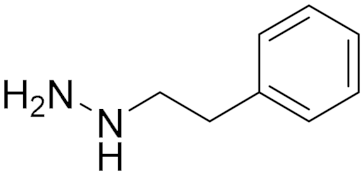 Phenelzine (5) IC50 = 0.9 μM Ki = 50 nM | In vivo studies [14] Phase Ib clinical trial ID: NCT03505528 [25] Phase II clinical trial, ID: NCT02217709 [17] Phase II clinical trial, ID: NCT01253642 [21] |
| Natural phenylpropanoid derivative | Nonselective MAO A/B inhibitor | 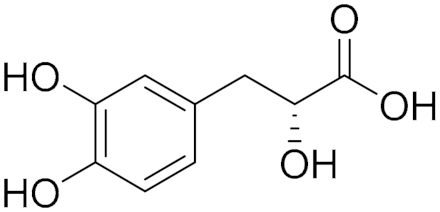 Danshensu (6) IC50 = 8.3 μM Ki = 34 μM | In vitro and in vivo studies [12] |
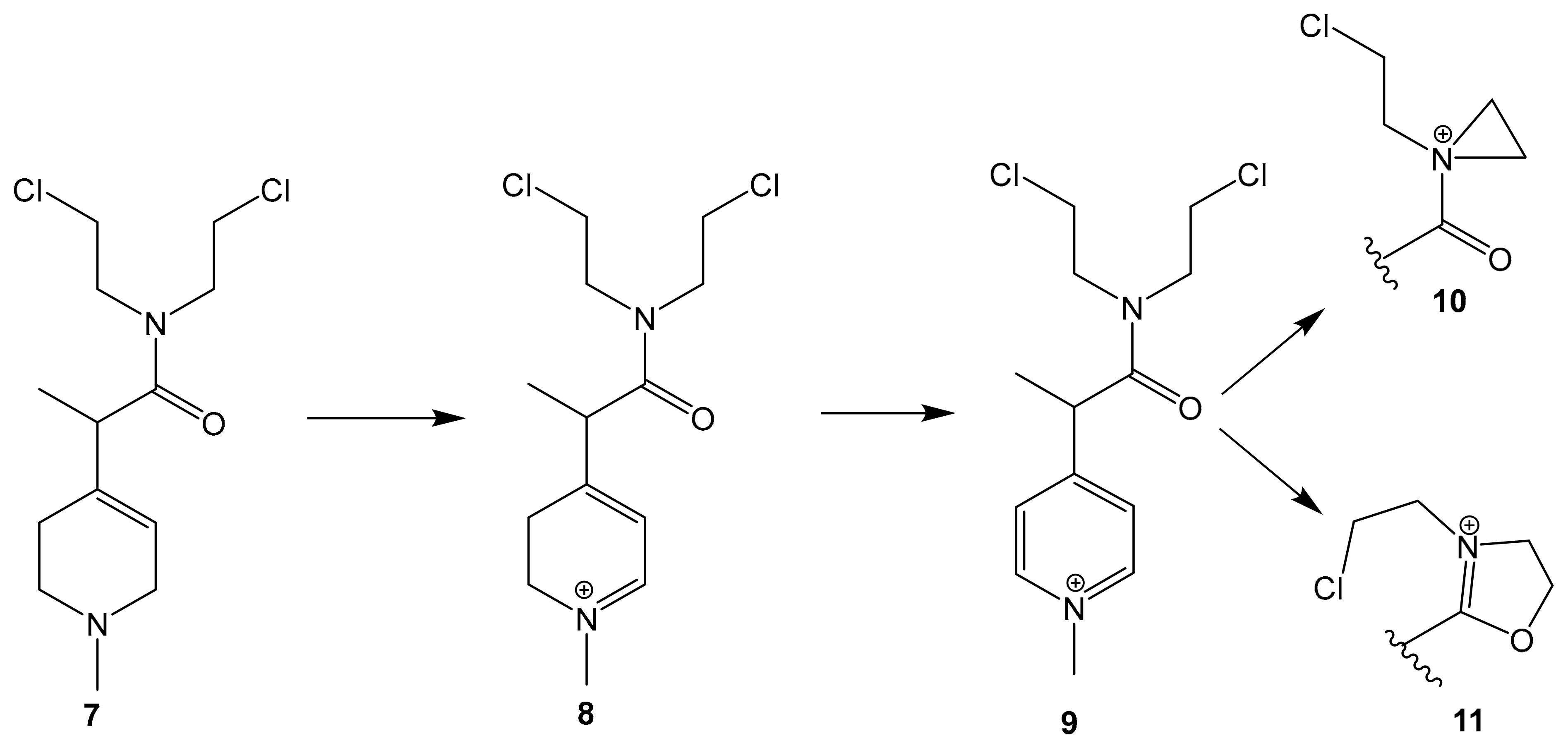
| Prodrug Synthetic pyridine derivative linked to N-mustard | Irreversible MAOB binding | 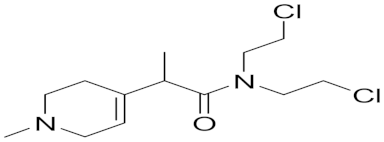 MP-MUS (7) IC50 = 80 μM | In vitro studies [69] |
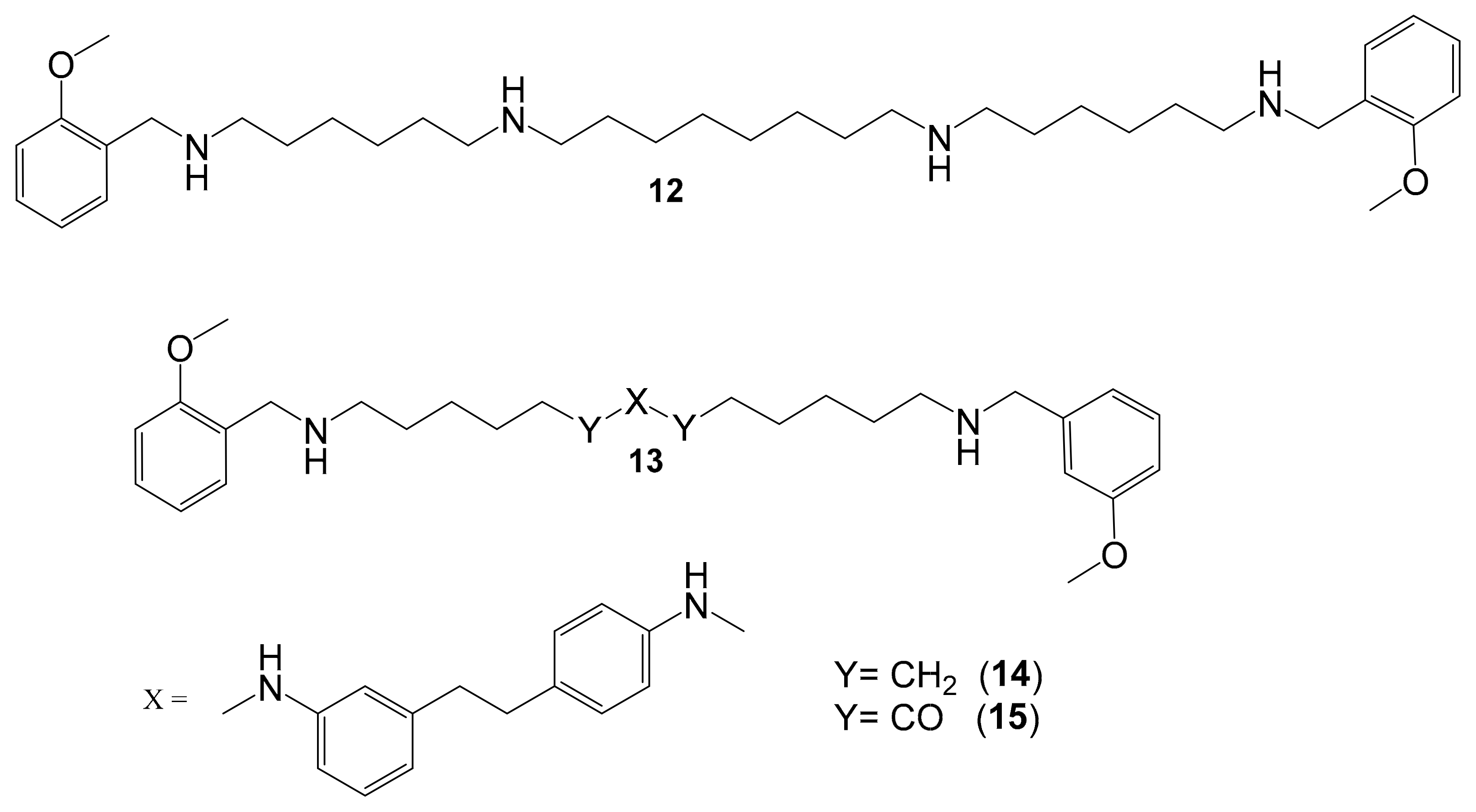
| Polyamine Derivatives | Nonselective MAO (A/B) inhibitor | 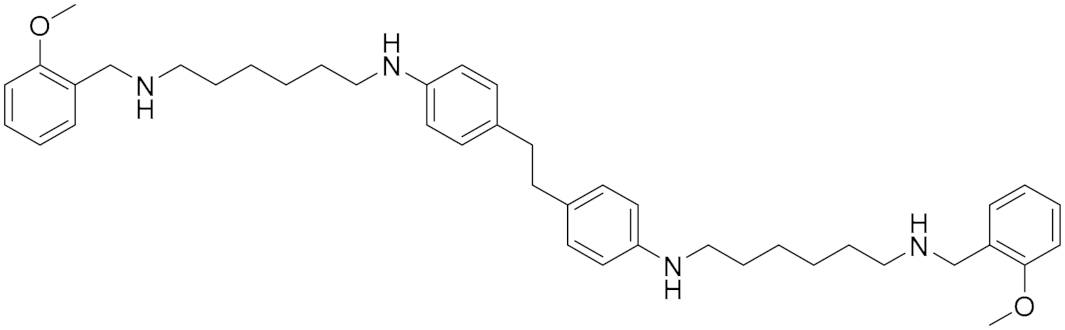 Compound (14) Compound (14)IC50 = 0.9 μM Ki = 0.3 μM. 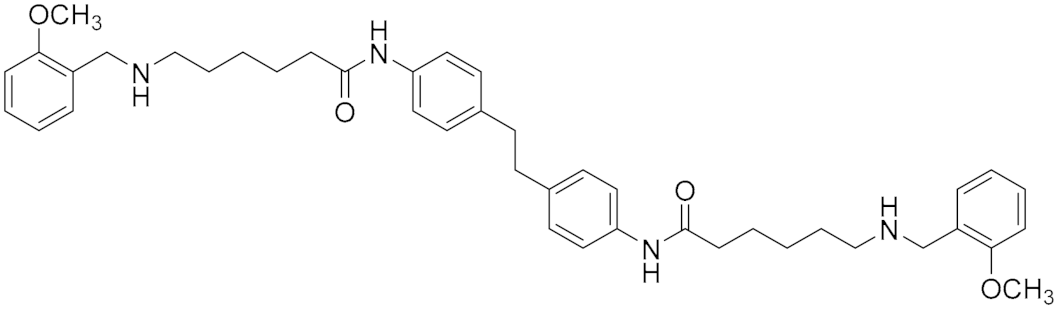 Compound (15) IC50 = 0.8 μM Ki = 0.2 μM | In vitro studies [5] |
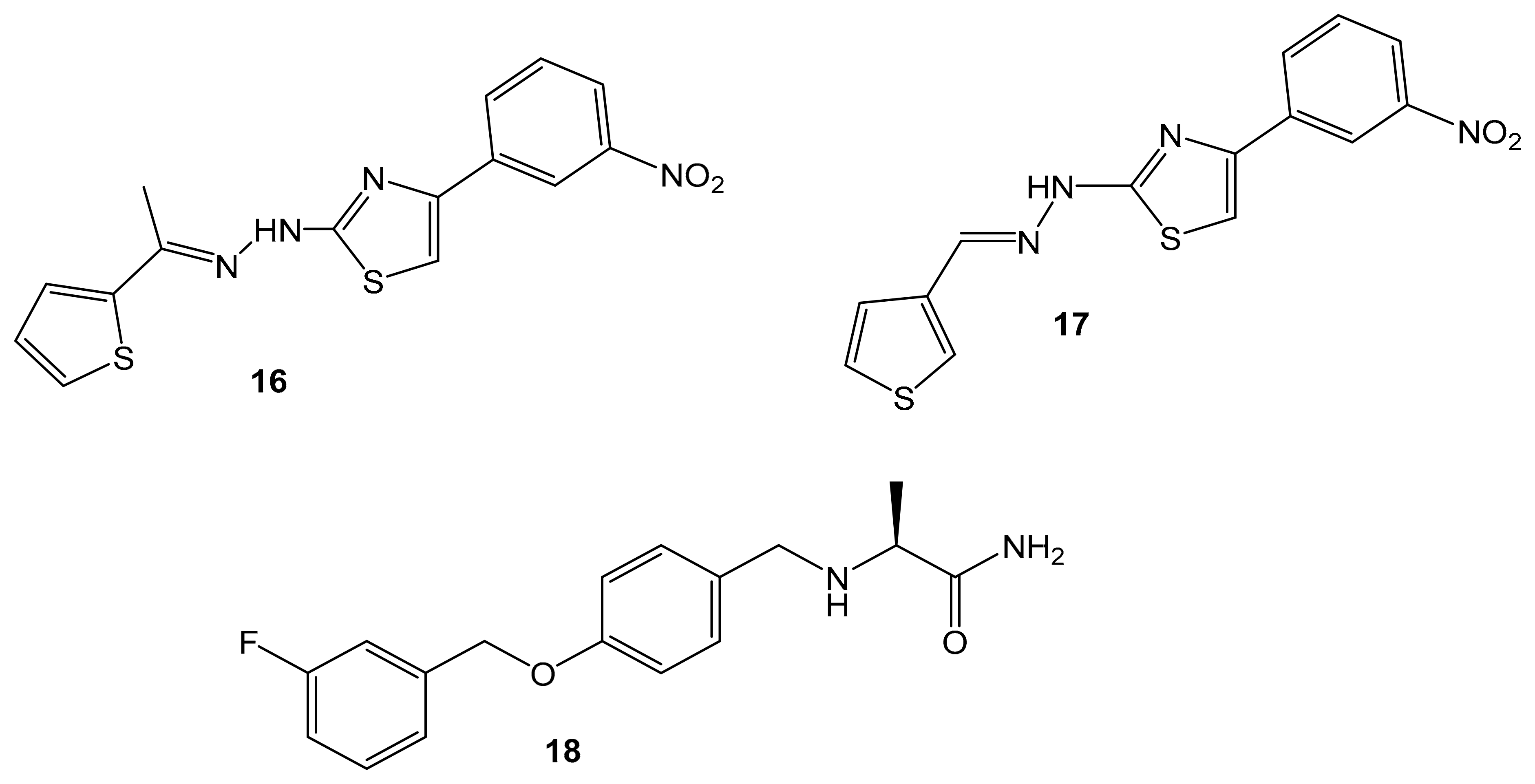
| Synthetic Hydrazothiazole Derivatives | Irreversible selective MAOB inhibitor | 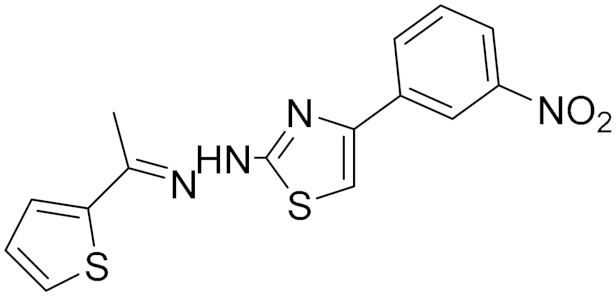 Compound (16) IC50 = 0.0068 μM Ki = 6.8 nM 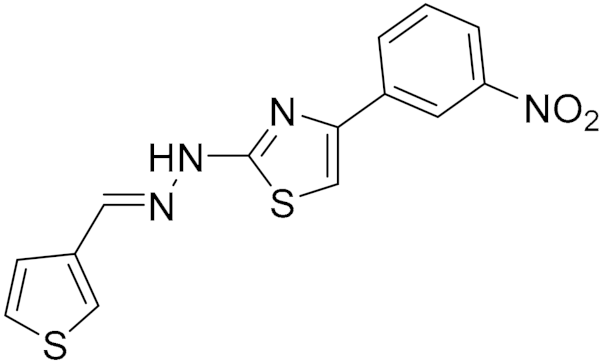 Compound (17) IC50 = 0.0025 μM Ki = 2.5 nM | In vitro studies [11] |
| Synthetic Chalcone Derivatives | Irreversible selective MAOB inhibitor | 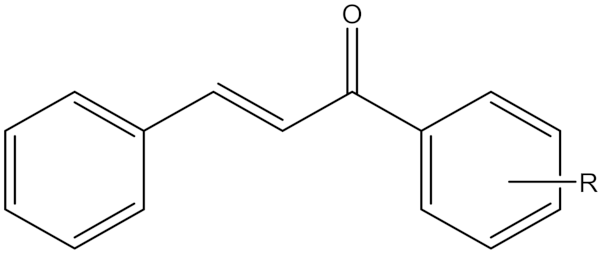 R = m-CF3, Compound (19) R = p -CF3, Compound (20) IC50(19) = 5 nM Ki (19) = 5.0 nM IC50(20) = 14.6 nM Ki (20) = 14.6 nM | In vitro studies reference [19] |
| Synthetic Chromone Derivatives | Irreversible selective MAOB inhibitor | 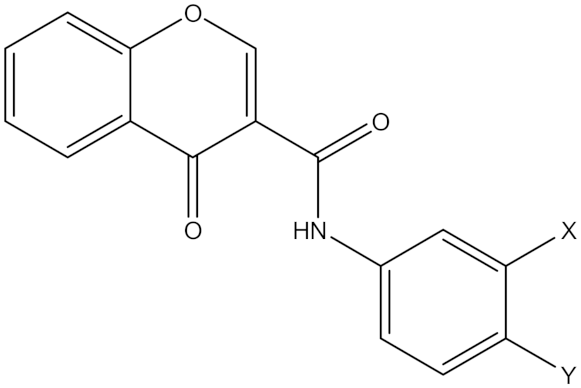 Compound (21), X = CH3, Y = CH3 Compound (22), X = CL, Y = H Ki (21) = 55.0 nM Ki (22) = 17 nM | In vitro studies [20] |
5.2. Cyclopropylamine Derivatives
5.3. Hydrazine Derivatives
5.4. Natural Phenylpropanoid Derivative
5.5. Synthetic Pyridine Derivative Linked to Nitrogen Mustard
5.6. Polyamine Derivatives
5.7. Synthetic Hydrazothiazole Derivatives
5.8. Synthetic Chalcone Derivatives
5.9. Synthetic Chromone Derivatives
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| Order Regarding Appearance in Text: | Abbreviation | |
| 1 | MAO-B | Monoamine oxidase B |
| 2 | ROS | Reactive oxygen species |
| 3 | MAO | Monoamine oxidase |
| 4 | FAD | Flavin adenine |
| 5 | MAO-A | Monoamine oxidase A |
| 6 | CNS | Central nervous system |
| 7 | ALDH | Aldehyde dehydrogenase |
| 8 | H2O2 | Hydrogen peroxide |
| 9 | NH3 | Ammonia |
| 10 | FADH2 | Dihydro-flavin adenine dinucleotide |
| 11 | R-CH2-CH3 | Amine neurotransmitter |
| 12 | O2 | Oxygen |
| 13 | BC | Bladder cancer |
| 14 | WHO | World Health Organization |
| 15 | HIF-1α factor | Hypoxia-inducible factor 1 |
| 16 | VEGF | Vascular endothelial growth factor |
| 17 | NF-kB | Nuclear factor kappa B subunit 1 |
| 18 | CRC | Colorectal cancer |
| 19 | BGA | Brain-gut axis |
| 20 | ENS | Enteric nervous system |
| 21 | LX-2 | Hepatic stellate cells |
| 22 | GGOH | Geranylgeraniol |
| 23 | GGA | Geranylgeranoic acid |
| 24 | siRNAs | Small interfering RNA |
| 25 | NSCLC | Non-small cell lung cancer |
| 26 | IR | Ionizing radiation |
| 27 | RCC | Renal cell carcinoma |
| 28 | ccRCC | Clear cell renal cell carcinoma |
| 29 | NN | Nearest neighbor |
| 30 | GLUT1 | Glucose transporter protein type 1 |
| 31 | HK2 | Hexokinase 2 |
| 32 | SOD | Superoxide dismutase |
| 33 | PARD | Poly (ADP-ribose) polymerase |
| 34 | LSD-1 | Lysine specific demethylase-1 |
| 35 | TCP | Tranylcypromine |
| 36 | ATRA | Retinoid all-Trans-Retinoic Acid |
| 37 | AML | Acute myeloid leukemia |
| 38 | DSS | Danshensu |
| 39 | TOM | Traditional Oriental Medicine |
| 40 | PARP | Poly ADP-ribose polymerase |
| 41 | FDA | Food and Drug Administration |
| 42 | MP-MUS | 1-Methyl-4-Phenyl-1,2,3,6-tetrahydropyridine-nitrogen Mustard |
| 46 | A2780 | Ovarian carcinoma |
| 47 | HT-29 | Colorectal adenocarcinoma |
| 48 | MSTO-211H | Biphasic mesothelioma |
References
- Ramsay, R.R. Molecular Aspects of Monoamine Oxidase B. Prog. Neuro Psychopharmacol. Biol. Psychiatry 2016, 69, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Sblano, S.; Boccarelli, A.; Mesiti, F.; Purgatorio, R.; de Candia, M.; Catto, M.; Altomare, C.D. A Second Life for MAO Inhibitors? From CNS Diseases to Anticancer Therapy. Eur. J. Med. Chem. 2024, 267, 116180. [Google Scholar] [CrossRef] [PubMed]
- Santin, Y.; Resta, J.; Parini, A.; Mialet-Perez, J. Monoamine Oxidases in Age-Associated Diseases: New Perspectives for Old Enzymes. Ageing Res. Rev. 2021, 66, 101256. [Google Scholar] [CrossRef] [PubMed]
- Shih, J.C. Monoamine Oxidase Isoenzymes: Genes, Functions and Targets for Behavior and Cancer Therapy. J. Neural Transm. 2018, 125, 1553–1566. [Google Scholar] [CrossRef]
- Nordio, G.; Piazzola, F.; Cozza, G.; Rossetto, M.; Cervelli, M.; Minarini, A.; Basagni, F.; Tassinari, E.; Dalla Via, L.; Milelli, A.; et al. From Monoamine Oxidase Inhibition to Antiproliferative Activity: New Biological Perspectives for Polyamine Analogs. Molecules 2023, 28, 6329. [Google Scholar] [CrossRef]
- Shih, J.C.; Chen, K. Regulation of MAO-A and MAO-B Gene Expression. Curr. Med. Chem. 2005, 11, 1995–2005. [Google Scholar] [CrossRef]
- Shih, J.C.; Chen, K.; Ridd, M.J. Monoamine Oxidase: From Genes to Behavior. Annu. Rev. Neurosci. 1999, 22, 197–217. [Google Scholar] [CrossRef]
- Sharpe, M.A.; Baskin, D.S. Monoamine Oxidase B Levels Are Highly Expressed in Human Gliomas and Are Correlated with the Expression of HiF-1α and with Transcription Factors Sp1 and Sp3. Oncotarget 2016, 7, 3379–3393. [Google Scholar] [CrossRef]
- Tomkins, B. 1 Introduction. Int. J. Press. Vessel. Pip. 1989, 40, 331–334. [Google Scholar] [CrossRef]
- Bortolato, M.; Chen, K.; Shih, J.C. Monoamine oxidase inactivation: From pathophysiology to therapeutics. Adv. Drug Deliv. Rev. 2008, 60, 1527–1533. [Google Scholar] [CrossRef]
- Marconi, G.D.; Gallorini, M.; Carradori, S.; Guglielmi, P.; Cataldi, A.; Zara, S. The Up-Regulation of Oxidative Stress as a Potential Mechanism of Novel MAO-B Inhibitors for Glioblastoma Treatment. Molecules 2019, 24, 2005. [Google Scholar] [CrossRef] [PubMed]
- Son, B.; Jun, S.Y.; Seo, H.J.; Youn, H.S.; Yang, H.J.; Kim, W.; Kim, H.K.; Kang, C.H.; Youn, B.H. Inhibitory Effect of Traditional Oriental Medicine-Derived Monoamine Oxidase B Inhibitor on Radioresistance of Non-Small Cell Lung Cancer. Sci. Rep. 2016, 6, 21986. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.T.; Jung, K.H.; Kim, S.K.; Choi, M.R.; Chai, Y.G. Effects of Pargyline on Cellular Proliferation in Human Breast Cancer Cells. Mol. Cell Toxicol. 2012, 8, 393–399. [Google Scholar] [CrossRef]
- Espina, V.; Liotta, L.; Chiechi, A.; Alessandra, R. Bone Modulators And Methods Therewith. World Patent WO US20120065166A1, 15 March 2012. [Google Scholar]
- Kormos, V.G.; Kálai, T.; Mangel, L.; Mátyus, P.; Helyes, A.T. Monoamine Oxidase b Inhibitors for Use in the Prevention or Treatment of Prostate Carcinoma. World Patent WO US2022/0273588A1, 1 August 2022. [Google Scholar]
- Youdim, M.B.H.; Gross, A.; Finberg, J.P.M. Rasagiline [N-Propargyl-1R(+)-Aminoindan], a Selective and Potent Inhibitor of Mitochondrial Monoamine Oxidase B. Br. J. Pharmacol. 2001, 132, 500–506. [Google Scholar] [CrossRef] [PubMed]
- Gross, M.E.; Agus, D.B.; Dorff, T.B.; Pinski, J.K.; Quinn, D.I.; Castellanos, O.; Gilmore, P.; Shih, J.C. Phase 2 Trial of Monoamine Oxidase Inhibitor Phenelzine in Biochemical Recurrent Prostate Cancer. Prostate Cancer Prostatic Dis. 2021, 24, 61–68. [Google Scholar] [CrossRef]
- Wang, L.C.; Chang, Y.Y.; Lee, I.C.; Kuo, H.C.; Tsai, M.Y. Systematic Review and Meta-Analysis of Chinese Herbal Medicine as Adjuvant Treatment in Advanced Non-Small Cell Lung Cancer Patients. Complement Ther. Med. 2020, 52, 102472. [Google Scholar] [CrossRef]
- Iacovino, L.G.; Pinzi, L.; Facchetti, G.; Bortolini, B.; Christodoulou, M.S.; Binda, C.; Rastelli, G.; Rimoldi, I.; Passarella, D.; Di Paolo, M.L.; et al. Promising Non-Cytotoxic Monosubstituted Chalcones to Target Monoamine Oxidase-B. ACS Med. Chem. Lett. 2021, 12, 1151–1158. [Google Scholar] [CrossRef]
- Reis, J.; Manzella, N.; Cagide, F.; Mialet-Perez, J.; Uriarte, E.; Parini, A.; Borges, F.; Binda, C. Tight-Binding Inhibition of Human Monoamine Oxidase B by Chromone Analogs: A Kinetic, Crystallographic, and Biological Analysis. J. Med. Chem. 2018, 61, 4203–4212. [Google Scholar] [CrossRef]
- NCT01253642; Phenelzine Sulfate and Docetaxel in Treating Patients with Prostate Cancer with Progressive Disease after First-Line Therapy with Docetaxel. ClinicalTrials.Gov: Bethesda, MD, USA, 2019.
- NCT02273102; Study of TCP-ATRA for Adult Patients with AML and MDS (TCP-ATRA). ClinicalTrials.Gov: Bethesda, MD, USA, 2020.
- NCT02261779; Phase I/II Trial of ATRA and TCP in Patients with Relapsed or Refractory AML and No Intensive Treatment Is Possible (TCP-AML). ClinicalTrials.Gov: Bethesda, MD, USA, 2017.
- NCT04586543; A Clinical Trial of Selegiline Plus Docetaxel for the Treatment of Metastatic, Castrate-Resistant Prostate Adenocarcinoma. ClinicalTrials.Gov: Bethesda, MD, USA, 2024.
- NCT03505528; An Early Phase Study of Abraxane Combined with Phenelzine Sulfate in Patients with Metastatic or Advanced Breast Cancer (Epi- PRIMED). ClinicalTrials.Gov: Bethesda, MD, USA, 2019.
- Chatterjee, R.; Chatterjee, J. ROS and Oncogenesis with Special Reference to EMT and Stemness. Eur. J. Cell Biol. 2020, 99, 151073. [Google Scholar] [CrossRef]
- Arfin, S.; Jha, N.K.; Jha, S.K.; Kesari, K.K.; Ruokolainen, J.; Roychoudhury, S.; Rathi, B.; Kumar, D. Oxidative Stress in Cancer Cell Metabolism. Antioxidants 2021, 10, 642. [Google Scholar] [CrossRef]
- Tasdogan, A.; Ubellacker, J.M.; Morrison, S.J. Redox Regulation in Cancer Cells during Metastasis. Cancer Discov. 2021, 11, 2682–2692. [Google Scholar] [CrossRef] [PubMed]
- Resta, J.; Santin, Y.; Roumigui, M.; Riant, E.; Lucas, A.; Couderc, B.; Binda, C.; Lluel, P.; Parini, A.; Mialet-perez, J. Monoamine Oxidase Inhibitors Prevent Glucose-Dependent Energy Production, Proliferation and Migration of Bladder Carcinoma Cells. Int. J. Mol. Sci. 2022, 23, 11747. [Google Scholar] [CrossRef] [PubMed]
- de Camargo, G.C.A.; Oliveira, G.; Alonso, J.C.C.; Ribeiro de Souza, B.; Silva Santos, B.N.N.S.; Ávila, M.; Roberto, I.M.; Duran, N.; Fávaro, W.J. Monoamine Oxidase as a Potential Target for the Development of New Therapeutic Strategies for Bladder Cancer. J. Clin. Oncol. 2023, 41, e16611. [Google Scholar] [CrossRef]
- Abdulghany, Z. Noah Determine MAO Gene Expression in Both Starved and Non-Starved Glioblastoma Cancer Cell Line. Iraqi J. Cancer Med. Genet. 2023, 16, 14–18. [Google Scholar] [CrossRef]
- Kaur, B.; Khwaja, F.W.; Severson, E.A.; Matheny, S.L.; Brat, D.J.; Van Meir, E.G. Hypoxia and the Hypoxia-Inducible-Factor Pathway in Glioma Growth and Angiogenesis. Neuro. Oncol. 2005, 7, 134–153. [Google Scholar] [CrossRef]
- Battaglin, F.; Jayachandran, P.; Strelez, C.; Lenz, A.; Algaze, S.; Soni, S.; Lo, J.H.; Yang, Y.; Millstein, J.; Zhang, W.; et al. Neurotransmitter Signaling: A New Frontier in Colorectal Cancer Biology and Treatment. Oncogene 2022, 41, 4769–4778. [Google Scholar] [CrossRef]
- Jarab, A.S.; Al-Qerem, W.; Almomani, N.; Abu Heshmeh, S.; Mukattash, T.L.; Al Hamarneh, Y.N. Colorectal Cancer Screening among the Public: Knowledge, Attitudes, and the Perceived Barriers. Int. J. Environ. Health Res. 2024, 34, 2578–2592. [Google Scholar] [CrossRef]
- Yang, Y.C.; Chien, M.H.; Lai, T.C.; Su, C.Y.; Jan, Y.H.; Hsiao, M.; Chen, C.L. Monoamine Oxidase b Expression Correlates with a Poor Prognosis in Colorectal Cancer Patients and Is Significantly Associated with Epithelial-to-Mesenchymal Transition-Related Gene Signatures. Int. J. Mol. Sci. 2020, 21, 2813. [Google Scholar] [CrossRef]
- Fan, N.; Wu, C.; Zhou, Y.; Wang, X.; Li, P.; Liu, Z.; Tang, B. Rapid Two-Photon Fluorescence Imaging of Monoamine Oxidase B for Diagnosis of Early-Stage Liver Fibrosis in Mice. Anal. Chem. 2021, 93, 7110–7117. [Google Scholar] [CrossRef]
- Tabata, Y.; Shidoji, Y. Hepatic Monoamine Oxidase B Is Involved in Endogenous Geranylgeranoic Acid Synthesis in Mammalian Liver Cells. J. Lipid Res. 2020, 61, 778–789. [Google Scholar] [CrossRef]
- Kery, M.; Papandreou, I. Emerging Strategies to Target Cancer Metabolism and Improve Radiation Therapy Outcomes. Br. J. Radiol. 2020, 93, 20200067. [Google Scholar] [CrossRef] [PubMed]
- Aljanabi, R.; Alsous, L.; Sabbah, D.A.; Gul, H.I.; Gul, M.; Bardaweel, S.K. Monoamine Oxidase (MAO) as a Potential Target for Anticancer Drug Design and Development. Molecules 2021, 26, 6019. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, J.; Yuan, C.; Luo, Y.; Li, Y.; Dai, P.; Sun, W.; Zhang, N.; Ren, J.; Zhang, J.; et al. Establishment of the Prognostic Index Reflecting Tumor Immune Microenvironment of Lung Adenocarcinoma Based on Metabolism-Related Genes. J. Cancer 2020, 11, 7101–7115. [Google Scholar] [CrossRef] [PubMed]
- di Meo, N.A.; Lasorsa, F.; Rutigliano, M.; Loizzo, D.; Ferro, M.; Stella, A.; Bizzoca, C.; Vincenti, L.; Pandolfo, S.D.; Autorino, R.; et al. Renal Cell Carcinoma as a Metabolic Disease: An Update on Main Pathways, Potential Biomarkers, and Therapeutic Targets. Int. J. Mol. Sci. 2022, 23, 14360. [Google Scholar] [CrossRef]
- Hodorová, I.; Rybárová, S.; Solár, P.; Benický, M.; Rybár, D.; Kováčová, Z.; Mihalik, J. Monoamine Oxidase B in Renal Cell Carcinoma. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2018, 24, 5422–5426. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Jensen, L.J.; Kuhn, M.; Stark, M.; Chaffron, S.; Creevey, C.; Muller, J.; Doerks, T.; Julien, P.; Roth, A.; Simonovic, M.; et al. STRING 8--a Global View on Proteins and Their Functional Interactions in 630 Organisms. Nucleic Acids Res. 2009, 37, D412–D416. [Google Scholar] [CrossRef]
- Vaupel, P.; Multhoff, G. The Warburg Effect: Historical Dogma Versus Current Rationale; Springer International Publishing: Cham, Switzerland, 2021; pp. 169–177. [Google Scholar]
- Coatrieux, C.; Sanson, M.; Negre-Salvayre, A.; Parini, A.; Hannun, Y.; Itohara, S.; Salvayre, R.; Auge, N. MAO-A-Induced Mitogenic Signaling Is Mediated by Reactive Oxygen Species, MMP-2, and the Sphingolipid Pathway. Free Radic. Biol. Med. 2007, 43, 80–89. [Google Scholar] [CrossRef]
- Binda, C.; Hubálek, F.; Li, M.; Edmondson, D.E.; Mattevi, A. Crystal Structure of Human Monoamine Oxidase B, a Drug Target Enzyme Monotopically Inserted into the Mitochondrial Outer Membrane. FEBS Lett. 2004, 564, 225–228. [Google Scholar] [CrossRef]
- Binda, C.; Li, M.; Hubálek, F.; Restelli, N.; Edmondson, D.E.; Mattevi, A. Insights into the Mode of Inhibition of Human Mitochondrial Monoamine Oxidase B from High-Resolution Crystal Structures. Proc. Natl. Acad. Sci. USA 2003, 100, 9750–9755. [Google Scholar] [CrossRef]
- Jia, W.Z.; Cheng, F.; Zhang, Y.J.; Ge, J.Y.; Yao, S.Q.; Zhu, Q. Rapid Synthesis of Flavone-based Monoamine Oxidase (MAO) Inhibitors Targeting Two Active Sites Using Click Chemistry. Chem. Biol. Drug Des. 2017, 89, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Murugan, N.A.; Zaleśny, R. Multiscale Modeling of Two-Photon Probes for Parkinson’s Diagnostics Based on Monoamine Oxidase B Biomarker. J. Chem. Inf. Model. 2020, 60, 3854–3863. [Google Scholar] [CrossRef] [PubMed]
- Hudspith, L.; Shmam, F.; Dalton, C.F.; Princivalle, A.; Turega, S.M. Neurotransmitter Selection by Monoamine Oxidase Isoforms, Dissected in Terms of Functional Groups by Mixed Double Mutant Cycles. Org. Biomol. Chem. 2019, 17, 8871–8877. [Google Scholar] [CrossRef] [PubMed]
- Knez, D.; Colettis, N.; Iacovino, L.G.; Sova, M.; Pišlar, A.; Konc, J.; Lešnik, S.; Higgs, J.; Kamecki, F.; Mangialavori, I.; et al. Stereoselective Activity of 1-Propargyl-4-Styrylpiperidine-like Analogues That Can Discriminate between Monoamine Oxidase Isoforms A and B. J. Med. Chem. 2020, 63, 1361–1387. [Google Scholar] [CrossRef]
- Nandigama, R.K.; Miller, J.R.; Edmondson, D.E. Loss of Serotonin Oxidation as a Component of the Altered Substrate Specificity in the Y444F Mutant of Recombinant Human Liver MAO A. Biochemistry 2001, 40, 14839–14846. [Google Scholar] [CrossRef]
- Geha, R.M.; Chen, K.; Wouters, J.; Ooms, F.; Shih, J.C. Analysis of Conserved Active Site Residues in Monoamine Oxidase A and B and Their Three-Dimensional Molecular Modeling. J. Biol. Chem. 2002, 277, 17209–17216. [Google Scholar] [CrossRef]
- Li, M.; Binda, C.; Mattevi, A.; Edmondson, D.E. Functional Role of the “Aromatic Cage” in Human Monoamine Oxidase B: Structures and Catalytic Properties of Tyr435 Mutant Proteins. Biochemistry 2006, 45, 4775–4784. [Google Scholar] [CrossRef]
- Alieva, I.N.; Mustafayeva, N.N.; Gojayev, N.M. Conformational Analysis of the N-Terminal Sequence Met1–Val60 of the Tyrosine Hydroxylase. J. Mol. Struct. 2006, 785, 76–84. [Google Scholar] [CrossRef]
- Son, S.-Y.; Ma, J.; Kondou, Y.; Yoshimura, M.; Yamashita, E.; Tsukihara, T. Structure of Human Monoamine Oxidase A at 2.2-Å Resolution: The Control of Opening the Entry for Substrates/Inhibitors. Proc. Natl. Acad. Sci. USA 2008, 105, 5739–5744. [Google Scholar] [CrossRef]
- Hubálek, F.; Binda, C.; Khalil, A.; Li, M.; Mattevi, A.; Castagnoli, N.; Edmondson, D.E. Demonstration of Isoleucine 199 as a Structural Determinant for the Selective Inhibition of Human Monoamine Oxidase B by Specific Reversible Inhibitors. J. Biol. Chem. 2005, 280, 15761–15766. [Google Scholar] [CrossRef]
- Ekström, F.; Gottinger, A.; Forsgren, N.; Catto, M.; Iacovino, L.G.; Pisani, L.; Binda, C. Dual Reversible Coumarin Inhibitors Mutually Bound to Monoamine Oxidase B and Acetylcholinesterase Crystal Structures. ACS Med. Chem. Lett. 2022, 13, 499–506. [Google Scholar] [CrossRef] [PubMed]
- Chemical Computing Group ULC. The Molecular Operating Environment (MOE); Version # 2022.02; Inc. Montreal Quebec: Montreal, QC, Canada, 2022; Available online: https://www.chemcomp.com/en/Products.htm (accessed on 20 November 2024).
- Pacureanu, L.; Bora, A.; Crisan, L. New Insights on the Activity and Selectivity of MAO-B Inhibitors through In Silico Methods. Int. J. Mol. Sci. 2023, 24, 9583. [Google Scholar] [CrossRef] [PubMed]
- Tábi, T.; Vécsei, L.; Youdim, M.B.; Riederer, P.; Szökő, É. Selegiline: A Molecule with Innovative Potential. J. Neural Transm. 2020, 127, 831–842. [Google Scholar] [CrossRef] [PubMed]
- Gholami, M.; Kaviani, N.; Motaghinejad, M.; Ulloa, L. Neuroprotective Effects of Selegiline Agent Methamphetamine-Prompted Mood-Related Behavior Disorder Mediated via 5-HT2 and D2 Receptors. Int. J. Prev. Med. 2023, 14, 78. [Google Scholar] [CrossRef]
- Magyar, K. The Pharmacology of Selegiline. Int. Rev. Neurobiol. 2011, 100, 65–84. [Google Scholar] [CrossRef]
- Ebadi, M.; Brown-Borg, H.; Ren, J.; Sharma, S.; Shavali, S.; El Refaey, H.; Carlson, E. Therapeutic Efficacy of Selegiline in Neurodegenerative Disorders and Neurological Diseases. Curr. Drug Targets 2006, 7, 1513–1529. [Google Scholar] [CrossRef]
- Ahmari, M.; Sharafi, A.; Mahmoudi, J.; Jafari-Anarkoli, I.; Gharbavi, M.; Hosseini, M.J. Selegiline (l-Deprenyl) Mitigated Oxidative Stress, Cognitive Abnormalities, and Histopathological Change in Rats: Alternative Therapy in Transient Global Ischemia. J. Mol. Neurosci. 2020, 70, 1639–1648. [Google Scholar] [CrossRef]
- Entzeroth, M.; Ratty, A.K. Monoamine Oxidase Inhibitors—Revisiting a Therapeutic Principle. Open J. Depress. 2017, 06, 31–68. [Google Scholar] [CrossRef]
- Lee, H.T.; Choi, M.R.; Doh, M.S.; Jung, K.H.; Chai, Y.G. Effects of the Monoamine Oxidase Inhibitors Pargyline and Tranylcypromine on Cellular Proliferation in Human Prostate Cancer Cells. Oncol. Rep. 2013, 30, 1587–1592. [Google Scholar] [CrossRef]
- Sharpe, M.A.; Livingston, A.D.; Gist, T.L.; Ghosh, P.; Han, J.; Baskin, D.S. Successful Treatment of Intracranial Glioblastoma Xenografts With a Monoamine Oxidase B-Activated Pro-Drug. EBioMedicine 2015, 2, 1122–1132. [Google Scholar] [CrossRef]
- Song, Y.; Yang, X.; Yu, B. Repurposing Antidepressants for Anticancer Drug Discovery. Drug Discov. Today 2022, 27, 1924–1935. [Google Scholar] [CrossRef] [PubMed]
- Callado, L.F.; Garibi, J.M.; Meana, J.J. Gliomas: Role of Monoamine Oxidase B in Diagnosis. In Tumors of the Central Nervous System; Springer: Berlin/Heidelberg, Germany, 2011; Volume 1, pp. 53–59. [Google Scholar] [CrossRef]
- Chen, D.; Chen, S.; Zhou, F.; Bo Chen, L.; Chen, M. Synergistic Effects of Tranylcypromine and NRF2 Inhibitor: A Repurposing Strategy for Effective Cancer Therapy. ChemMedChem 2023, 18, e202300282. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, S.; Ricken, R.; Adli, M. Tranylcypromine in Mind (Part I): Review of Pharmacology. Eur. Neuropsychopharmacol. 2017, 27, 697–713. [Google Scholar] [CrossRef] [PubMed]
- Tayari, M.M.; Santos, H.G.; Kwon, D.; Bradley, T.J.; Thomassen, A.; Chen, C.; Dinh, Y.; Perez, A.; Zelent, A.; Morey, L.; et al. Clinical Responsiveness to All-Trans Retinoic Acid Is Potentiated by LSD1 Inhibition and Associated with a Quiescent Transcriptome in Myeloid Malignancies. Clin. Cancer Res. 2021, 27, 1893–1903. [Google Scholar] [CrossRef]
- Matveychuk, D.; MacKenzie, E.M.; Kumpula, D.; Song, M.S.; Holt, A.; Kar, S.; Todd, K.G.; Wood, P.L.; Baker, G.B. Overview of the Neuroprotective Effects of the MAO-Inhibiting Antidepressant Phenelzine. Cell Mol. Neurobiol. 2022, 42, 225–242. [Google Scholar] [CrossRef]
- Prasanna, T.; Malik, L.; McCuaig, R.D.; Tu, W.J.; Wu, F.; Lim, P.S.; Tan, A.H.Y.; Dahlstrom, J.E.; Clingan, P.; Moylan, E.; et al. A Phase 1 Proof of Concept Study Evaluating the Addition of an LSD1 Inhibitor to Nab-Paclitaxel in Advanced or Metastatic Breast Cancer (EPI-PRIMED). Front. Oncol. 2022, 12, 862427. [Google Scholar] [CrossRef]
- Tang, Y.; Wang, M.; Le, X.; Meng, J.; Huang, L.; Yu, P.; Chen, J.; Wu, P. Antioxidant and Cardioprotective Effects of Danshensu (3-(3, 4-Dihydroxyphenyl)-2-Hydroxy-Propanoic Acid from Salvia Miltiorrhiza) on Isoproterenol-Induced Myocardial Hypertrophy in Rats. Phytomedicine 2011, 18, 1024–1030. [Google Scholar] [CrossRef]
- Reily, C.; Mitchell, T.; Chacko, B.K.; Benavides, G.A.; Murphy, M.P.; Darley-Usmar, V.M. Mitochondrially Targeted Compounds and Their Impact on Cellular Bioenergetics. Redox Biol. 2013, 1, 86–93. [Google Scholar] [CrossRef]
- Dixit, P.; Jeyaseelan, C.; Gupta, D. Nitrogen Mustard: A Promising Class of Anti-Cancer Chemotherapeutics-a Review. Biointerface Res. Appl. Chem. 2022, 13, 135–161. [Google Scholar] [CrossRef]
- Sharpe, M.A.; Raghavan, S.; Baskin, D.S. PAM-OBG: A Monoamine Oxidase B Specific Prodrug That Inhibits MGMT and Generates DNA Interstrand Crosslinks, Potentiating Temozolomide and Chemoradiation Therapy in Intracranial Glioblastoma. Oncotarget 2018, 9, 23923–23943. [Google Scholar] [CrossRef]
- Minarini, A.; Milelli, A.; Tumiatti, V.; Rosini, M.; Bolognesi, M.L.; Melchiorre, C. Synthetic Polyamines: An Overview of Their Multiple Biological Activities. Amino Acids 2009, 38, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Emanuela, B.; Minarini, A.; Tumiatti, V.; Milelli, A.; Lunelli, M.; Pegoraro, M.; Rizzoli, V.; Di Paolo, M.L. Synthetic Polyamines as Potential Amine Oxidase Inhibitors: A Preliminary Study. Amino Acids 2012, 42, 913–928. [Google Scholar] [CrossRef] [PubMed]
- Secci, D.; Carradori, S.; Petzer, A.; Guglielmi, P.; D’Ascenzio, M.; Chimenti, P.; Bagetta, D.; Alcaro, S.; Zengin, G.; Petzer, J.P.; et al. 4-(3-Nitrophenyl)Thiazol-2-Ylhydrazone Derivatives as Antioxidants and Selective HMAO-B Inhibitors: Synthesis, Biological Activity and Computational Analysis. J. Enzyme Inhib. Med. Chem. 2019, 34, 597–612. [Google Scholar] [CrossRef] [PubMed]
- Abbruzzese, G.; Barone, P.; Lopiano, L.; Stocchi, F. The Current Evidence for the Use of Safinamide for the Treatment of Parkinson’s Disease. Drug Des. Dev. Ther. 2021, 15, 2507–2517. [Google Scholar] [CrossRef]
- Stocchi, F.; Antonini, A.; Berg, D.; Bergmans, B.; Jost, W.; Katzenschlager, R.; Kulisevsky, J.; Odin, P.; Valldeoriola, F.; Ray Chaudhuri, K. Safinamide in the Treatment Pathway of Parkinson’s Disease: A European Delphi Consensus. NPJ Park. Dis. 2022, 8, 17. [Google Scholar] [CrossRef]
- Mathew, B.; Mathew, G.E.; Ucar, G.; Joy, M.; Nafna, E.K.; Lohidakshan, K.K.; Suresh, J. Monoamine Oxidase Inhibitory Activity of Methoxy-Substituted Chalcones. Int. J. Biol. Macromol. 2017, 104, 1321–1329. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alsaad, I.; Abdel Rahman, D.M.A.; Al-Tamimi, O.; Alhaj, S.A.; Sabbah, D.A.; Hajjo, R.; Bardaweel, S.K. Targeting MAO-B with Small-Molecule Inhibitors: A Decade of Advances in Anticancer Research (2012–2024). Molecules 2025, 30, 126. https://doi.org/10.3390/molecules30010126
Alsaad I, Abdel Rahman DMA, Al-Tamimi O, Alhaj SA, Sabbah DA, Hajjo R, Bardaweel SK. Targeting MAO-B with Small-Molecule Inhibitors: A Decade of Advances in Anticancer Research (2012–2024). Molecules. 2025; 30(1):126. https://doi.org/10.3390/molecules30010126
Chicago/Turabian StyleAlsaad, Iyman, Diana M. A. Abdel Rahman, Ola Al-Tamimi, Shayma’a A. Alhaj, Dima A. Sabbah, Rima Hajjo, and Sanaa K. Bardaweel. 2025. "Targeting MAO-B with Small-Molecule Inhibitors: A Decade of Advances in Anticancer Research (2012–2024)" Molecules 30, no. 1: 126. https://doi.org/10.3390/molecules30010126
APA StyleAlsaad, I., Abdel Rahman, D. M. A., Al-Tamimi, O., Alhaj, S. A., Sabbah, D. A., Hajjo, R., & Bardaweel, S. K. (2025). Targeting MAO-B with Small-Molecule Inhibitors: A Decade of Advances in Anticancer Research (2012–2024). Molecules, 30(1), 126. https://doi.org/10.3390/molecules30010126