Synthesis of (2R,5R)-2-Amino-5-Hydroxyhexanoic Acid by Intramolecular Cycloaddition

Abstract
:Introduction
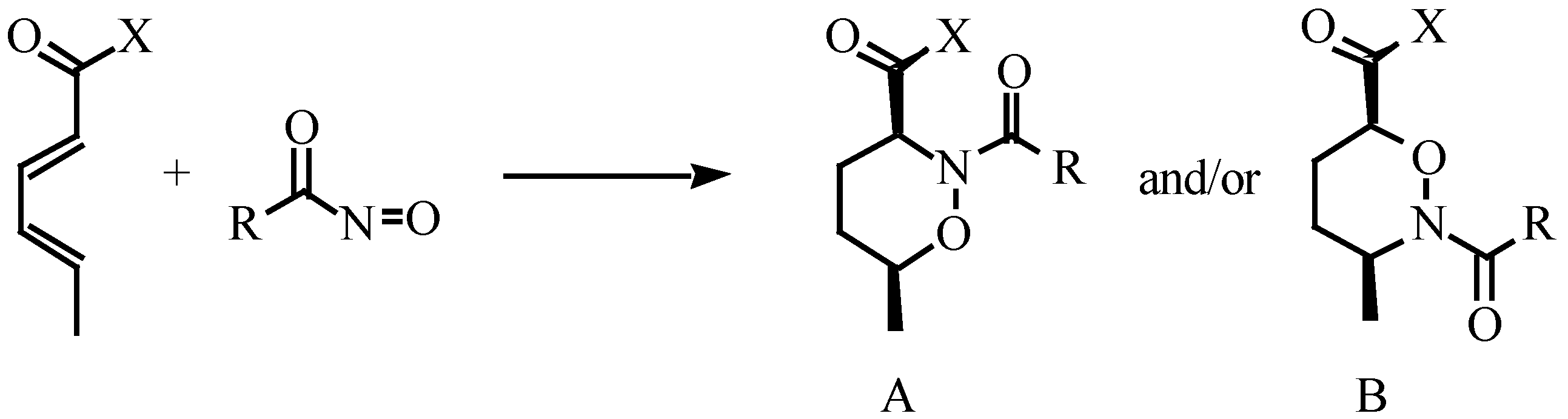
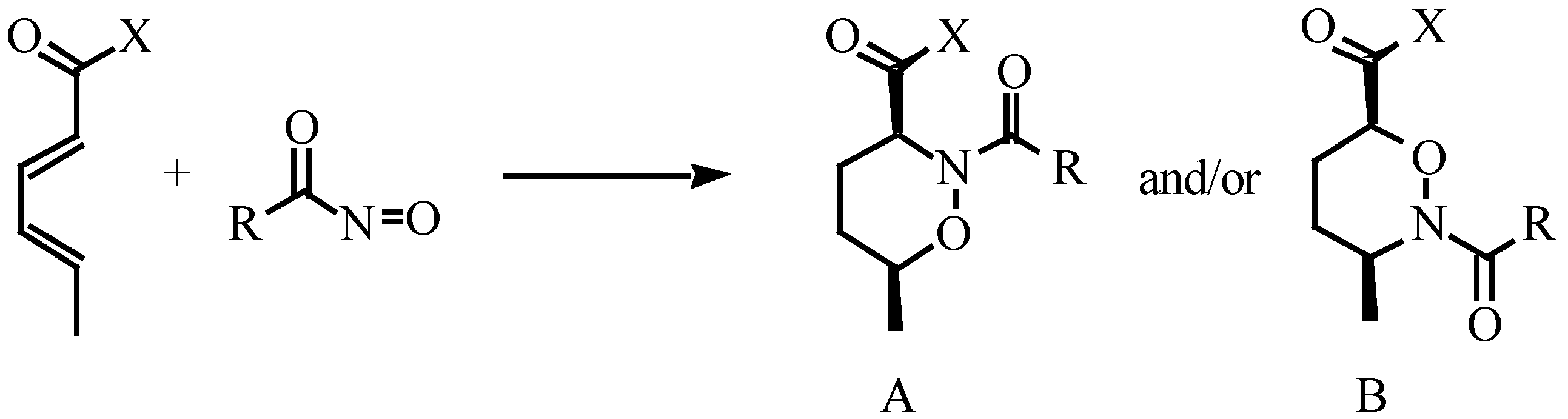
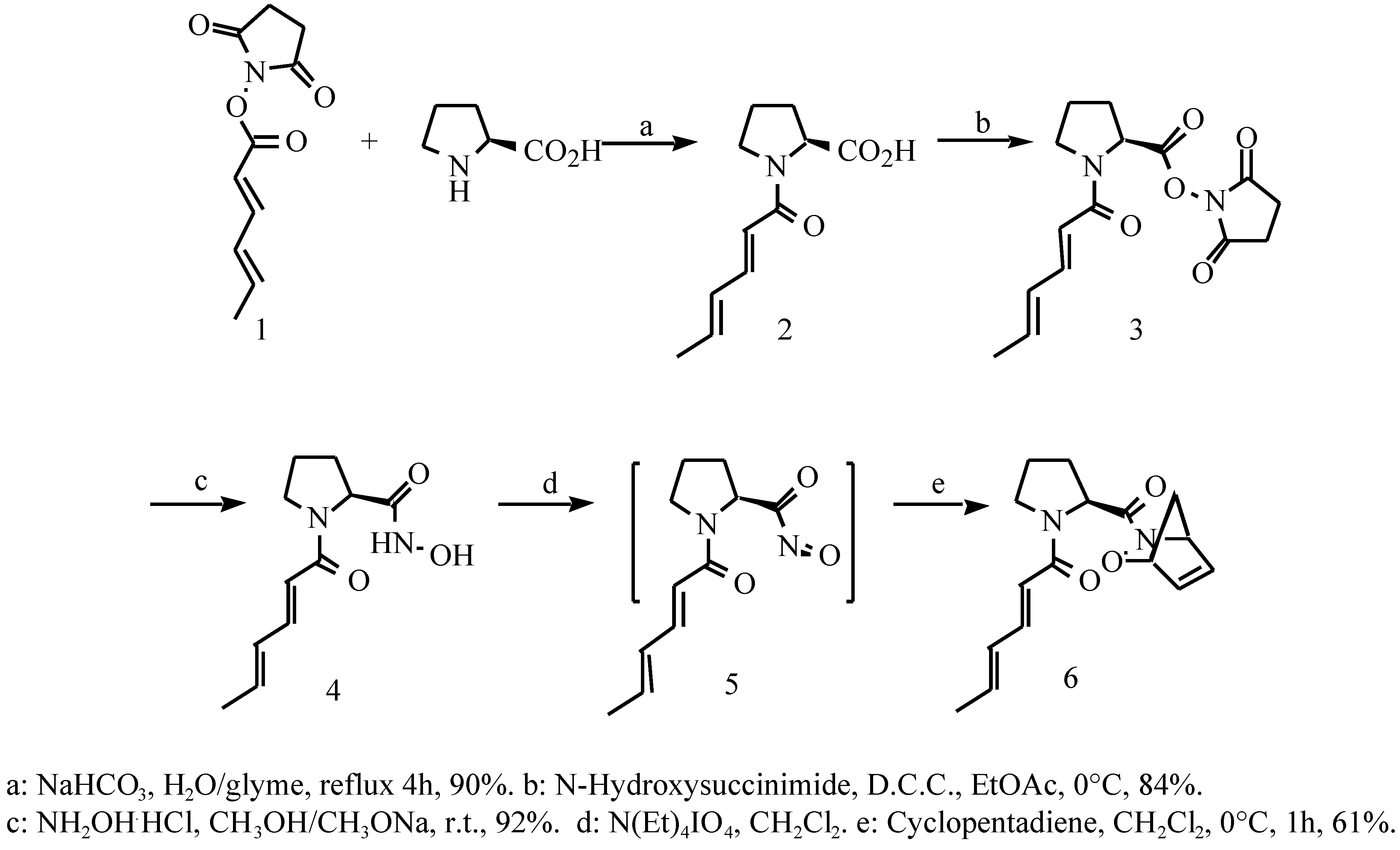
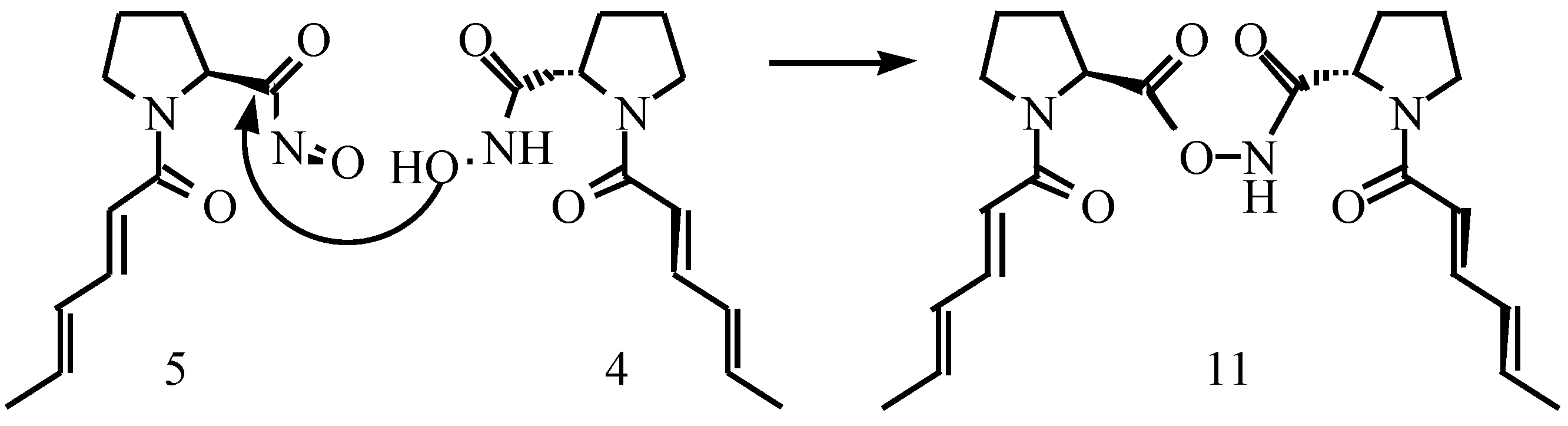
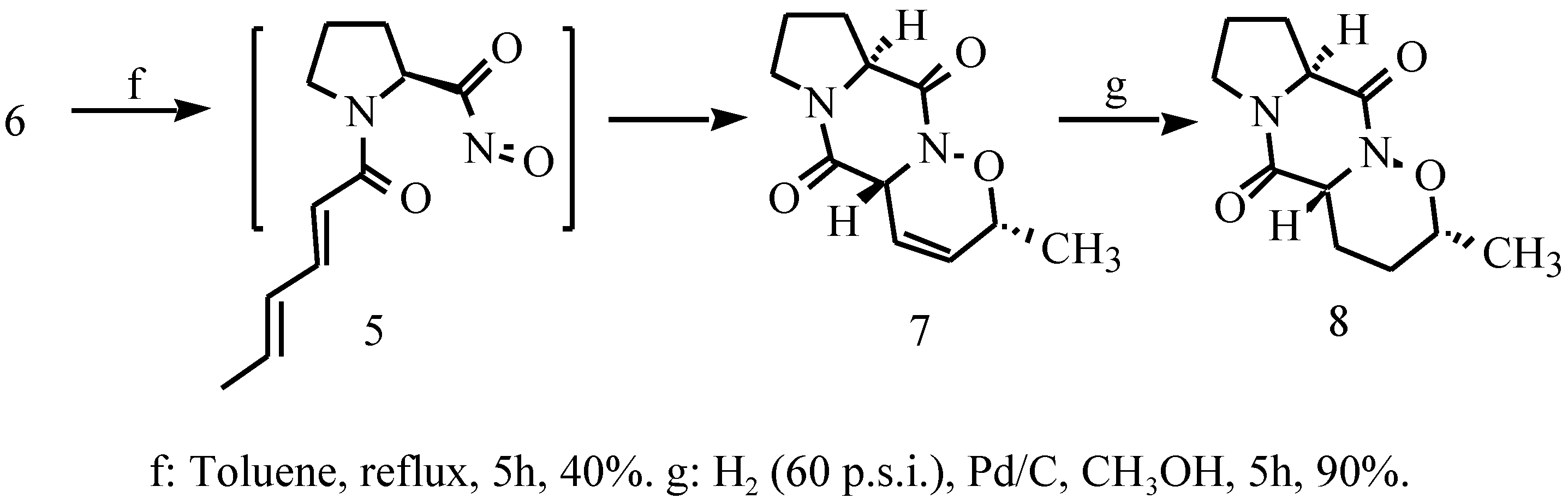
Results and Discussions

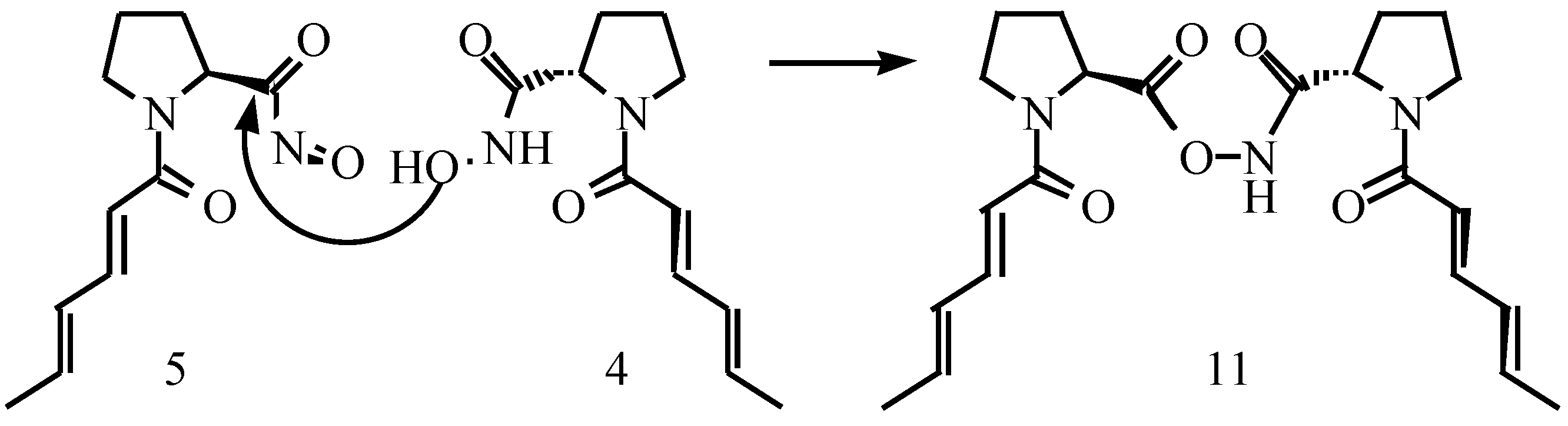
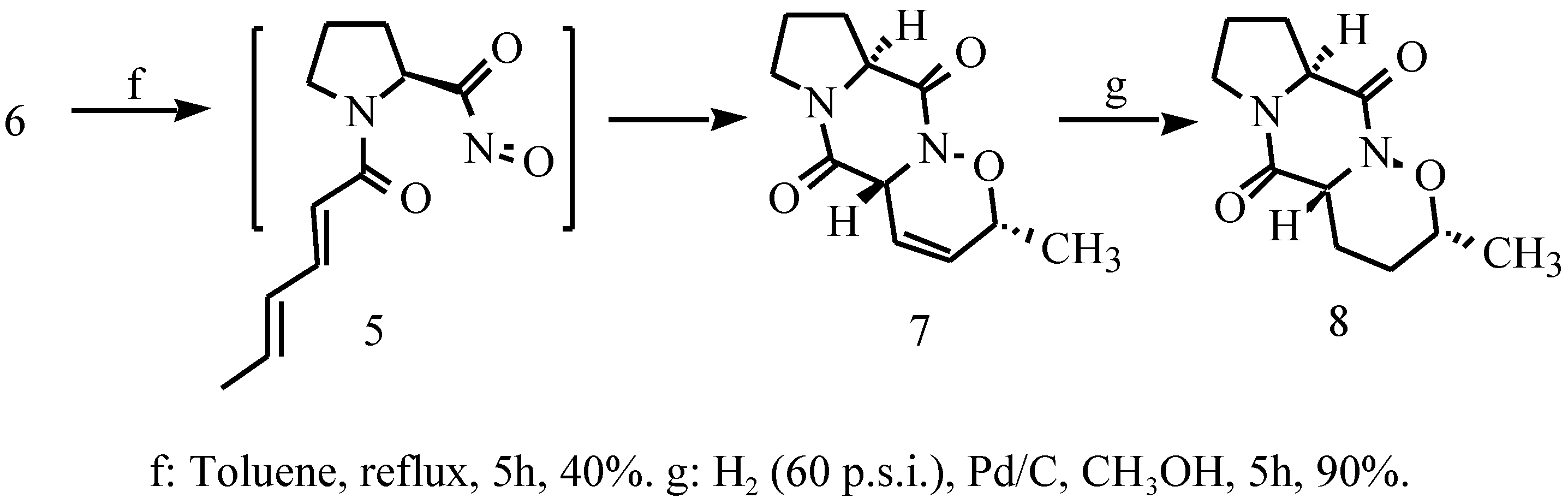

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
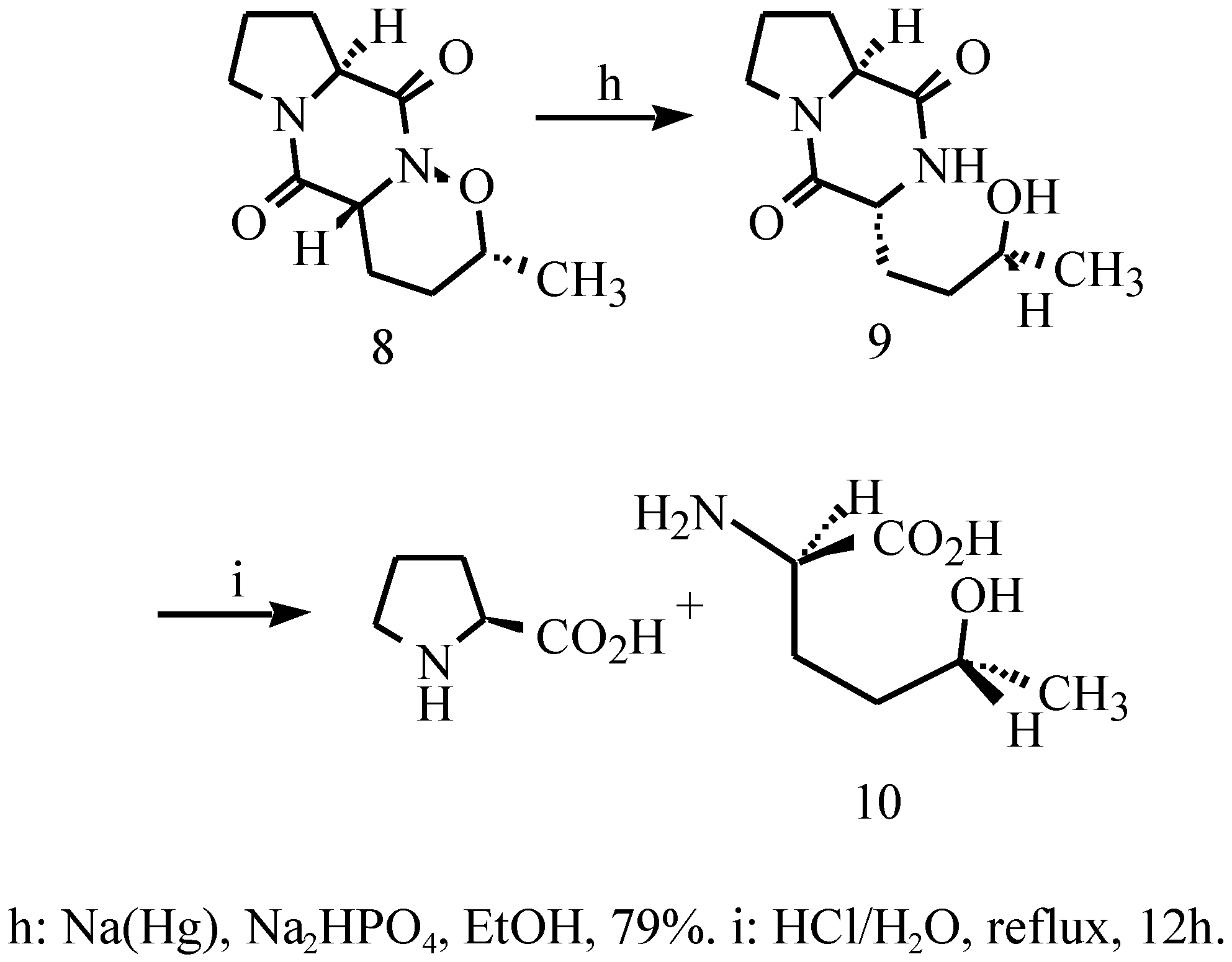{kind=link}
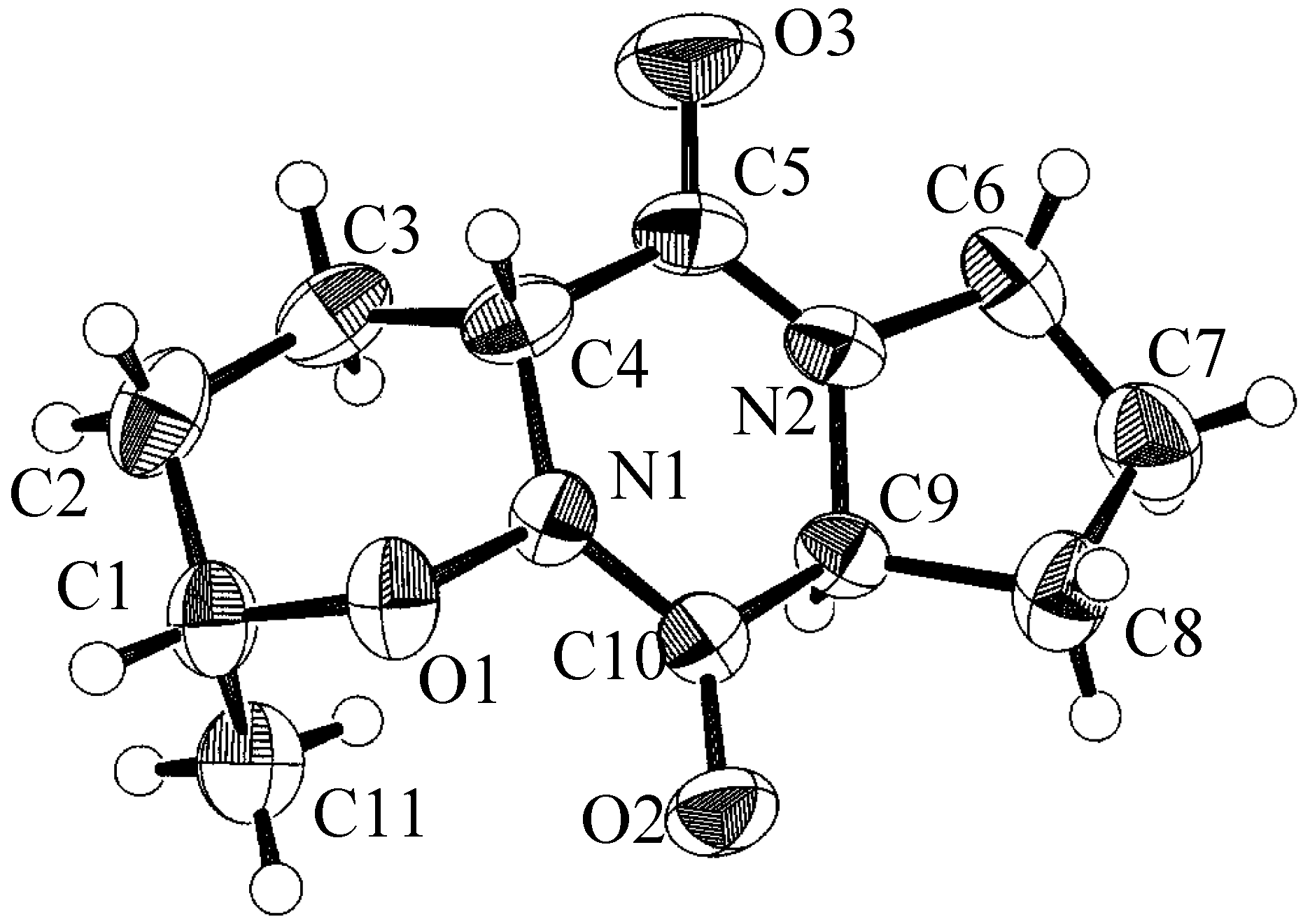
| atom | x | y | z | B(eq) |
| O(1) | 0.2999(3) | 0.1010(1) | 0.5877(5) | 3.4(1) |
| O(2) | 0.0560(3) | 0.0643(1) | 0.7896(6) | 4.2(1) |
| O(3) | 0.2797(4) | 0.2422(1) | 1.191(1) | 7.1(2) |
| O(4) | 0.6924(3) | -0.0034(2) | 0.3352(7) | 6.0(2) |
| N(1) | 0.2349(3) | 0.1242(1) | 0.8077(7) | 2.9(1) |
| N(2) | 0.1141(4) | 0.1777(1) | 1.2110(7) | 3.4(1) |
| C(1) | 0.4349(4) | 0.0755(2) | 0.6691(8) | 3.6(2) |
| C(2) | 0.5329(5) | 0.1189(2) | 0.778(1) | 4.6(2) |
| C(3) | 0.4619(5) | 0.1503(2) | 0.995(1) | 4.2(2) |
| C(4) | 0.3142(4) | 0.1683(2) | 0.9114(9) | 3.5(2) |
| C(5) | 0.2343(5) | 0.1983(2) | 1.121(1) | 4.0(2) |
| C(6) | 0.0131(5) | 0.2056(2) | 1.382(1) | 4.5(2) |
| C(7) | -0.1222(6) | 0.1743(2) | 1.375(1) | 5.3(2) |
| C(8) | -0.1050(5) | 0.1344(2) | 1.151(1) | 4.5(2) |
| C(9) | 0.0572(4) | 0.1247(1) | 1.1457(8) | 3.0(2) |
| C(10) | 0.1147(4) | 0.1019(2) | 0.8962(8) | 2.9(2) |
| C(11) | 0.4079(5) | 0.0299(2) | 0.846(1) | 4.6(2) |
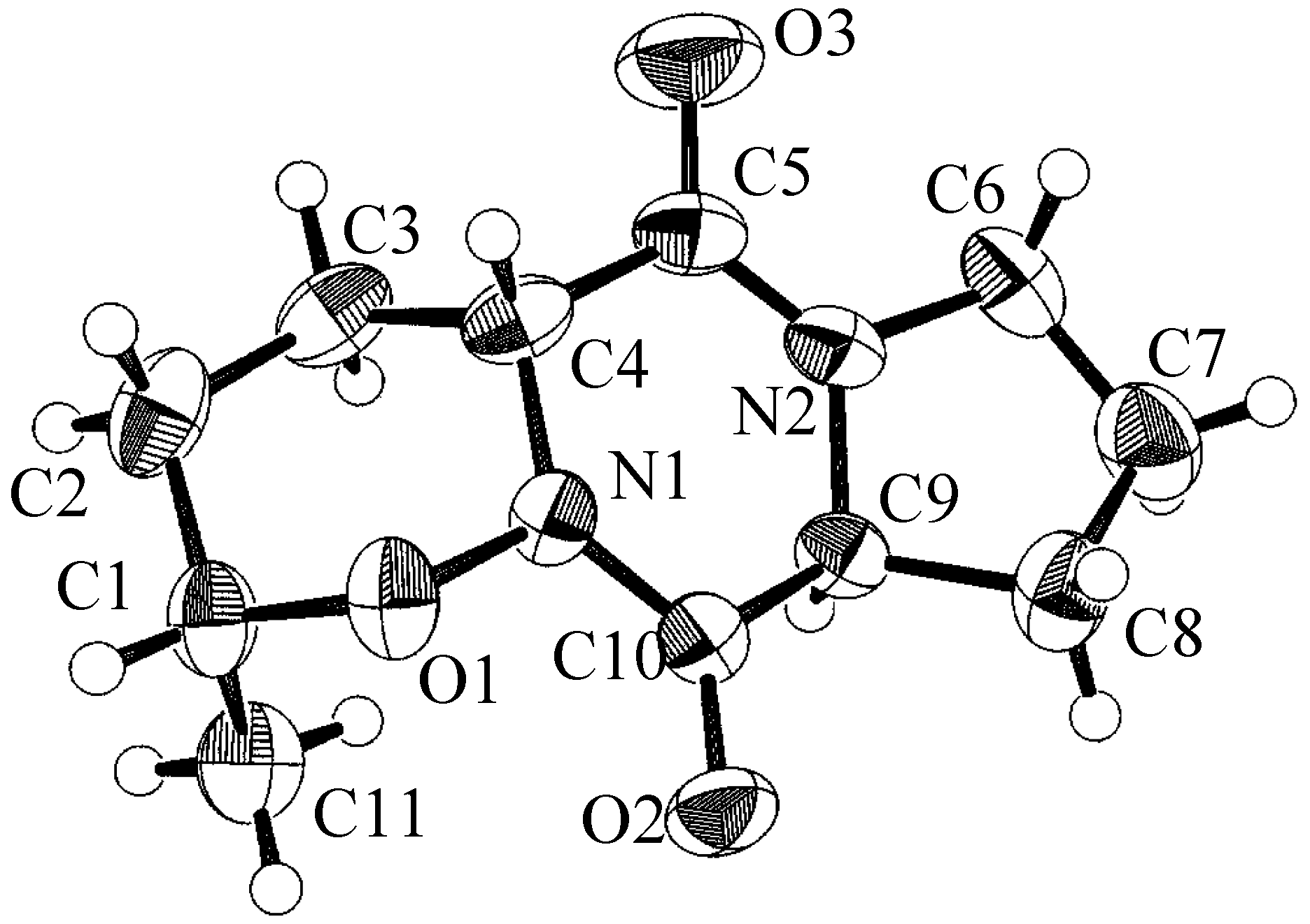
Conclusion
Experimental Section
General
Materials
Synthesis of the compounds
Sorbyl N-hydroxysuccinimide ester (1)
N-Sorbyl-L-proline (2)
N-Sorbyl L-proline N-hydroxysuccinimide ester (3)
N-Sorbyl L-proline hydroxamic acid (4)
Tetraethylammonium periodate
2-N-Sorbyl-L-prolinyl-1-oxa-2-azatricyclo[2,2,1]hepta-4- ene (6)
3,10-Diaza-5-methyl-4-oxatricyclo[8,4,03,7,0]deca-6-ene- 2,9-dione (7)
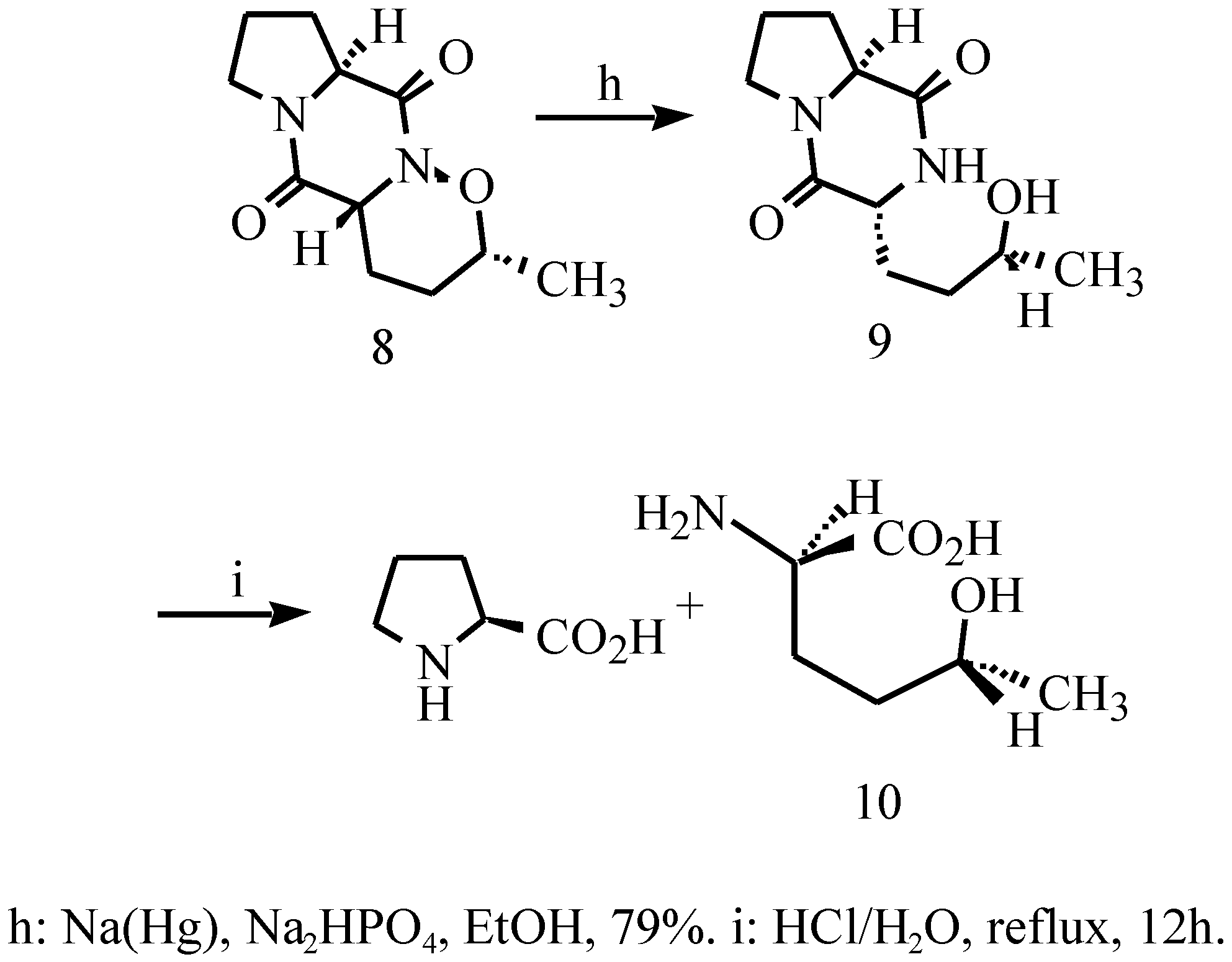
3,10-Diaza-5-methyl-4-oxatricyclo[8,4,03,7,0]deca-2,9- dione (8)
3-(3-hydroxy-1-butyl)-pyrrolo[1,2-e]pyrazine-2,5-dione (9)
(2R,5R)-2-Amino-5-hydroxyhexanoic acid (10)
References and Notes
- Corey, E.J.; Ensley, H.E. Preparation of an Optically Active Prostaglandin Intermediate Via Asymmetric Induction. J. Am. Chem. Soc. 1975, 97, 6908–6909. [Google Scholar] [CrossRef] [PubMed]
- Oppolzer, W.; Chapuis, C.; Depuis, D.; Guo, M. Asymmetric Diels-Alder Reactions of Neopentyl- Ester-Shielded Acrylates and Allenic Esters: Syntheses of (-)-Norbornenone and (-)-β-Santalene. Helv. Chim. Acta 1985, 68, 2100–2114. [Google Scholar] [CrossRef]
- Stork, G; Nakamura, E. A Simplified Total Synthesis of Cytochalasins Via an Intramolecular Diels-Alder Reaction. J. Am. Chem. Soc. 1983, 105, 5510–5512. [Google Scholar] [CrossRef]
- Hirama, M.; Uei, M. Chiral Total Synthesis of Compactin. J. Am. Chem. Soc. 1982, 104, 4251–4253. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Papahadjis, D.P.; Claremon, D.A.; Dolle, R.E. Total Synthesis of Ionophore Antibiotic X-14547-A. 1. Enantioselective Synthesis of the Tetrahydropyran and Tetrahydroindan Building Blocks. J. Am. Chem. Soc. 1981, 103, 6967–6969. [Google Scholar] [CrossRef]
- Sheradsky, T.; Milvitskaya, J.; Pollak, I.E. Intramolecular Oxidative Diels-Alder Reaction of N- Sorbyl-L-proline Acylhydrazines. Tetrahedron Lett. 1991, 32, 133–136. [Google Scholar] [CrossRef]
- Kibayashi, C; Aoyagi, S. Nitrogenous Natural Products Synthesis Via N-Acylnitroso Diels-Alder Methodology. Synlett 1995, 873–879. [Google Scholar] [CrossRef]
- Martin, S.F.; Hartmann, M.; Josey, J.A. Diastereoselective [4+2] Cycloadditions of Acyl Nitroso Compounds. Tetrahedron Lett. 1992, 33, 3583–3586, and corrigendum: Tetrahedron Lett. 1993, 34, 2852. [Google Scholar] [CrossRef]
- Kirby, G.W.; MacKinnon, J.W.M. Cycloadducts of C-Nitrosocarbonyl Compounds and Ergosteryl Acetate; [3+3] Sigmatropic Rearrangements of N- Aroyl-3,6-dihydro-1,2-oxazines. J. Chem. Soc. Perkin 1 1985, 887–897. [Google Scholar] [CrossRef]
- Kirby, G.W.; McGuigan, H.; MacKinnon, J.W.M.; McLean, D.; Sharma, R.P. Formation and Reactions of C-Nitrosoformate Esters, a New Class of Transient Dienophiles. J. Chem. Soc. Perkin 1 1985, 1437–1442. [Google Scholar] [CrossRef]
- X-ray diffraction data was measured with either an ENRAF-NONIUS CAD-4 automatic diffractometer or a Phillips four circle computer controlled diffractometer. The crystallographic computing was done on a VAX 900 computer using TAXAN analysis software.
- Formula C11H16N2O3; M 224.26; space group P212121; a (Å) 9.317(2); b (Å) 25.216(5); c (Å) 5.122(2); V (Å3) 1203.4(7); z 4; ρ(g cm-3) 1.34; m MoKα(cm-1) 9.6; no. of unique reflections 1271; no. of reflections with I≥3σI 1046; R 0.050; Rw 0.067.
- Keck, G.E.; Fleming, S.; Nickell, D.; Weider, P. Mild and Efficient Methods for the Reductive Cleavage of Nitrogen-Oxygen Bonds. Synth. Commun. 1979, 9, 281–286. [Google Scholar] [CrossRef]
- Sample Availability: available from the authors.
© 1998 MDPI. All rights reserved. Molecules http://www.mdpi.org/molecules/
Share and Cite
Sheradsky, T.; Silcoff, E.R. Synthesis of (2R,5R)-2-Amino-5-Hydroxyhexanoic Acid by Intramolecular Cycloaddition. Molecules 1998, 3, 80-87. https://doi.org/10.3390/30300080
Sheradsky T, Silcoff ER. Synthesis of (2R,5R)-2-Amino-5-Hydroxyhexanoic Acid by Intramolecular Cycloaddition. Molecules. 1998; 3(3):80-87. https://doi.org/10.3390/30300080
Chicago/Turabian StyleSheradsky, Tuvia, and Elliad R. Silcoff. 1998. "Synthesis of (2R,5R)-2-Amino-5-Hydroxyhexanoic Acid by Intramolecular Cycloaddition" Molecules 3, no. 3: 80-87. https://doi.org/10.3390/30300080
APA StyleSheradsky, T., & Silcoff, E. R. (1998). Synthesis of (2R,5R)-2-Amino-5-Hydroxyhexanoic Acid by Intramolecular Cycloaddition. Molecules, 3(3), 80-87. https://doi.org/10.3390/30300080