4-Aryldihydropyrimidines via the Biginelli Condensation: Aza-Analogs of Nifedipine-Type Calcium Channel Modulators

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Introduction


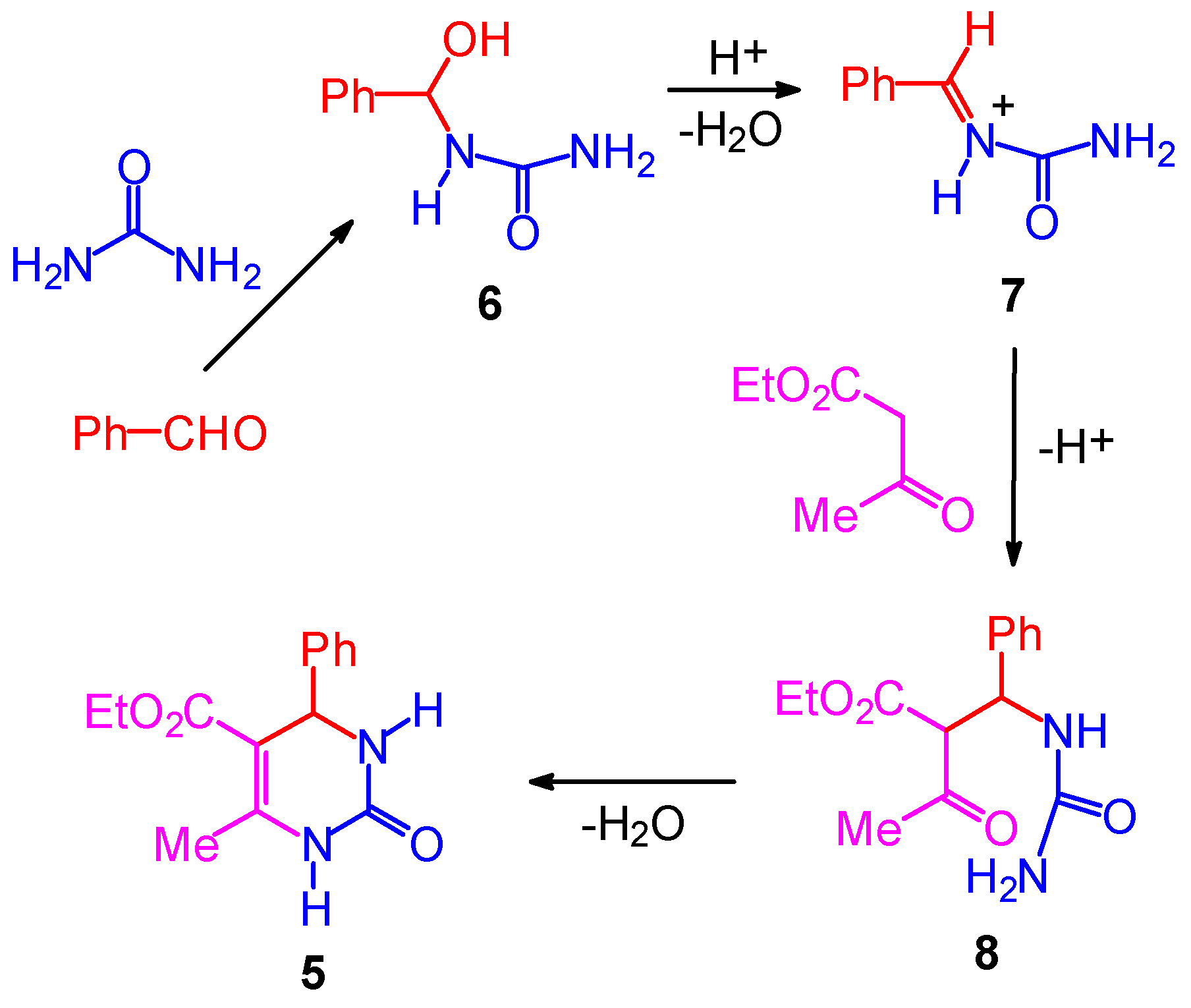
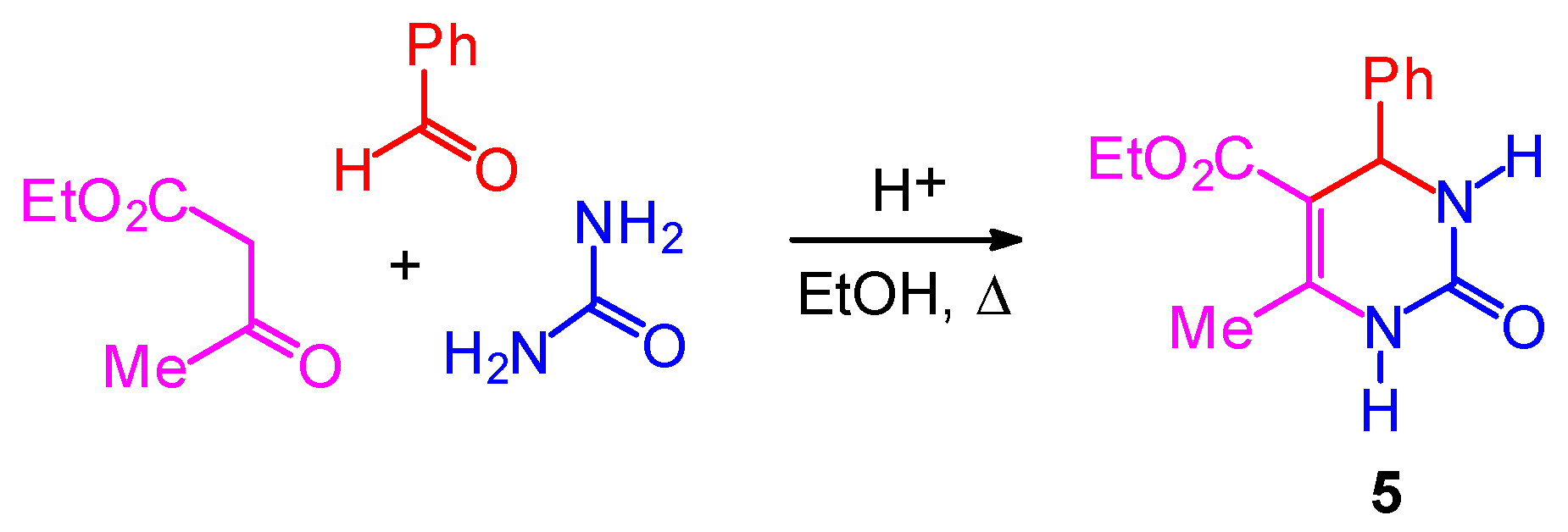
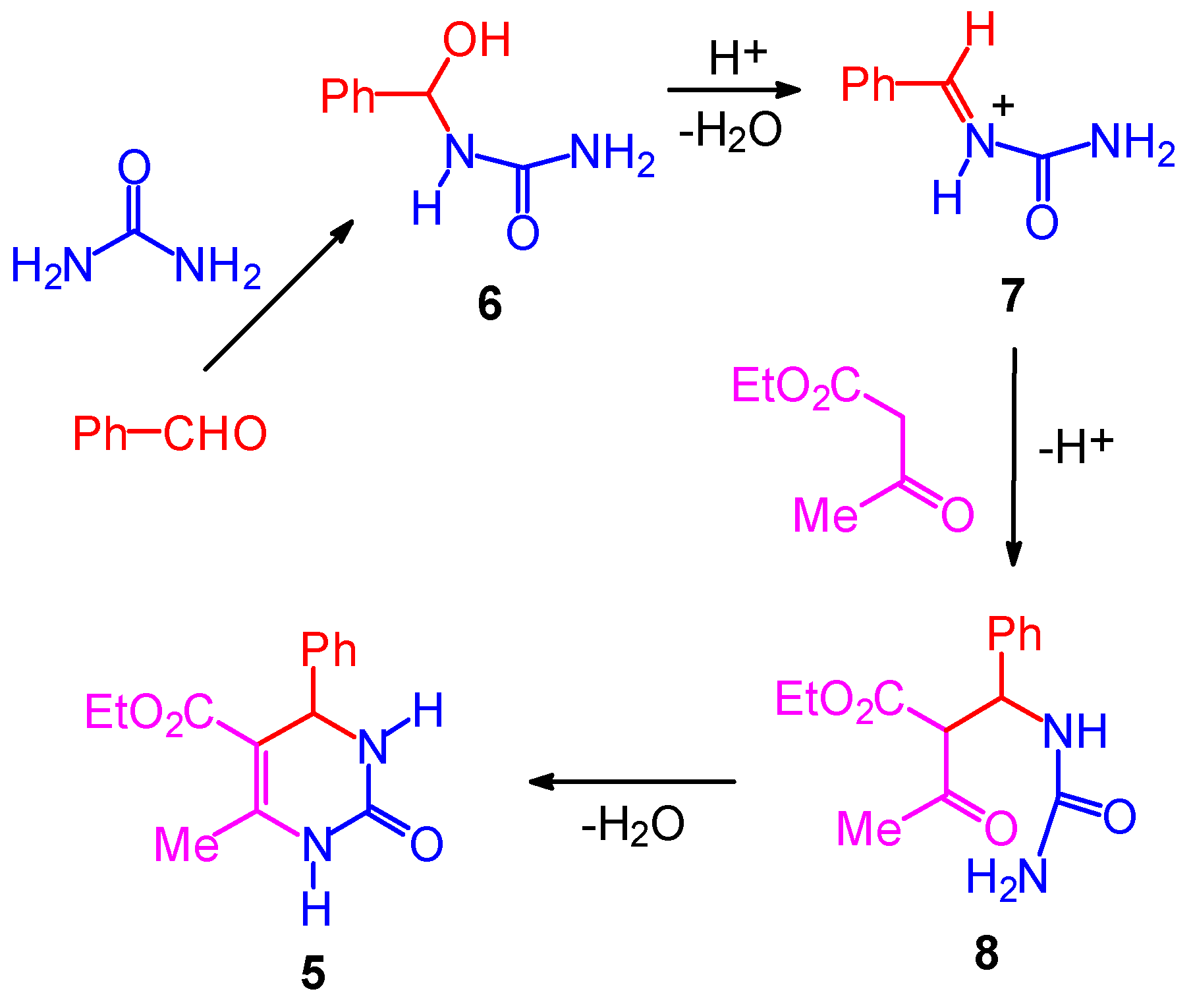
The Mechanism of the Biginelli Reaction
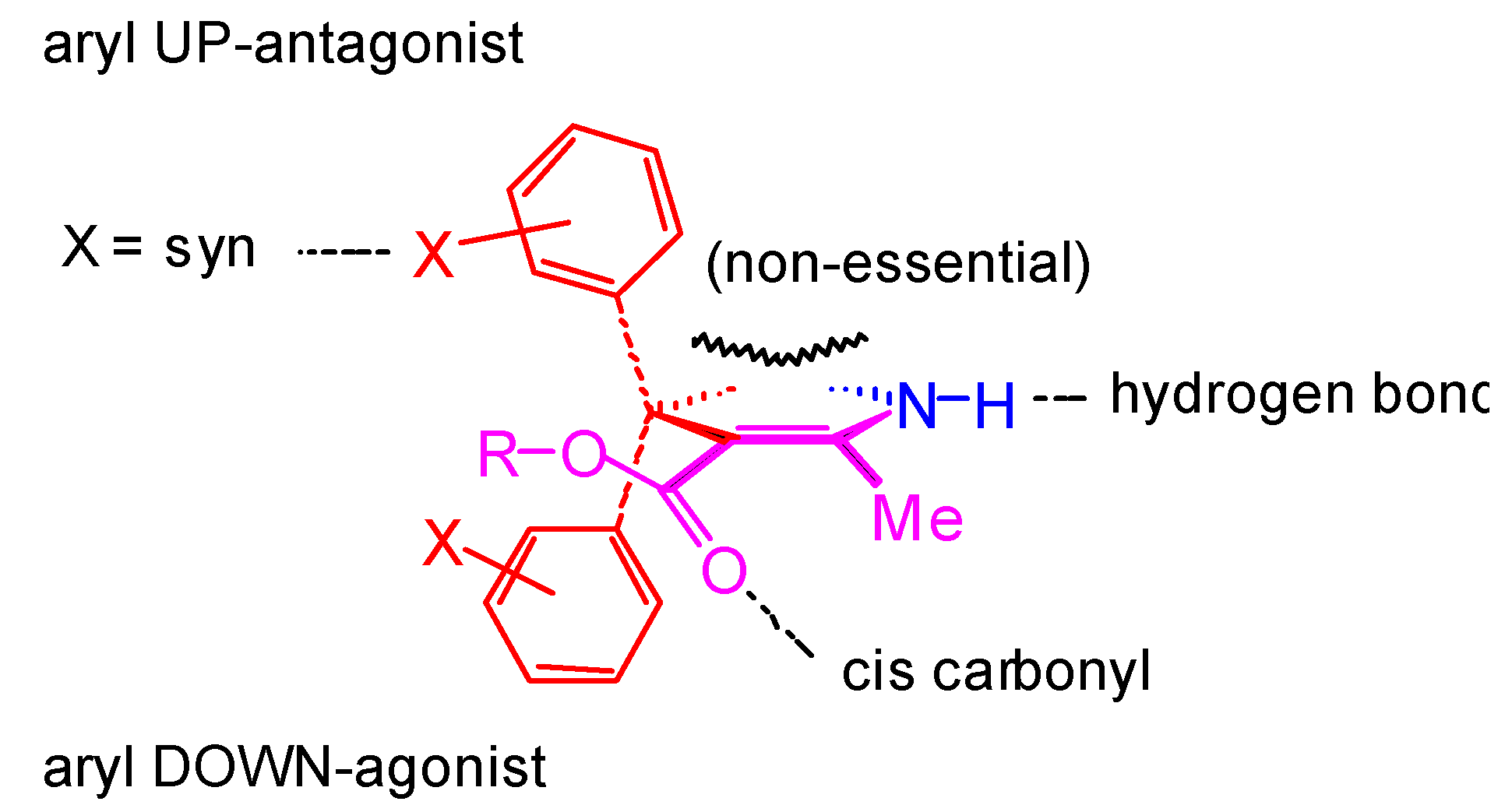
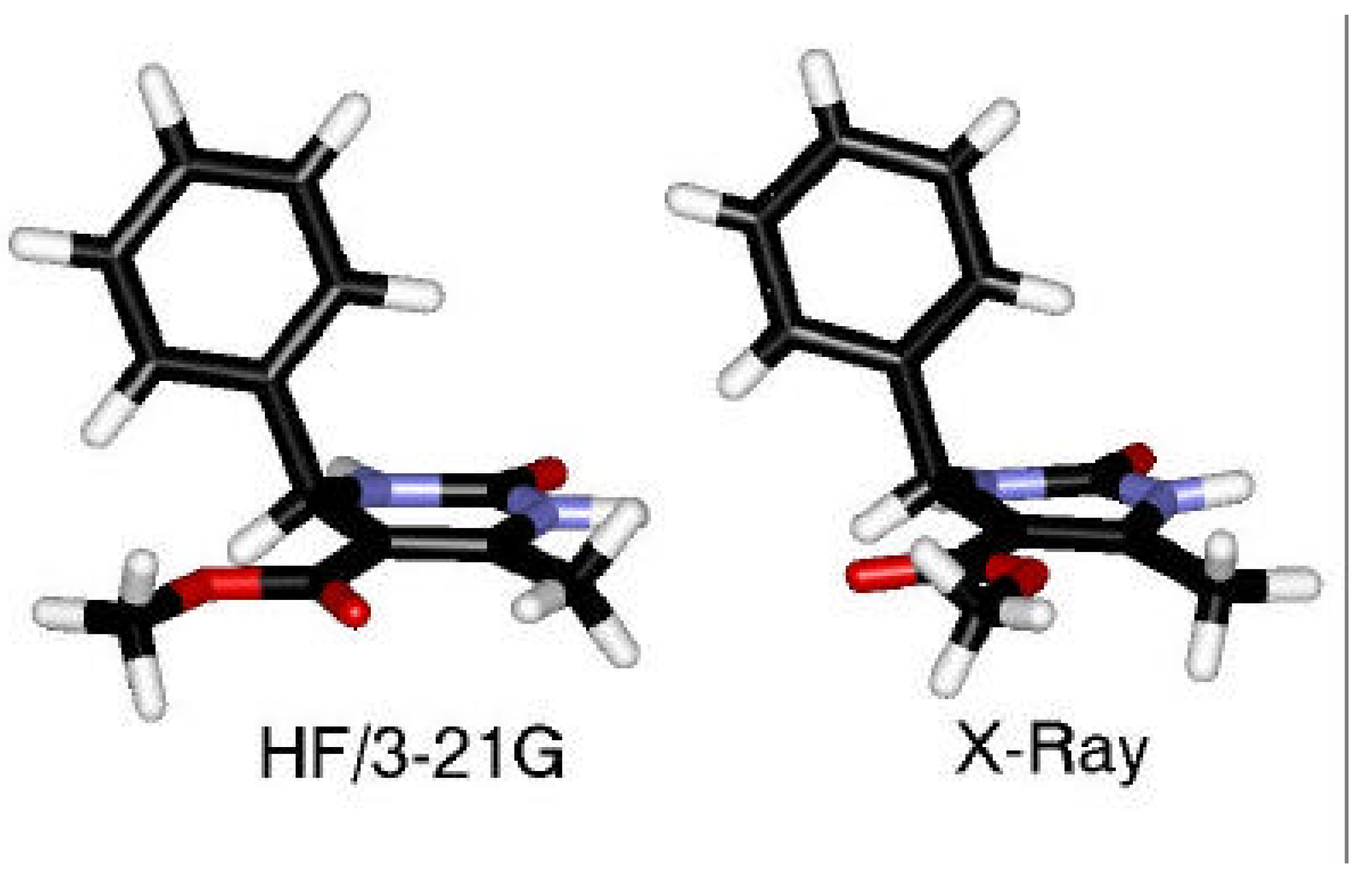
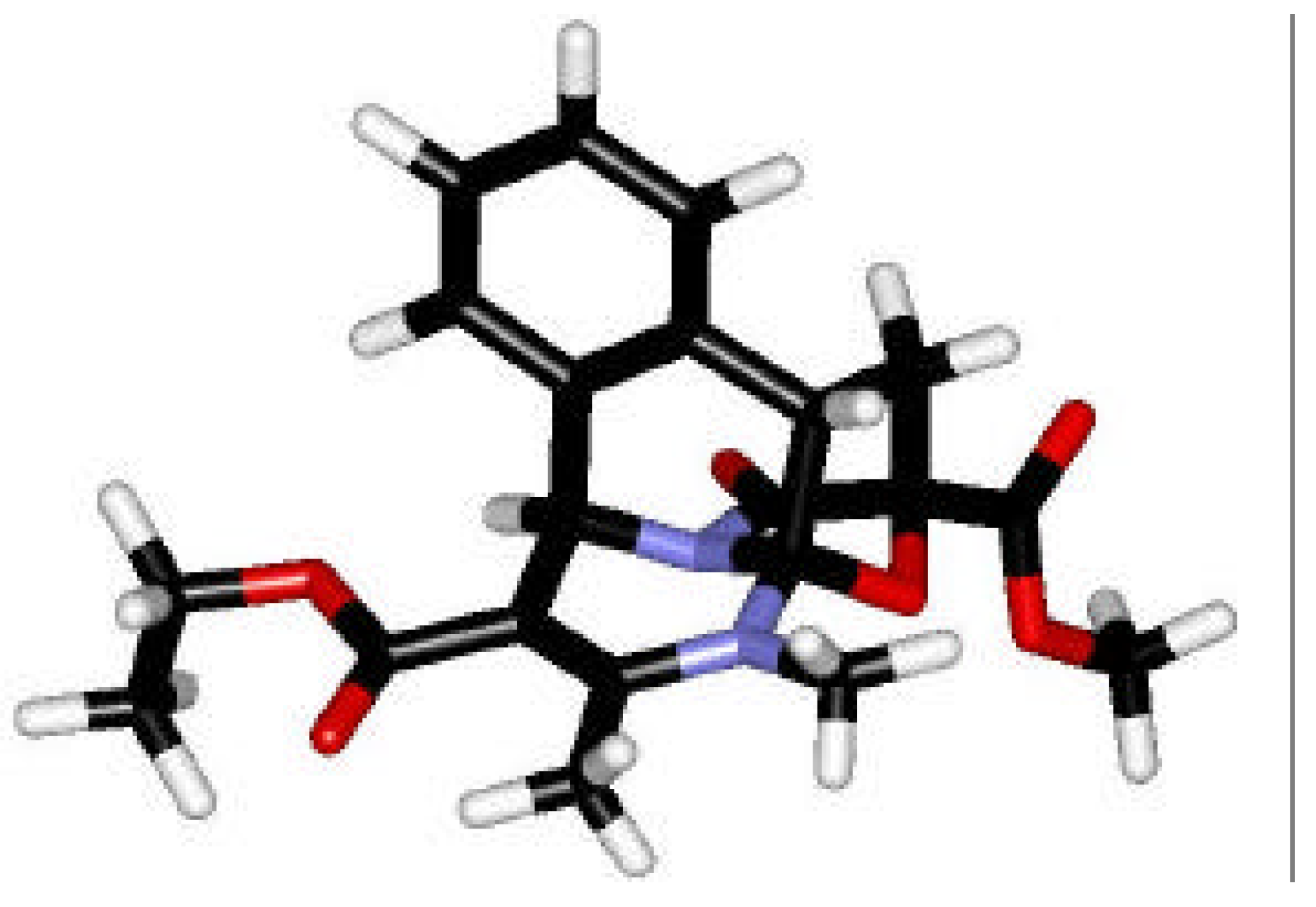
Conformational Studies

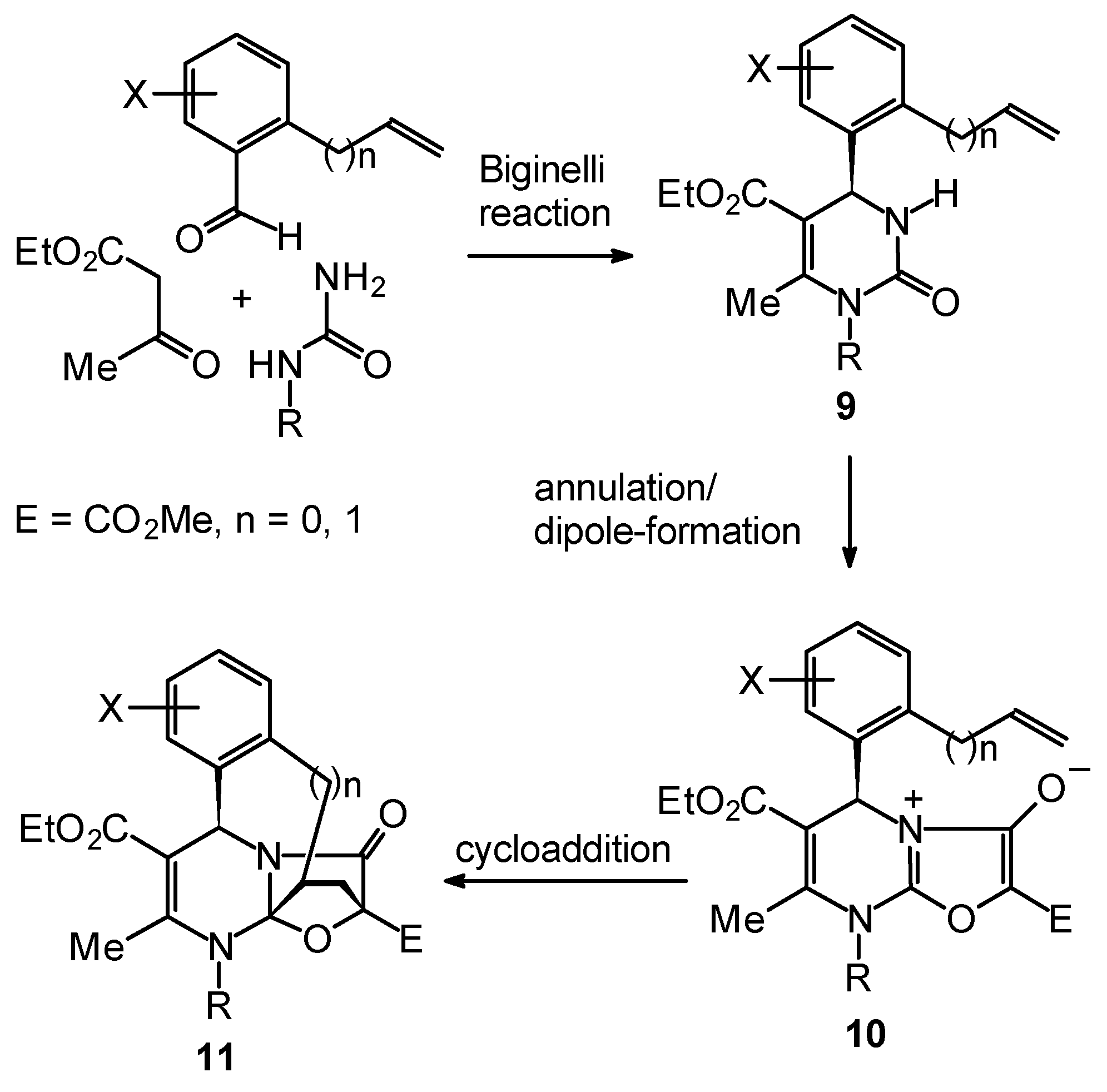
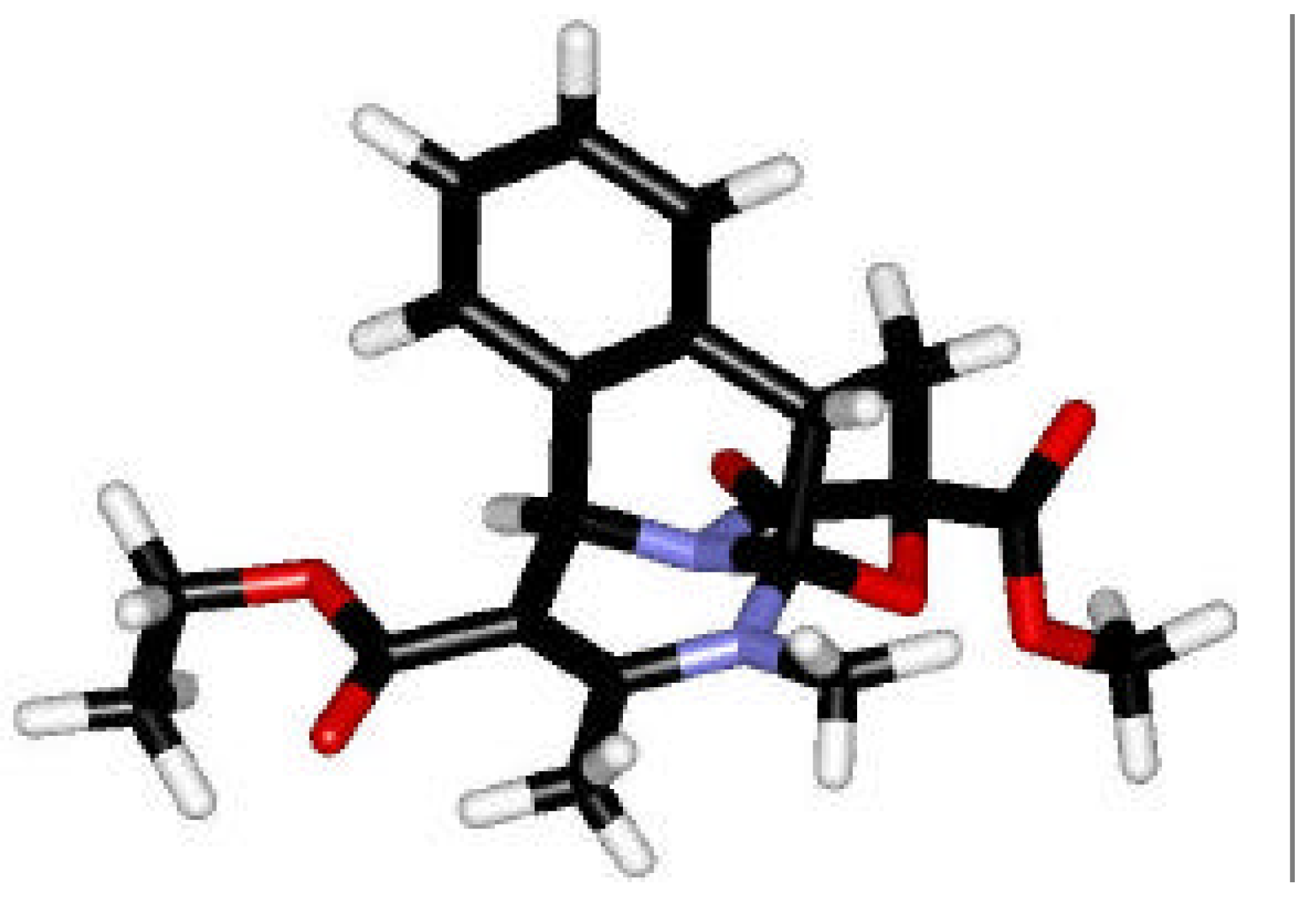
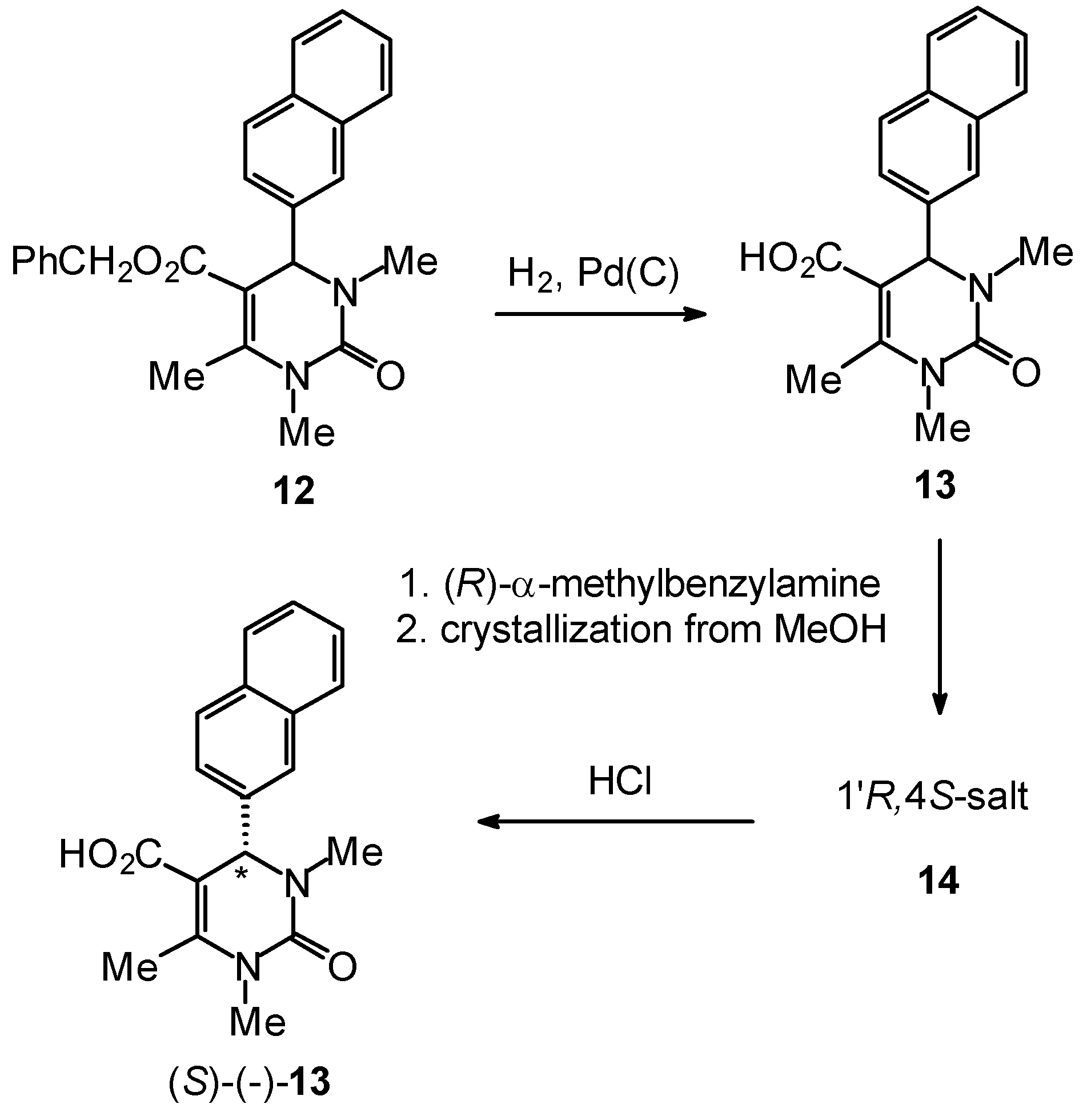
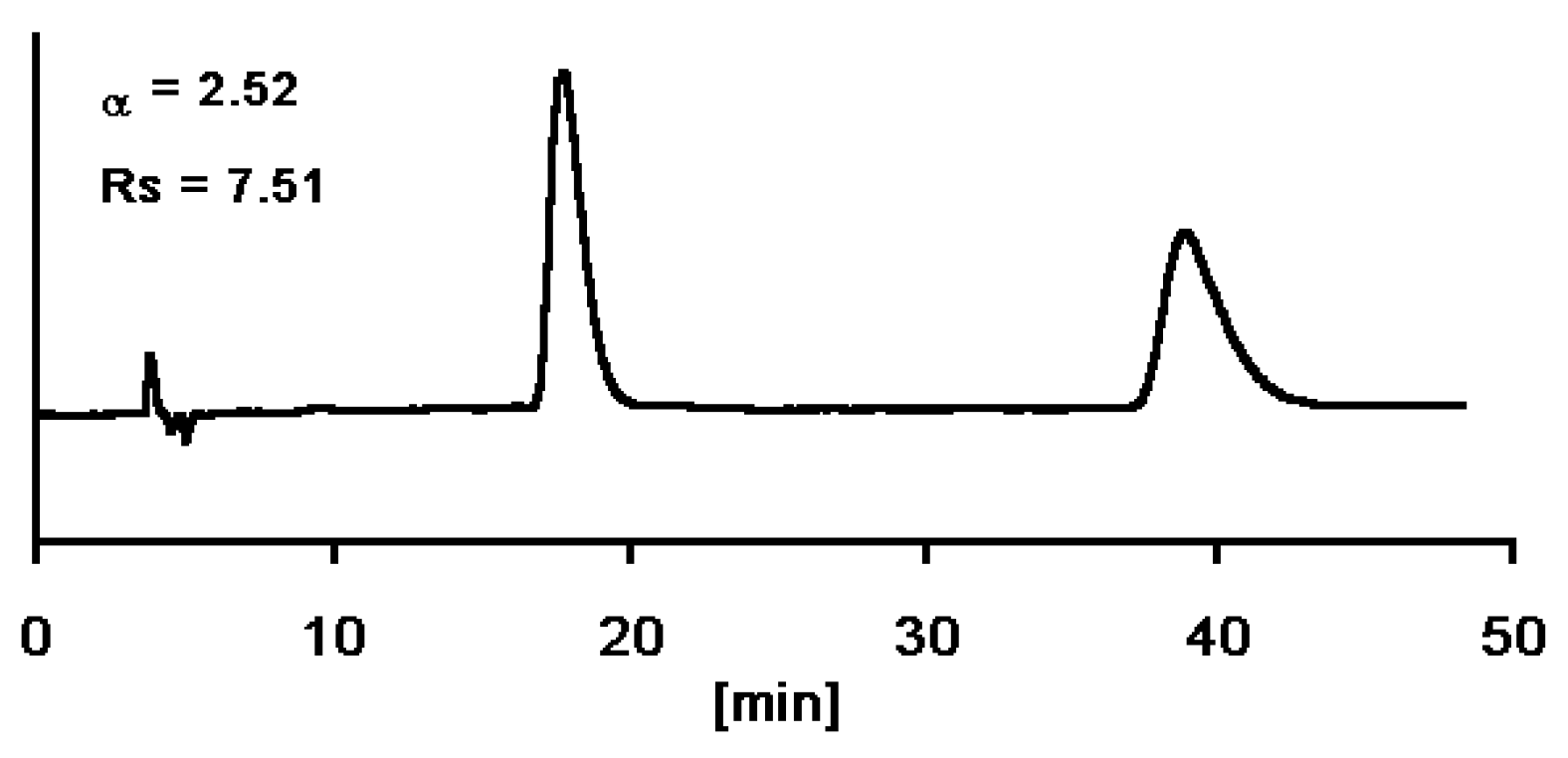
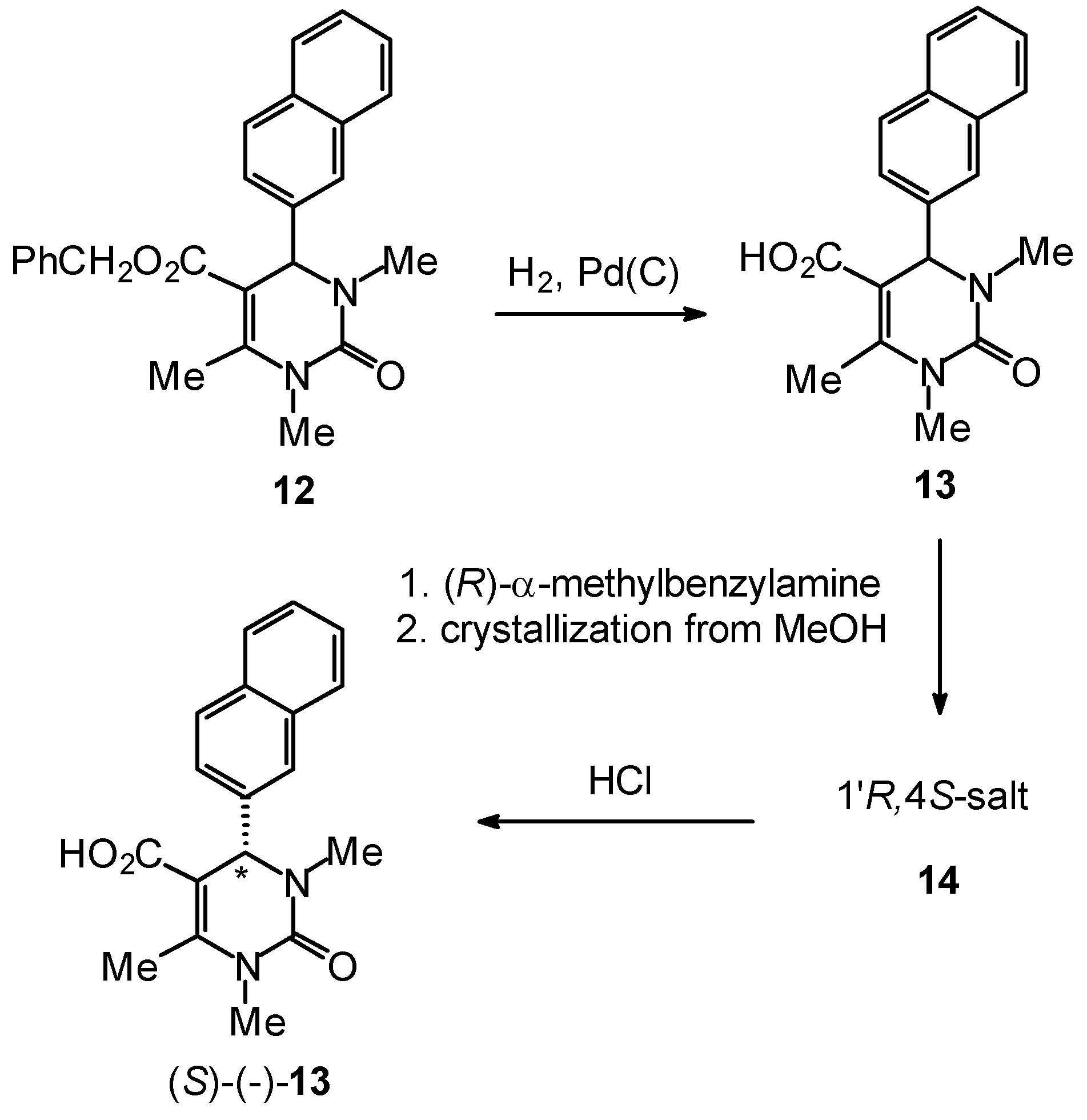
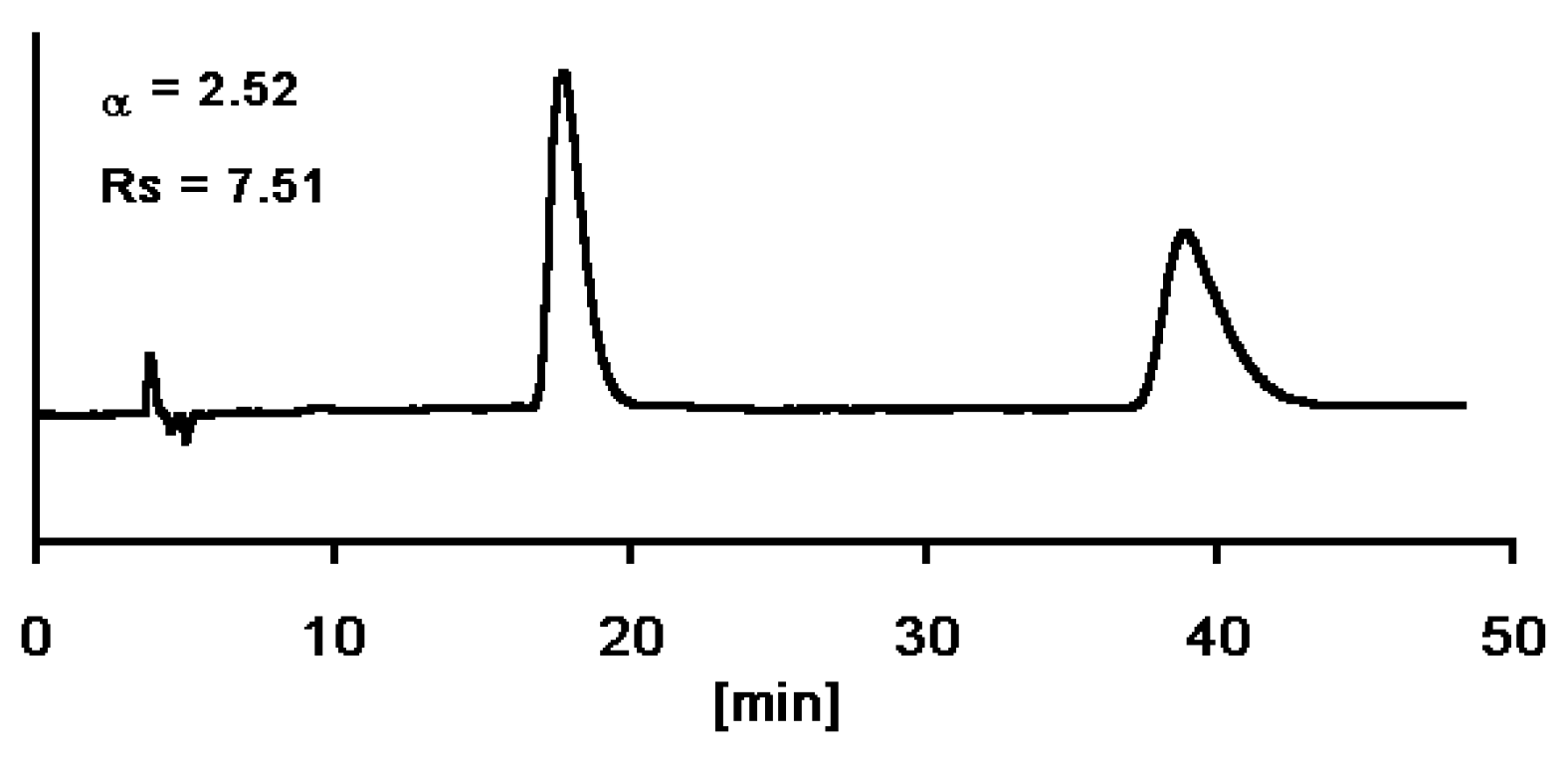
Enantiomerically Pure Dihydropyrimidines
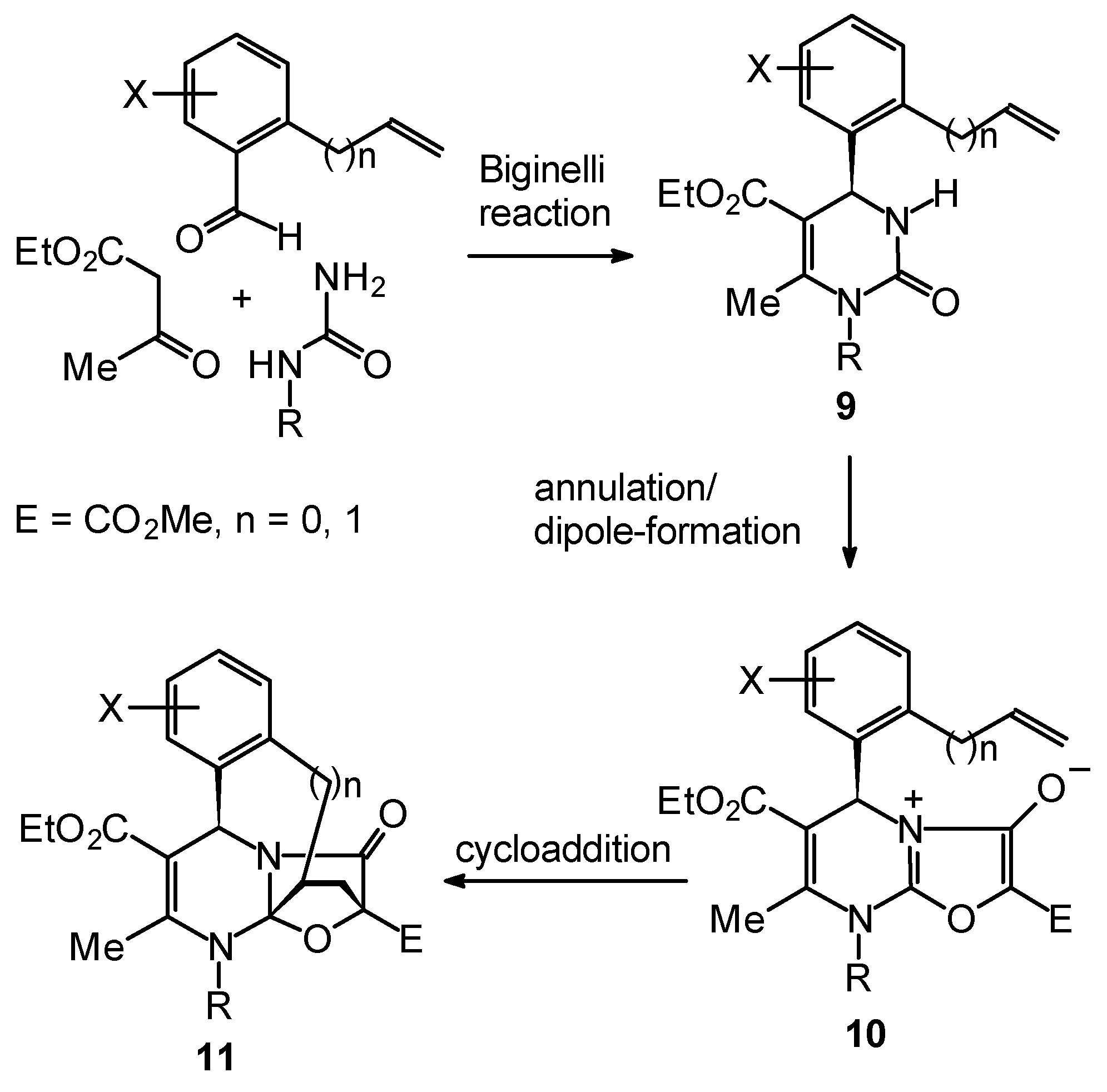
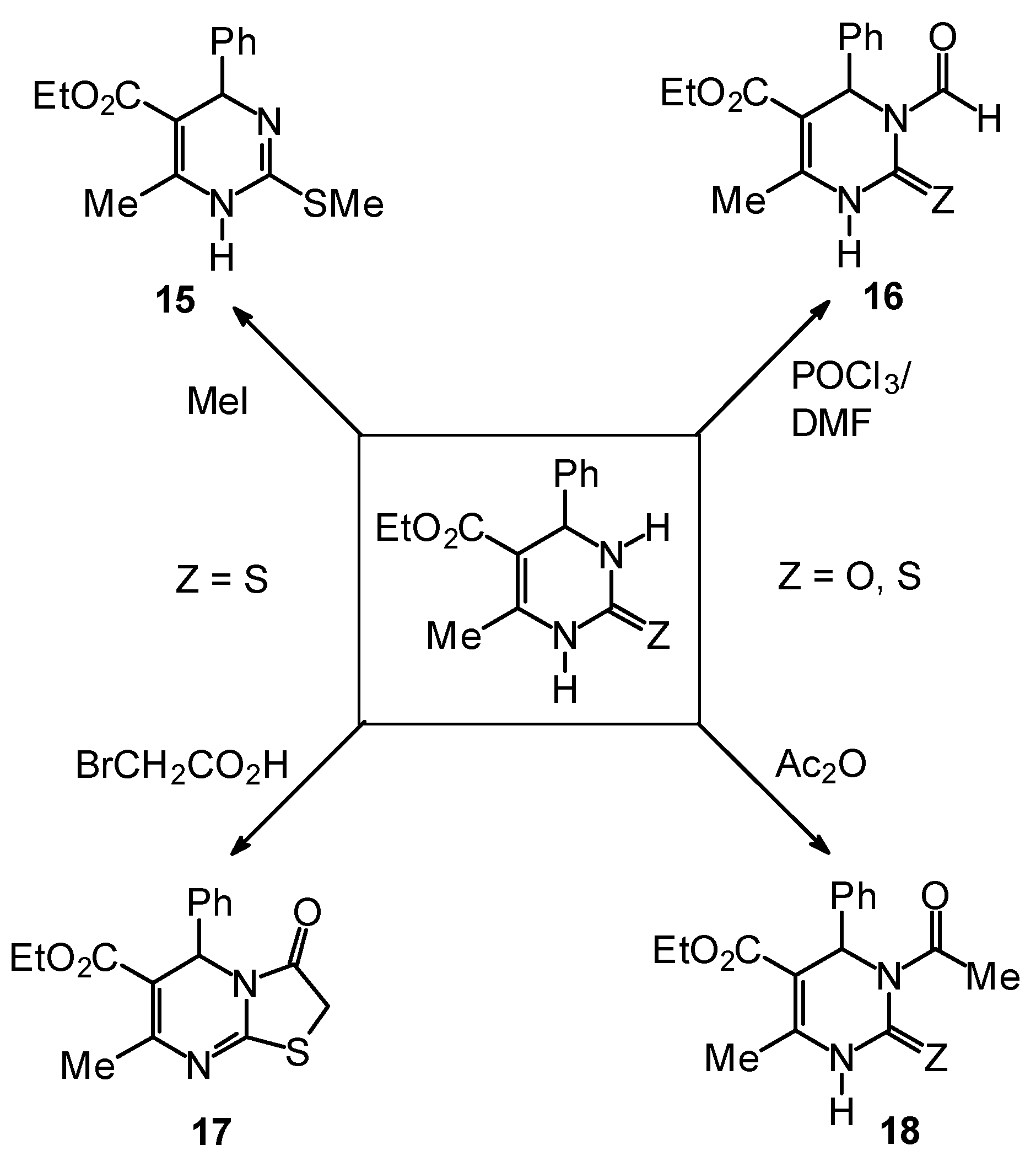
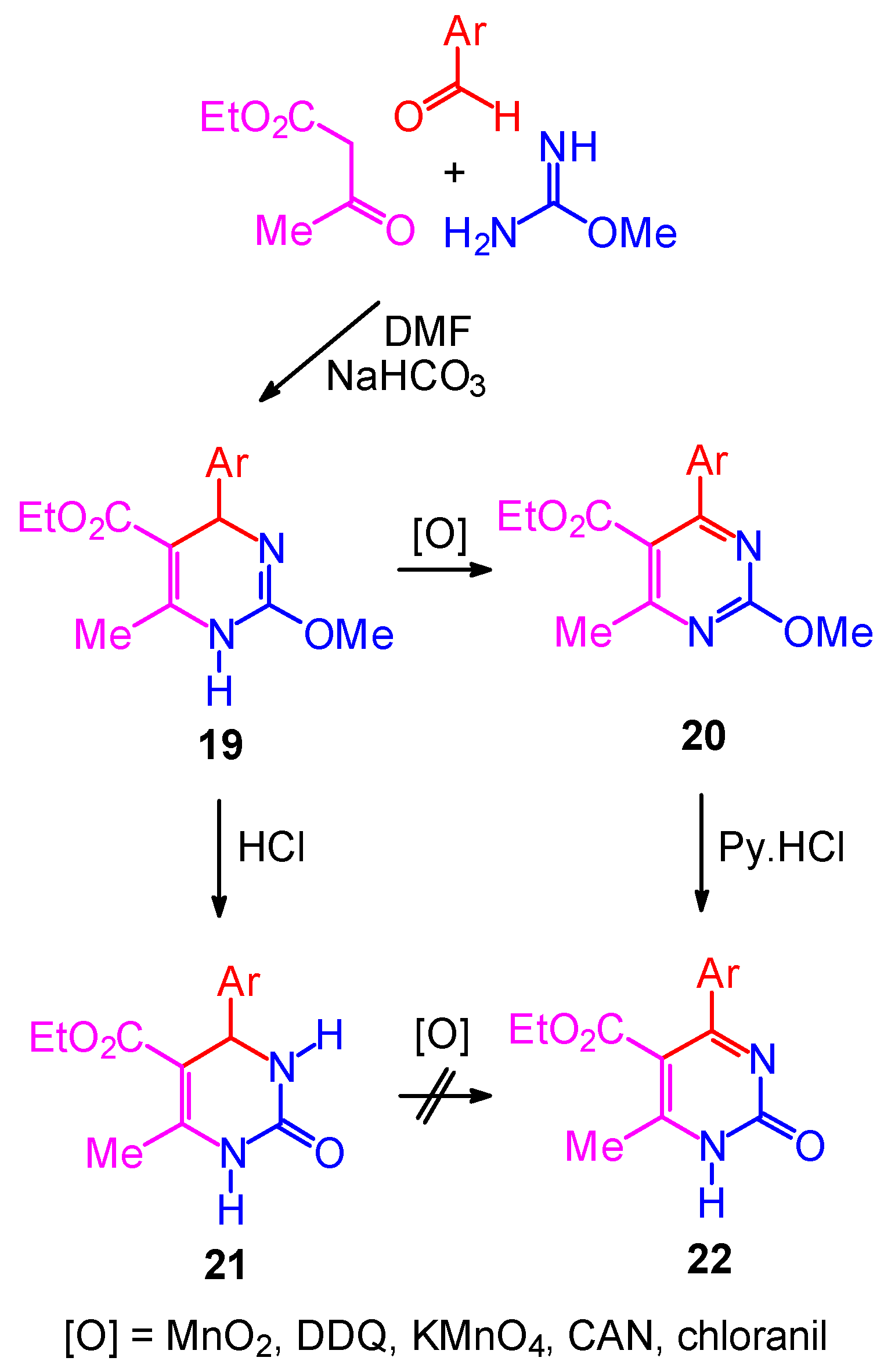
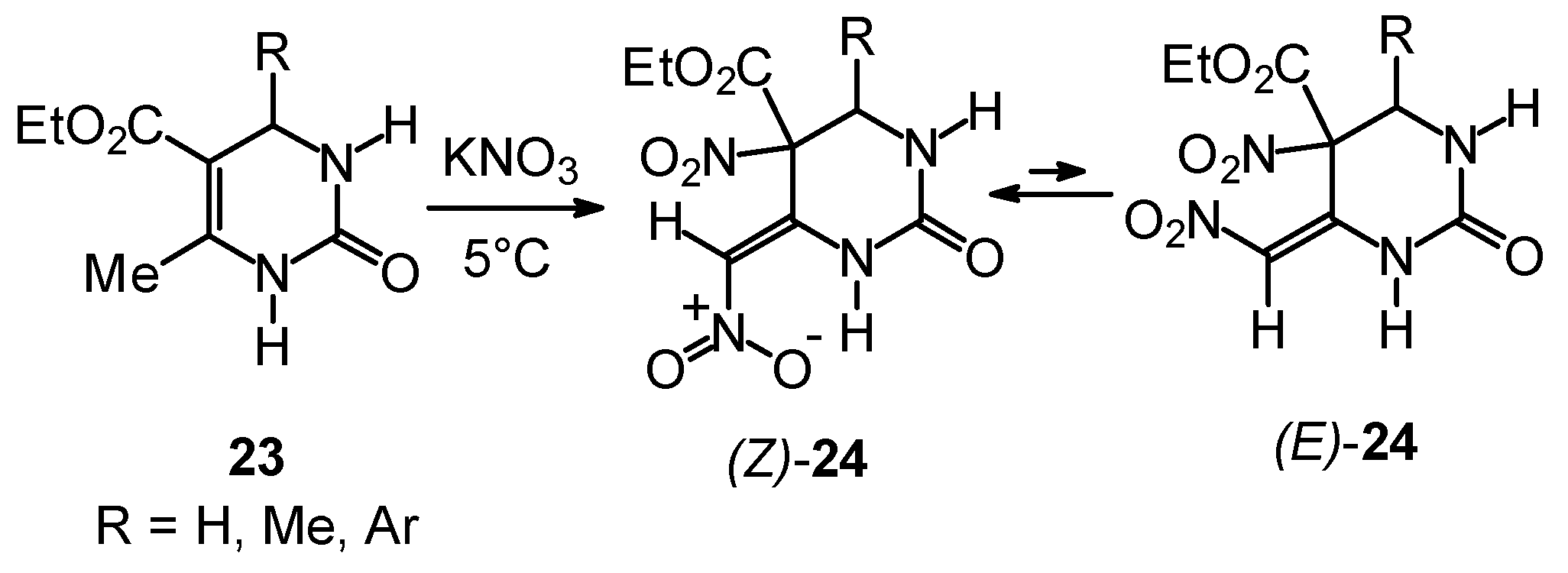
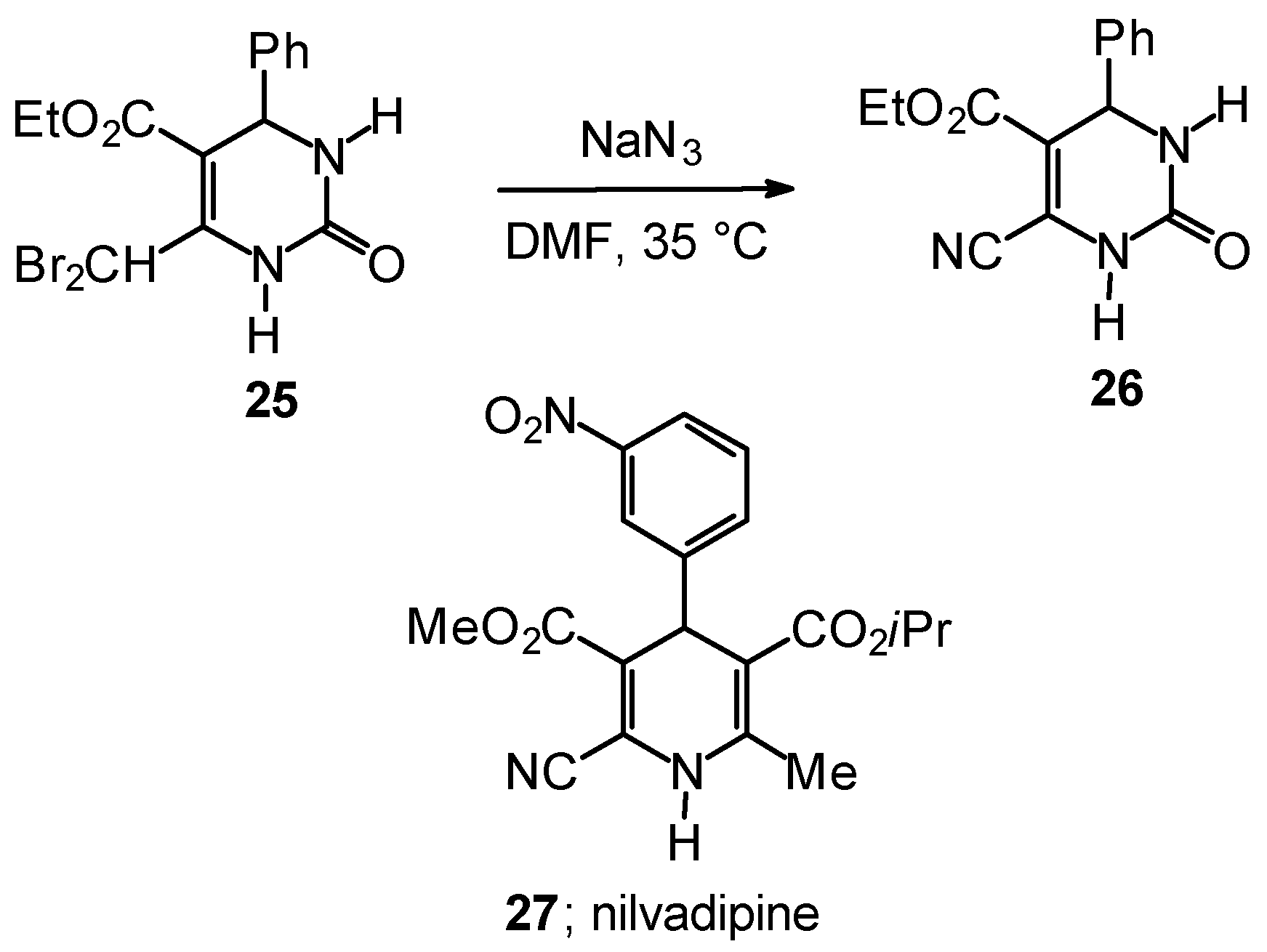
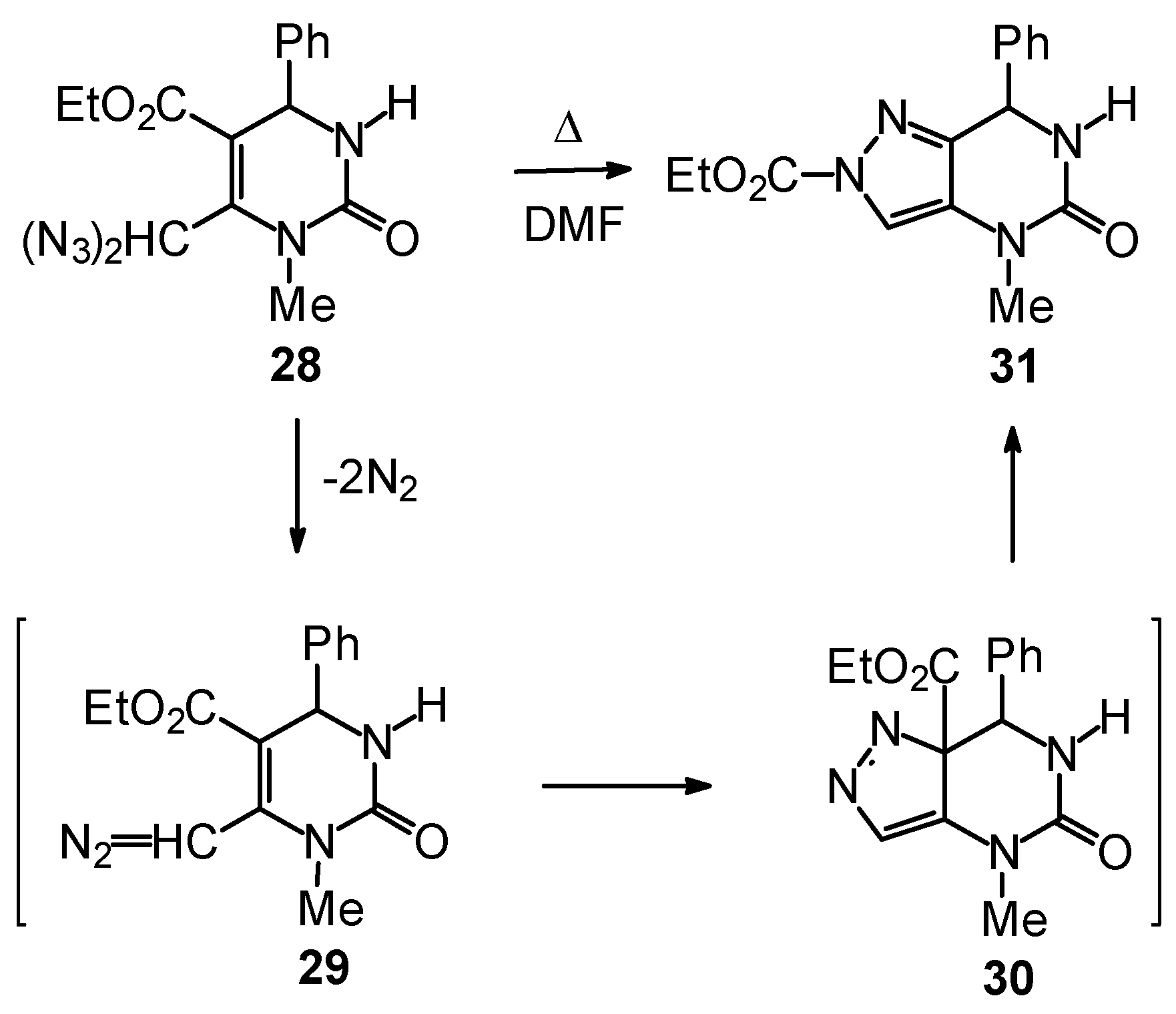
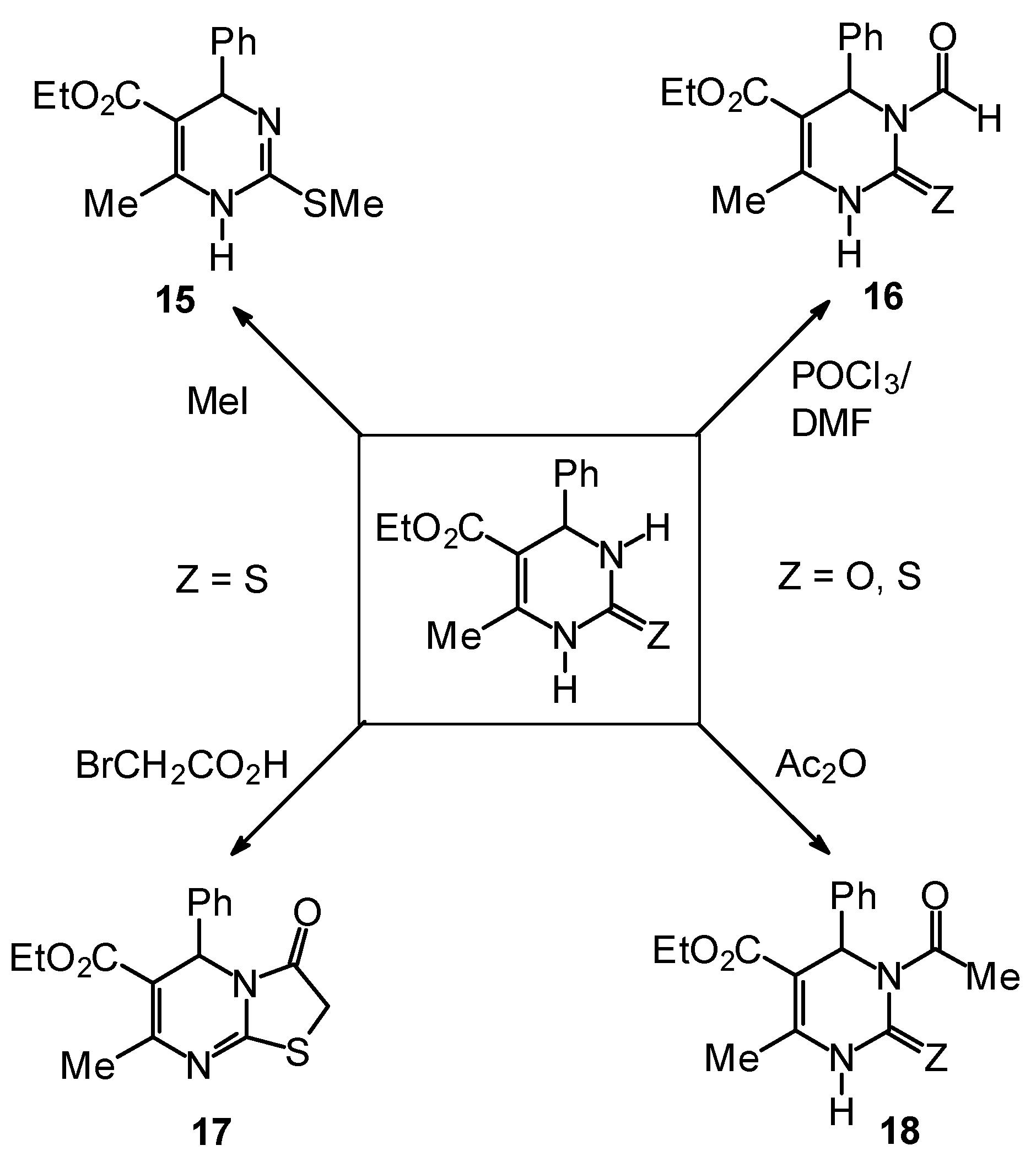
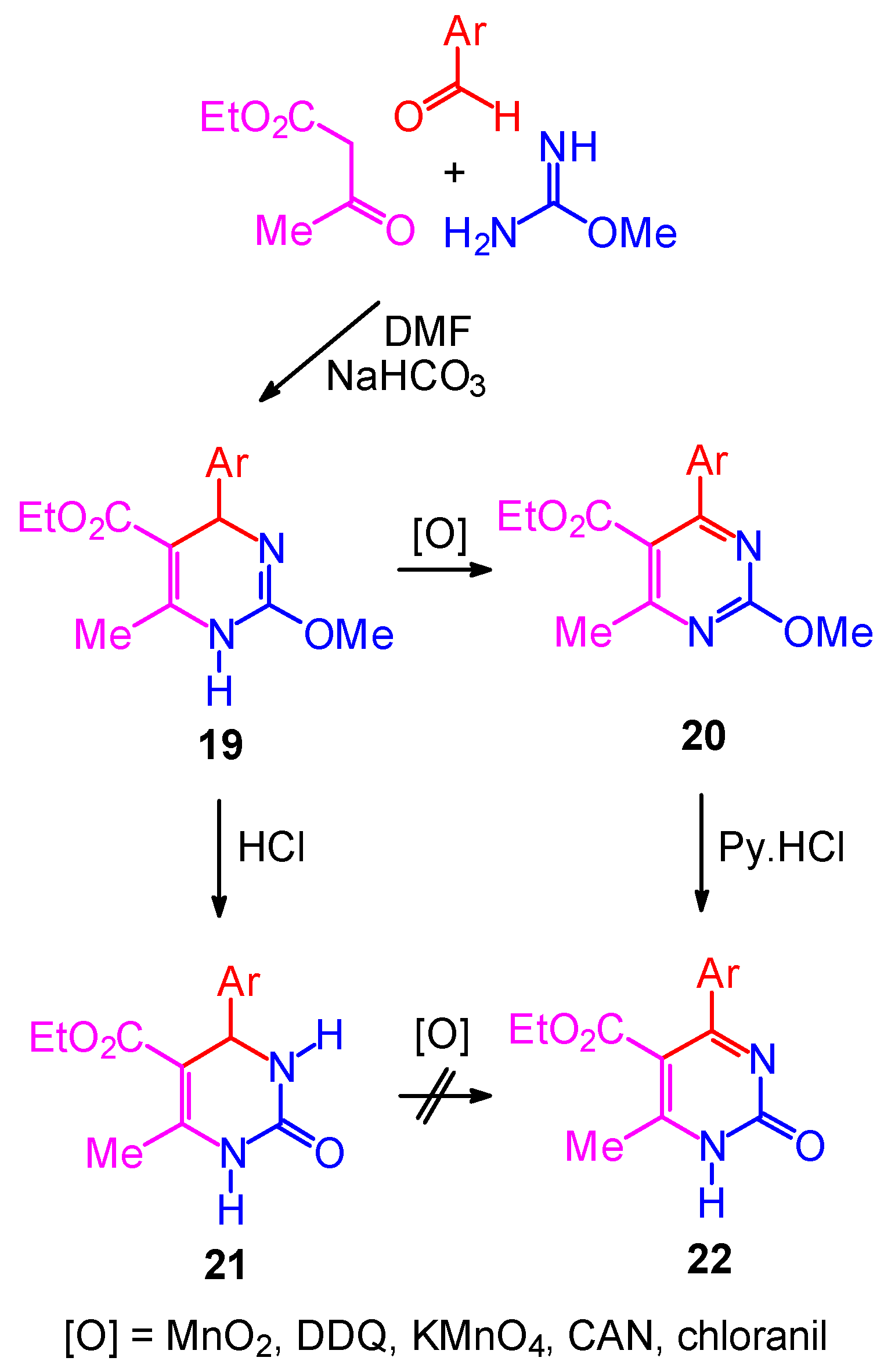
Synthetic Studies
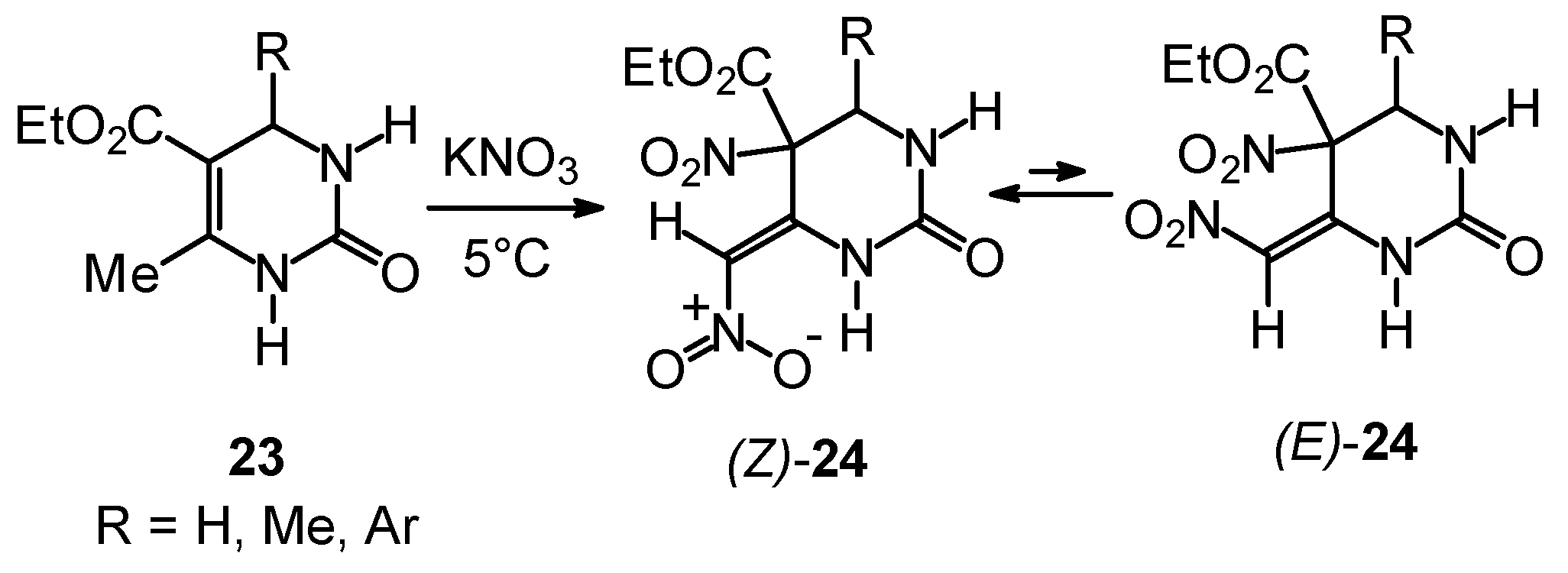
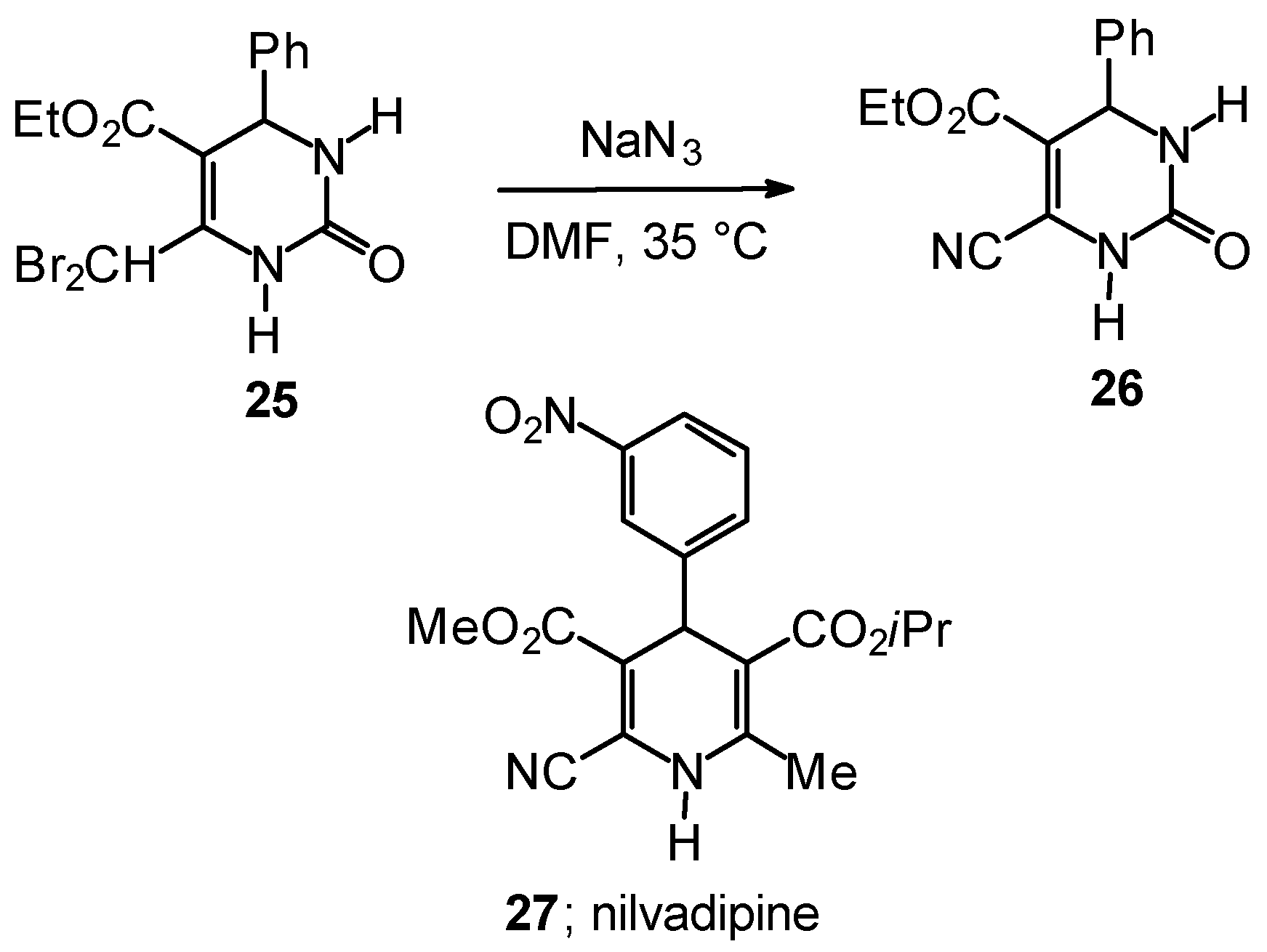
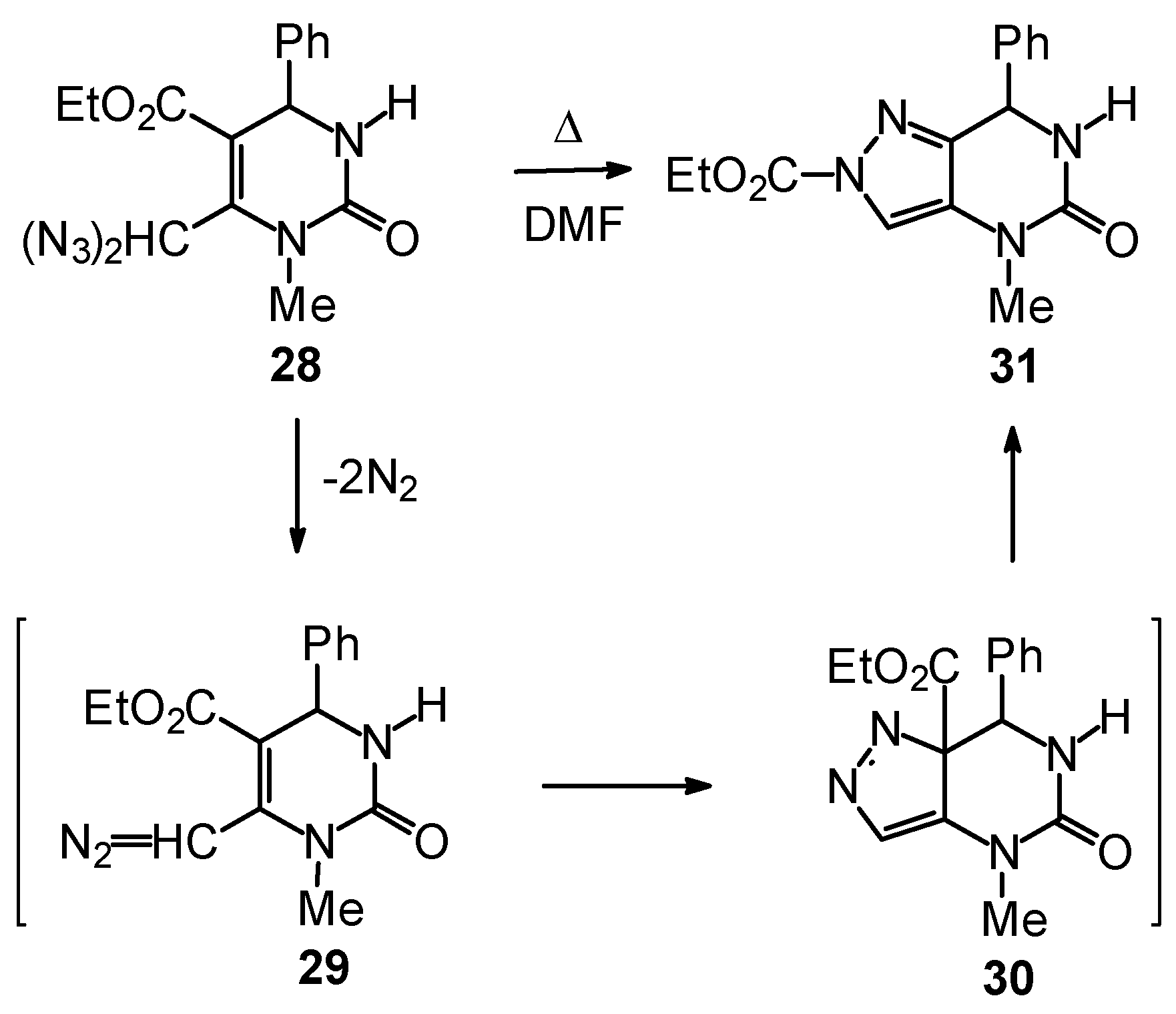
Outlook
Acknowledgements
References and Notes
- Janis, R. A.; Silver, P. J.; Triggle, D. J. Adv. Drug Res. 1987, 16, 309.
- Bossert, F.; Vater, W. Med. Res. Rev. 1989, 9, 291. [CrossRef]
- Cho, H.; Ueda, M.; Shima, K.; Mizuno, A.; Hayashimatsu, M.; Ohnaka, Y.; Takeuchi, Y.; Hamaguchi, M.; Aisaka, K.; Hidaka, T.; Kawai, M.; Takeda, M.; Ishihara, T.; Funahashi, K.; Satah, F.; Morita, M.; Noguchi, T. J. Med. Chem. 1989, 32, 2399. [CrossRef]
- Atwal, K.; Rovnyak, G. C.; Schwartz, J.; Moreland, S.; Hedberg, A.; Gougoutas, J. Z.; Malley, M. F.; Floyd, D. M. J. Med. Chem. 1990, 33, 1510. [CrossRef]
- Atwal, K. S.; Rovnyak, G. C.; Kimball, S. D.; Floyd, D. M.; Moreland, S.; Swanson, B. N.; Gougoutas, J. Z.; Schwartz, J.; Smillie, K. M.; Malley, M. F. J. Med. Chem. 1990, 33, 2629. [CrossRef]
- Atwal, K. S.; Swanson, B. N.; Unger, S. E.; Floyd, D. M.; Moreland, S.; Hedberg, A.; O´Reilly, B. C. J. Med. Chem. 1991, 34, 806. [CrossRef]
- Rovnyak, G. C.; Atwal, K. S.; Hedberg, A.; Kimball, S. D.; Moreland, S.; Gougoutas, J. Z.; O´Reilly, B. C.; Schwartz, J.; Malley, M. F. J. Med. Chem. 1992, 35, 3254. [CrossRef] [PubMed]
- Grover, G. J.; Dzwonczyk, S.; McMullen, D. M.; Normadinam, C. S.; Sleph, P. G.; Moreland, S. J. J. Cardiovasc. Pharmacol. 1995, 26, 289. Negwer, M. Organic-Chemical Drugs and their Synonyms; Akademie Verlag: Berlin, 1994; p. 2558. [Google Scholar]
- Rovnyak, G. C.; Kimball, S. D.; Beyer, B.; Cucinotta, G.; DiMarco, J. D.; Gougoutas, J.; Hedberg, A.; Malley, M.; McCarthy, J. P.; Zhang, R.; Moreland, S. J. Med. Chem. 1995, 38, 119. Triggle, D. J.; Padmanabhan, S. Chemtracts: Org. Chem. 1995, 8, 191.
- For reviews of the Hantzsch synthesis and related reactions, see: Sausins, A.; Duburs, G. Heterocycles 1988, 27, 269. Stout, D. M.; Meyers, A. J. Chem. Rev. 1982, 82, 223. Eisner, U.; Kuthan, J. Chem. Rev. 1972, 72, 1.
- Biginelli, P. Gazz. Chim. Ital. 1893, 23, 360.
- Kappe, C. O. Tetrahedron 1993, 49, 6937.
- O’Reilly, B. C.; Atwal, K. S. Heterocycles 1987, 26, 1185. Atwal, K S.; O’Reilley, B. C.; Gougoutas, J. Z.; Malley, M. F. Heterocycles 1987, 26, 1189. Atwal, K. S.; Rovnyak, G. C.; O’Reilly, B. C.; Schwartz, J. J. Org. Chem. 1989, 54, 5898. Zavyalov, S. I.; Kulikova, L. B. Khim.-Farm. Zh. 1992, 26, 116. Sár, C. P.; Hankovszky, O. H.; Jerkovich, G.; Pallagi, I.; Hideg, K. ACH - Models Chem. 1994, 131, 363. Gupta, R.; Gupta, A. K.; Paul, S.; Kachroo, P. L. Ind. J. Chem. 1995, 34B, 151. Hu, E. H.; Sidler, D. R.; Dolling, U. H. PCT WO 97 21,687.
- Wipf, P.; Cunningham, A. Tetrahedron Lett. 1995, 36, 7819. Studer, A.; Hadida, S.; Ferritto, R.; Kim, S.-Y.; Jeger, P.; Wipf, P.; Curran, D. P. Science 1997, 275, 823. Studer, A.; Jeger, P.; Wipf, P.; Curran, D. P. J. Org. Chem. 1997, 62, 2917. Robinett, L. D.; Yager, K. M.; Phelan, J. C. 211th National Meeting of the American Chemical Society, New Orleans, 1996; American Chemical Society: Washington, DC, 1996. ORGN 122.
- Crambine alkaloids, see: Snider, B. B.; Shi, Z. J. Org. Chem. 1993, 58, 3828. and references cited therein. Batzelladine alkaloids, see: Patil, A. D.; Kumar, N. V.; Kokke, W. C.; Bean, M. F.; Freyer, A. J.; DeBrosse, C.; Mai, S.; Truneh, A.; Faulkner, D. J.; Carte, B.; Breen, A. L.; Hertzberg, R. P.; Johnson, R. K.; Westley, J. W.; Potts, B. C. M. J. Org. Chem. 1995, 60, 1182. Patil, A. D.; Freyer, A. J.; Taylor, P. B.; Carte, B.; Zuber, G.; Johnson, R.; Faulkner, D. J. J. Org. Chem. 1997, 62, 1814. Patil, A. D.; Freyer, A. J.; Cheng, J.; Snider, B. Tetrahedron Lett. 1996, 37, 6977. Rama Rao, A. V.; Gurjar, M. K.; Vasudevan, J. J. Chem. Soc., Chem. Commun. 1995, 1369. Ptilomycalin A and related alkaloids, see: Overman, L. E.; Rabinowitz, M. H. J. Org. Chem. 1993, 58, 3235. Overman, L. E.; Rabinowitz, M. H.; Renhowe, P. A. J. Am. Chem. Soc. 1995, 117, 2657. Palagiano, E.; De Marino, S.; Minale, L.; Riccio, R.; Zollo, F.; Iorizzi, M.; Carre, J. B.; Debitus, C.; Lucarain, L.; Provost, J. Tetrahedron 1995, 51, 3675.
- Folkers, K.; Johnson, T. B. J. Am. Chem. Soc. 1933, 55, 3784. [CrossRef]
- Sweet, F.; Fissekis, J. D. J. Am. Chem. Soc. 1973, 95, 8741. [CrossRef]
- Kappe, C. O. J. Org. Chem. 1997, 62, 7201. [CrossRef]
- Dixon, L. A. Encyclopedia of Reagents in Organic Chemistry; Wiley: New York, 1995; Vol. 6, p. 4166. [Google Scholar]
- Marson, C. M.; Grabowska, U.; Fallah, A.; Walsgrove, T.; Eggleston, D. S.; Baures, P. W. J. Org. Chem. 1994, 59, 291. [CrossRef]
- Falsone, S. F.; Kappe, C. O. unpublished results.
- Kappe, C. O.; Fabian, W. M. F.; Semones, M. A. Tetrahedron 1997, 53, 2803.
- Goldman, S.; Stoltefuss, J. Angew. Chem., Int. Ed. Engl. 1991, 30, 1559.
- Bikker, J. A.; Weaver, D. F. J. Mol. Struct. (Theochem) 1993, 281, 173. Bikker, J. A.; Weaver, D. F. Can. J. Chem. 1992, 70, 2249. Gaudio, A. C.; Korolkovas, A.; Takahata, Y. J. Mol. Struct. 1994, 303, 255. Shishkin, O. V.; Timofeeva, T. V.; Desenko, S. M.; Orlov, V. D.; Lindeman, S. V.; Struchkov, Y. T. Russ. Chem. Bull. 1993, 42, 1160. Langs, D. A.; Kwon, Y. W.; Strong, P. D.; Triggle, D. J. J. Comput.-Aided. Mol. Design 1991, 5, 95. Cozzi, P.; Carganico, G.; Fusar, D.; Grossoni, M.; Menichineri, M.; Pinciroli, V.; Tonani, R.; Vaghi, F.; Salvati, P. J. Med. Chem. 1993, 36, 2964. Palmer, R. B.; Andersen, N. H. Tetrahedron 1996, 52, 9665.
- Fabian, W. M. F.; Semones, M. A.; Kappe, C. O. J. Mol. Struct. (Theochem) 1998, in press.
- Kappe, C. O.; Peters, K.; Peters, E.-M. J. Org. Chem. 1997, 62, 2786. For related work on isomünchnones, see: Kappe, C. O. Tetrahedron Lett. 1997, 38, 3323.
- Kappe, C. O.; Uray, G.; Roschger, P.; Lindner, W.; Kratky, C.; Keller, W. Tetrahedron 1992, 26, 5473.
- Schulte, M.; Kinkel, J. N.; Nicoud, R.-M.; Charton, F. Chem. Ing. Tech. 1996, 68, 670. Stinson, S. C. Chem. Eng. News 1995, 44. ibid. 1994 September 19; issue. 38. Schulte, M.; Derwenskus, K.-H.; Ditz, R. Nachr. Chem. Tech. Lab. 1997, 45, 487.
- Kleidernigg, O. P.; Kappe, C. O. Tetrahedron: Asymmetry 1997, 8, 2057.
- Sausins, A.; Duburs, G. Heterocycles 1988, 27, 291.
- Kappe, C. O.; Roschger, P. J. Heterocycl. Chem. 1989, 26, 55. [CrossRef]
- Bossert, F.; Meyer, H.; Wehinger, E. Angew. Chem. Int. Ed. Engl. 1981, 20, 762. De Matteis, F.; Hollands, C.; Gibbs, A. H.; De Sa, N.; Rizzardini, M. FEBS Lett. 1982, 145, 87. Augusto, O.; Beilan, H. S.; De Montellano, O. P. R. J. Biol. Chem. 1982, 257, 1288. Böcker, R. H.; Guengerich, F. P. J. Med. Chem. 1986, 29, 1596. Mc Cluskey, S. A.; Riddick, D. S.; Mackie, J. E.; Kimmette, S. M.; Withney, R. A.; Marks, G. S. Can. J. Physiol. Pharmacol. 1992, 70, 1069.
- Vanden Eynde, J. J.; Audiart, N.; Canonne, V.; Michel, S.; van Haverbeke, Y.; Kappe, C. O. Heterocycles 1997, 45, 1967.
- Khromov-Borisov, N. V.; Savchenko, A. M. Zh. Obschch. Khim. 1952, 22, 1680.
- Kappe, C. O.; Wagner, U. G. Heterocycles 1989, 29, 761.
- Kappe, C. O. Liebigs Ann. Chem. 1990, 505.
- Kappe, C. O.; Färber, G. J. Chem. Soc., Perkin Trans. 1 1991, 1342.
- For a review on the chemistry of geminal diazides, see: Kappe, T.; Kappe, C. O. Progress in Heterocyclic Chemistry; Suschitzky, H., Gribble, G. W., Eds.; Pergamon Press: Oxford, 1996; Vol. 8, Chapter 1. [Google Scholar]
- Nagarathanm, D.; Chiu, G.; Dhar, T. G. M.; Wong, W. C.; Marzabadi, M. R.; Gluchowski, C. L.; Lagu, B.; Miao, S. W. PCT Int. Appl. WO 96 14,846. Kettmann, V.; Drimal, J.; Svetlik, J. Pharmazie 1996, 51, 10. Kattmann, V.; Svetlik, J. Acta Cryst. 1996, C52, 1496. Alajarin, R.; Vaquero, J. J.; Alvarez- Builla, J.; De Casa-Juana, M. F.; Sunkel, C.; Priego, J. G.; Gomez-Sal, P.; Torres, R. Bioorg. Med. Chem. 1994, 2, 323. Alajaran, R.; Jordan, P.; Vaquero, J. J.; Alvarez-Builla, J. Synthesis 1995, 389. Gupta, R.; Sudan, S.; Kachroo, P. L. Ind. J. Chem. 1996, 35B, 985. Sudan, S.; Gupta, R.; Bani, S.; Singh, G. B.; Jain, S. M.; Kachroo, P. L. J. Ind. Chem. Soc. 1996, 73, 431. Ghorab, M. M.; Mohamed, Y. A.; Mohamed, S. A.; Ammar, Y. A. Phosphorus, Sulfur Silicon Relat. Elem. 1996, 108, 249. Sharaf, M. A. F.; Aal, F. A. A.; Fattah, A. M. A.; Khalik, A. M. R. A. J. Chem. Res. (S) 1996, 354. Rehani, R.; Shah, A. C.; Arya, V. P. Ind. J. Chem. 1994, 33B, 775. Sherif, S. M.; Youssef, M. M.; Mobarak, K.; Abdel-Fattah, A.-S. M. Tetrahedron 1993, 49, 9561. Remmenikov, G. Ya.; Shavaran, S. S.; Boldyrev, I. V.; Kurilenko, L. K.; Klebanov, B. M.; Kukhar, V. P. Khim.-Farm. Zh. 1991, 25, 35. Remennikov, G. Ya.; Boldyrev, I. V.; Kravchenko, S. A.; Pirozhenko, V. V. Khim. Geterotsikl. Soedin. 1993, 1290. Remnenikov, G. Y.; Boldyrev, I. V.; Krachenko, S. A.; Pirozhenko, V. V. Khim. Geterotsikl. Soedin. 1993, 1398. Schleifer, K.-J. Pharm. Pharmacol. Lett. 1995, 5, 162.
Biographical Sketch
 C. Oliver Kappe was born in Graz, Austria in 1965. He received his diploma degree (1989) and doctoral degree (1992) from the Karl-Franzens-University in Graz where he worked with Professor Gert Kollenz on cycloaddition and rearrangement reactions of acylketenes. After periods of postdoctoral research work with Professor Curt Wentrup at the University of Queensland in Brisbane, Australia (1993-1994), and with Professor Albert Padwa at Emory University in Atlanta, USA (1994-1996) he moved back to the University of Graz in 1996 to start his independent academic career. He received the Dissertation Award of the Austrian Chemical Society in 1993, the Erwin-Schrödinger Fellowship of the Austrian Science Fund in 1994 and an APART Fellowship of the Austrian Academy of Sciences in 1996. He is currently a member of the Advisory Board of the International Society of Heterocyclic Chemistry (1998-1999) and is coauthor of more than 50 publications in the field of synthetic and mechanistic organic chemistry. His current research interests include dihydropyrimidine chemistry, N-ylide chemistry, and pseudopericyclic reactions.
C. Oliver Kappe was born in Graz, Austria in 1965. He received his diploma degree (1989) and doctoral degree (1992) from the Karl-Franzens-University in Graz where he worked with Professor Gert Kollenz on cycloaddition and rearrangement reactions of acylketenes. After periods of postdoctoral research work with Professor Curt Wentrup at the University of Queensland in Brisbane, Australia (1993-1994), and with Professor Albert Padwa at Emory University in Atlanta, USA (1994-1996) he moved back to the University of Graz in 1996 to start his independent academic career. He received the Dissertation Award of the Austrian Chemical Society in 1993, the Erwin-Schrödinger Fellowship of the Austrian Science Fund in 1994 and an APART Fellowship of the Austrian Academy of Sciences in 1996. He is currently a member of the Advisory Board of the International Society of Heterocyclic Chemistry (1998-1999) and is coauthor of more than 50 publications in the field of synthetic and mechanistic organic chemistry. His current research interests include dihydropyrimidine chemistry, N-ylide chemistry, and pseudopericyclic reactions.- Sample Availability: Available from MDPI.
© 1998 MDPI. All rights reserved
Share and Cite
Kappe, C.O. 4-Aryldihydropyrimidines via the Biginelli Condensation: Aza-Analogs of Nifedipine-Type Calcium Channel Modulators. Molecules 1998, 3, 1-9. https://doi.org/10.3390/30100001
Kappe CO. 4-Aryldihydropyrimidines via the Biginelli Condensation: Aza-Analogs of Nifedipine-Type Calcium Channel Modulators. Molecules. 1998; 3(1):1-9. https://doi.org/10.3390/30100001
Chicago/Turabian StyleKappe, C. Oliver. 1998. "4-Aryldihydropyrimidines via the Biginelli Condensation: Aza-Analogs of Nifedipine-Type Calcium Channel Modulators" Molecules 3, no. 1: 1-9. https://doi.org/10.3390/30100001
APA StyleKappe, C. O. (1998). 4-Aryldihydropyrimidines via the Biginelli Condensation: Aza-Analogs of Nifedipine-Type Calcium Channel Modulators. Molecules, 3(1), 1-9. https://doi.org/10.3390/30100001