
Carbonic Anhydrases: Different Active Sites, Same Metal Selectivity Rules




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. αCA
2.2. βCA
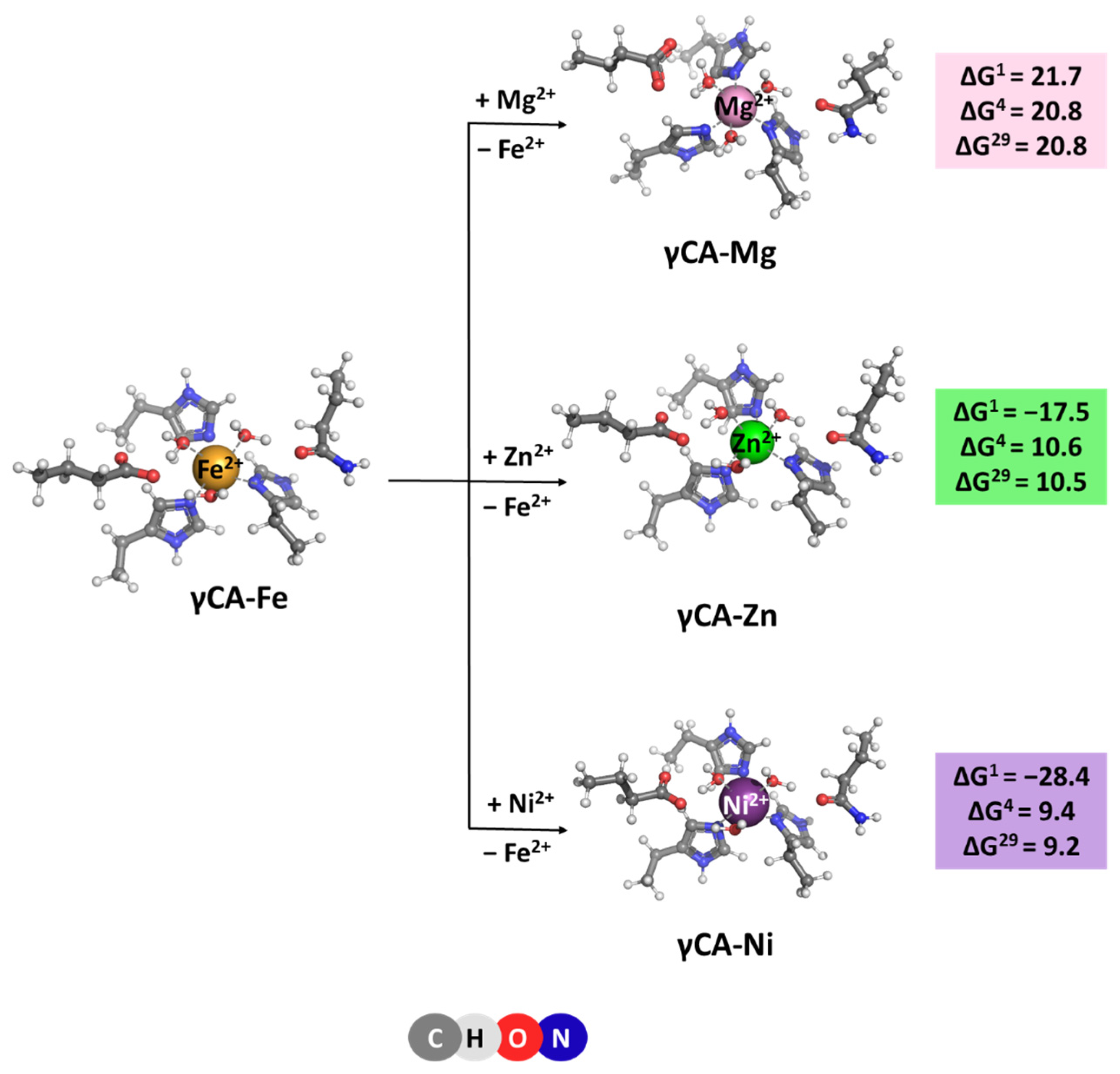
2.3. γCA
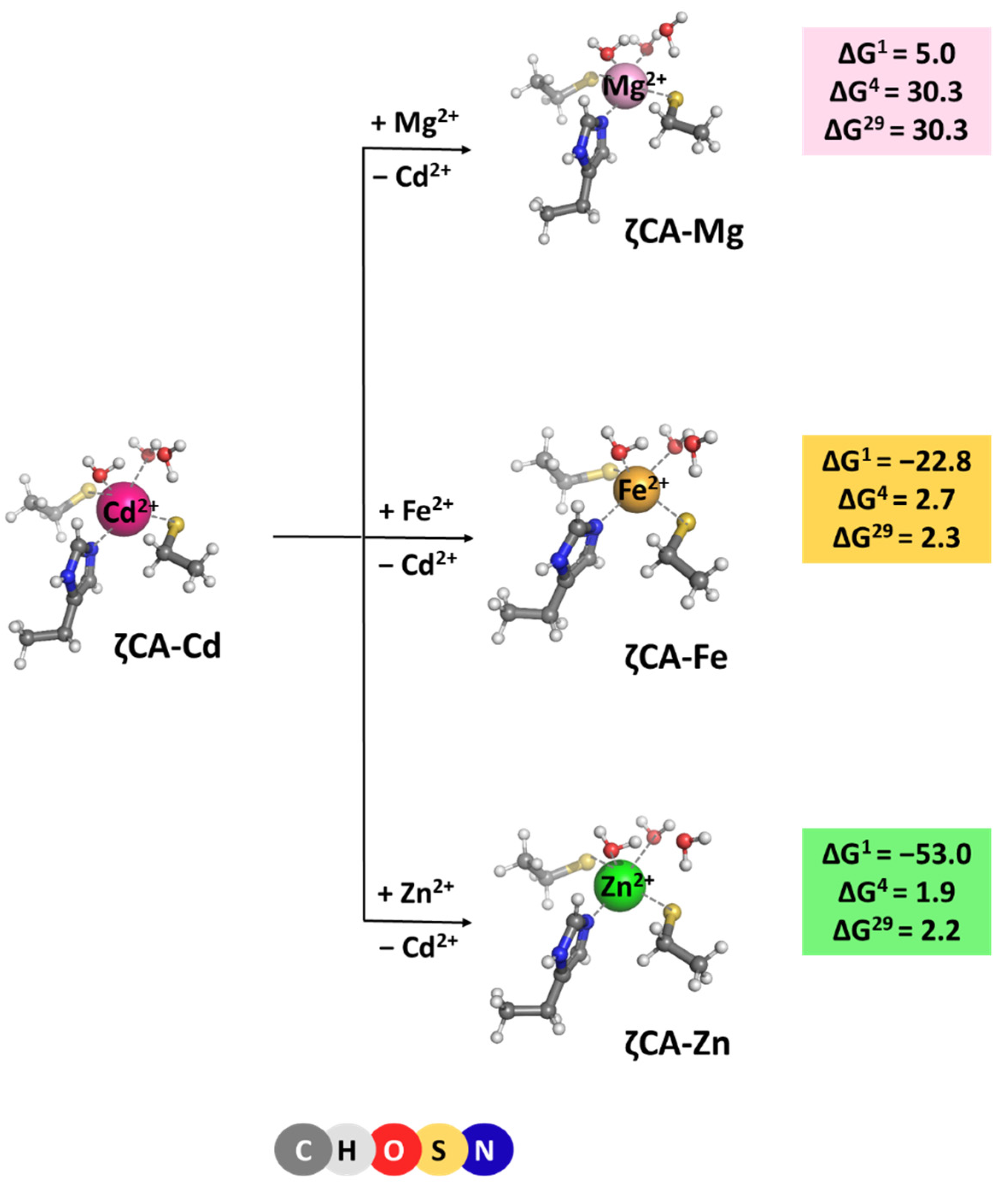
2.4. ζCA
3. Methods
3.1. Models Used
3.2. DFT/PCM Calculations
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Di Fiore, A.; D’Ambrosio, K.; Ayoub, J.; Alterio, V.; De Simone, G. α-Carbonic Anhydrases; Elsevier Inc.: London, UK, 2019; ISBN 9780128164761. [Google Scholar]
- Henry, R.P. Multiple Roles of Carbonic Anhydrase in Cellular Transport and Metabolism. Annu. Rev. Physiol. 1996, 58, 523–538. [Google Scholar] [CrossRef]
- Supuran, C.T. Carbonic Anhydrases: Catalytic and Inhibition Mechanisms, Distribution and Physio-Logical Roles. In Carbonic Anhydrases; Taylor & Francis: Abingdon, UK, 2004. [Google Scholar]
- Occhipinti, R.; Boron, W.F. Role of Carbonic Anhydrases and Inhibitors in Acid–Base Physiology: Insights from Mathematical Modeling. Int. J. Mol. Sci. 2019, 20, 3841. [Google Scholar] [CrossRef]
- DiMario, R.J.; Clayton, H.; Mukherjee, A.; Ludwig, M.; Moroney, J.V. Plant Carbonic Anhydrases: Structures, Locations, Evolution, and Physiological Roles. Mol. Plant 2017, 10, 30–46. [Google Scholar] [CrossRef]
- Ferraroni, M. γ-Carbonic Anhydrases. In Carbon Anhydrases Biochemistry and Pharmacology of an Evergreen Pharmaceutical Target; Elsevier: Amsterdam, The Netherlands, 2019; pp. 79–105. [Google Scholar] [CrossRef]
- Alterio, V.; Langella, E.; Viparelli, F.; Vullo, D.; Ascione, G.; Dathan, N.A.; Morel, F.M.M.; Supuran, C.T.; De Simone, G.; Monti, S.M. Structural and Inhibition Insights into Carbonic Anhydrase CDCA1 from the Marine Diatom Thalassiosira weissflogii. Biochimie 2012, 94, 1232–1241. [Google Scholar] [CrossRef]
- Xu, Y.; Feng, L.; Jeffrey, P.D.; Shi, Y.; Morel, F.M.M. Structure and Metal Exchange in the Cadmium Carbonic Anhydrase of Marine Diatoms. Nature 2008, 452, 56–61. [Google Scholar] [CrossRef]
- Lindskog, S. Structure and Mechanism of Carbonic Anhydrase. Pharmacol. Ther. 1997, 74, 1–20. [Google Scholar] [CrossRef]
- Fisher, Z.; Hernandez Prada, J.A.; Tu, C.; Duda, D.; Yoshioka, C.; An, H.; Govindasamy, L.; Silverman, D.N.; McKenna, R. Structural and Kinetic Characterization of Active-Site Histidine as a Proton Shuttle in Catalysis by Human Carbonic Anhydrase II. Biochemistry 2005, 44, 1097–1105. [Google Scholar] [CrossRef]
- Kimber, M.S. The Active Site Architecture of Pisum Sativum β-Carbonic Anhydrase Is a Mirror Image of That of Alpha -Carbonic Anhydrases. EMBO J. 2000, 19, 1407–1418. [Google Scholar] [CrossRef]
- Iverson, T.M.; Alber, B.E.; Kisker, C.; Ferry, J.G.; Rees, D.C. A Closer Look at the Active Site of γ-Class Carbonic Anhydrases: High-Resolution Crystallographic Studies of the Carbonic Anhydrase from Methanosarcina thermophila. Biochemistry 2000, 39, 9222–9231. [Google Scholar] [CrossRef]
- Nikolova, V.; Angelova, S.; Markova, N.; Dudev, T. Gallium as a Therapeutic Agent: A Thermodynamic Evaluation of the Competition between Ga3+ and Fe3+ Ions in Metalloproteins. J. Phys. Chem. B 2016, 120, 2241–2248. [Google Scholar] [CrossRef]
- Dudev, T.; Cheshmedzhieva, D.; Doudeva, L. Competition between Abiogenic Al3+ and Native Mg2+, Fe2+ and Zn2+ Ions in Protein Binding Sites: Implications for Aluminum Toxicity. J. Mol. Model. 2018, 24, 55. [Google Scholar] [CrossRef]
- Dudev, T.; Grauffel, C.; Lim, C. How Pb2+ Binds and Modulates Properties of Ca2+-Signaling Proteins. Inorg. Chem. 2018, 57, 14798–14809. [Google Scholar] [CrossRef]
- Dudev, T.; Lim, C. Ion Selectivity Strategies of Sodium Channel Selectivity Filters. Acc. Chem. Res. 2014, 47, 3580–3587. [Google Scholar] [CrossRef]
- Dudev, T.; Lim, C. Competition among Metal Ions for Protein Binding Sites: Determinants of Metal Ion Selectivity in Proteins. Chem. Rev. 2014, 114, 538–556. [Google Scholar] [CrossRef]
- Schmidt, S.B.; Eisenhut, M.; Schneider, A. Chloroplast Transition Metal Regulation for Efficient Photosynthesis. Trends Plant Sci. 2020, 25, 817–828. [Google Scholar] [CrossRef]
- Yruela, I. Transition Metals in Plant Photosynthesis. Metallomics 2013, 5, 1090–1109. [Google Scholar] [CrossRef]
- Bardi, U. Extracting Minerals from Seawater: An Energy Analysis. Sustainability 2010, 2, 980–992. [Google Scholar] [CrossRef]
- Bazzi, A.O. Heavy Metals in Seawater, Sediments and Marine Organisms in the Gulf of Chabahar, Oman Sea. J. Oceanogr. Mar. Sci. 2014, 5, 20–29. [Google Scholar] [CrossRef][Green Version]
- Berman, H.; Henrick, K.; Nakamura, H. Announcing the Worldwide Protein Data Bank. Nat. Struct. Biol. 2003, 10, 980. [Google Scholar] [CrossRef]
- Berman, H.; Henrick, K.; Nakamura, H.; Markley, J.L. The Worldwide Protein Data Bank (WwPDB): Ensuring a Single, Uniform Archive of PDB Data. Nucleic Acids Res. 2007, 35, 2006–2008. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D. 01; Gaussian Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 Other Function. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Andrae, D.; Häußermann, U.; Dolg, M.; Stoll, H.; Preuß, H. Energy-Adjusted Ab Initio Pseudopotentials for the Second and Third Row Transition Elements. Theor. Chim. Acta 1990, 77, 123–141. [Google Scholar] [CrossRef]
- Dudev, T.; Nikolova, V. Determinants of Fe2+ over M2+ (M = Mg, Mn, Zn) Selectivity in Non-Heme Iron Proteins. Inorg. Chem. 2016, 55, 12644–12650. [Google Scholar] [CrossRef]
- Dudev, T.; Lim, C. Determinants of K+ vs Na+ Selectivity in Potassium Channels. J. Am. Chem. Soc. 2009, 131, 8092–8101. [Google Scholar] [CrossRef]
- Li, L.; Li, C.; Zhang, Z.; Alexov, E. On the Dielectric “Constant” of Proteins: Smooth Dielectric Function for Macromolecular Modeling and Its Implementation in DelPhi. J. Chem. Theory Comput. 2013, 9, 2126–2136. [Google Scholar] [CrossRef]
- Mertz, E.L.; Krishtalik, L.I. Low Dielectric Response in Enzyme Active Site. Proc. Natl. Acad. Sci. USA 2000, 97, 2081–2086. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys.Chem B 2009, 113, 6378–6396. [Google Scholar]
- Friedman, H.L.; Krishnan, C.V. Water: A Comprehensive Treatise; Franks, F., Ed.; Plenum Press: New York, NY, USA, 1973; Volume 3. [Google Scholar]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kircheva, N.; Angelova, S.; Dudev, T. Carbonic Anhydrases: Different Active Sites, Same Metal Selectivity Rules. Molecules 2024, 29, 1995. https://doi.org/10.3390/molecules29091995
Kircheva N, Angelova S, Dudev T. Carbonic Anhydrases: Different Active Sites, Same Metal Selectivity Rules. Molecules. 2024; 29(9):1995. https://doi.org/10.3390/molecules29091995
Chicago/Turabian StyleKircheva, Nikoleta, Silvia Angelova, and Todor Dudev. 2024. "Carbonic Anhydrases: Different Active Sites, Same Metal Selectivity Rules" Molecules 29, no. 9: 1995. https://doi.org/10.3390/molecules29091995
APA StyleKircheva, N., Angelova, S., & Dudev, T. (2024). Carbonic Anhydrases: Different Active Sites, Same Metal Selectivity Rules. Molecules, 29(9), 1995. https://doi.org/10.3390/molecules29091995