Purity Assessment of Tripropyl Phosphate through Mass Balance and 1H and 31P Quantitative Nuclear Magnetic Resonance

Abstract
1. Introduction
2. Results and Discussion
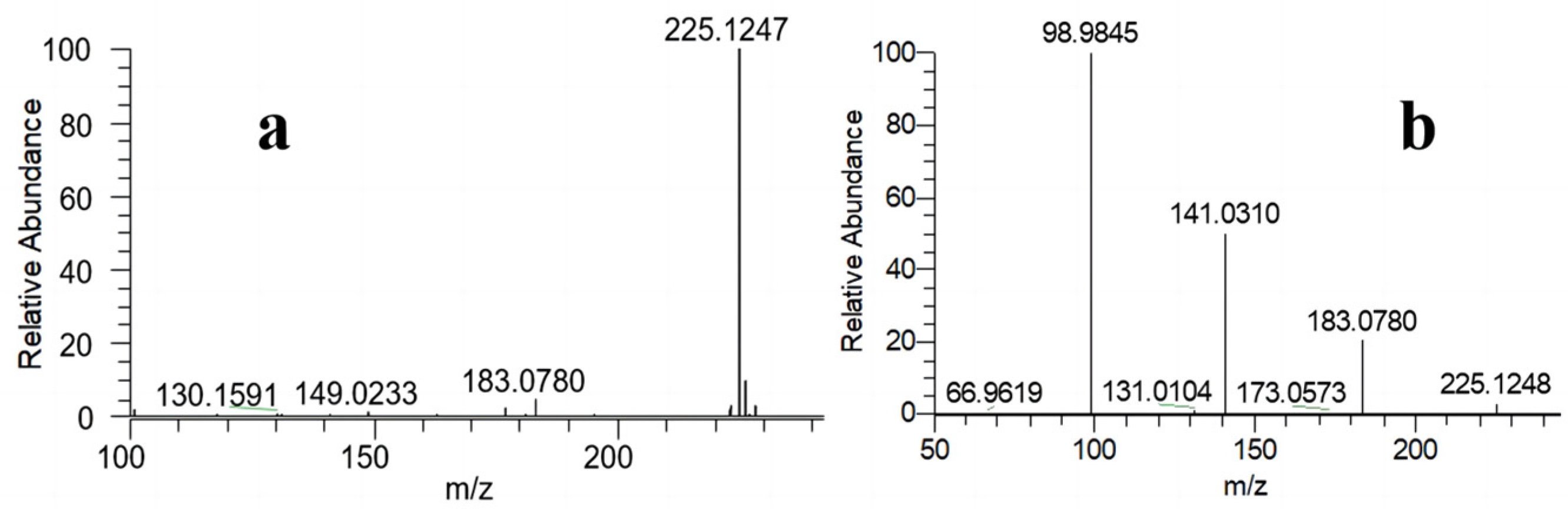
2.1. Qualitative Characterization
2.2. Quantitative Analysis by MB
2.2.1. Determination of TnPP and Its Structurally Related Impurities by GC-FID
2.2.2. Water Determination
2.2.3. Inorganic Impurity Determination
2.2.4. Residual Organic Solvent Determination
2.2.5. Mass Fraction by MB
2.3. Quantitative Analysis by qNMR
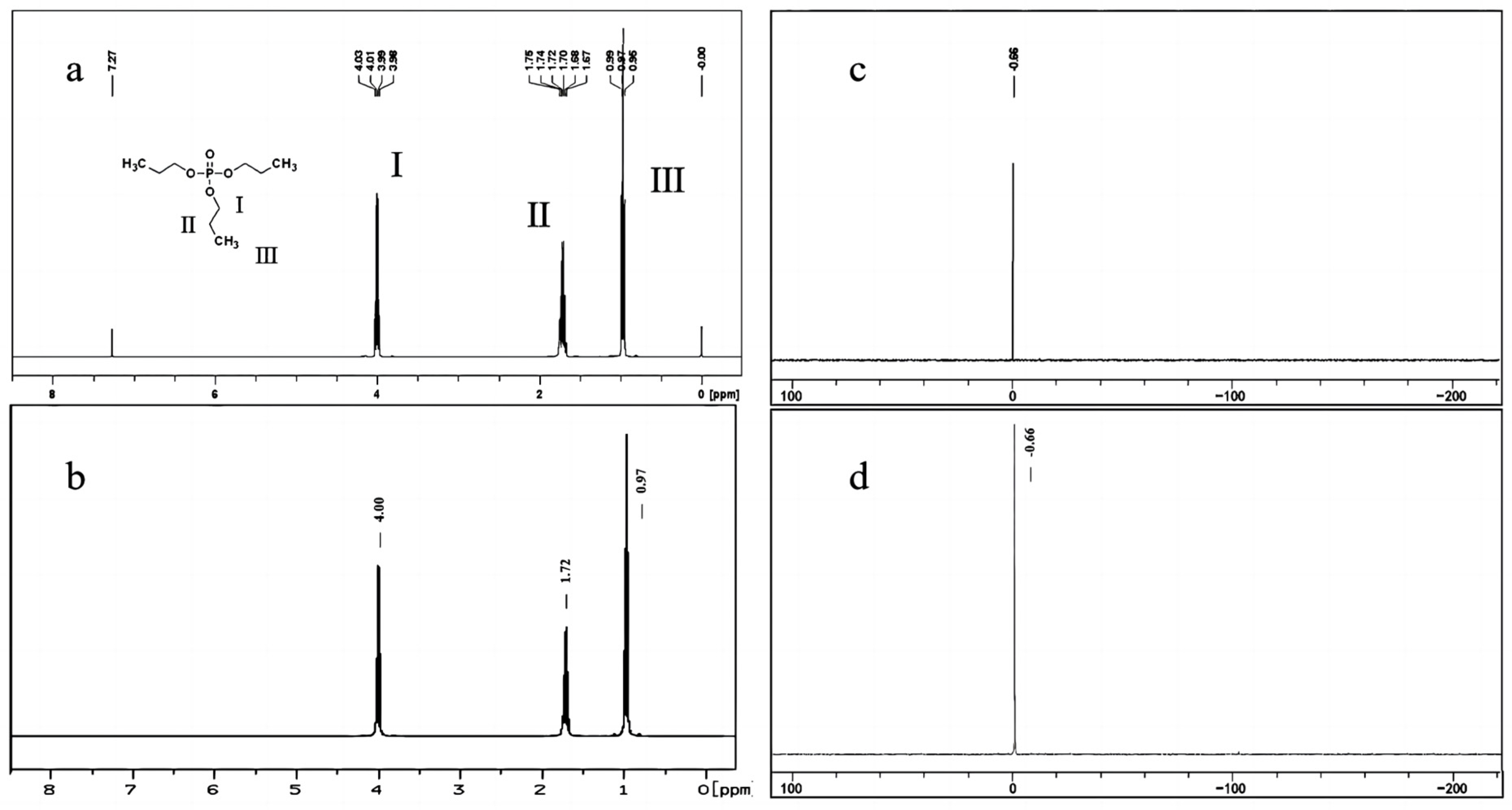
2.3.1. Quantitative Analysis by 1H-qNMR
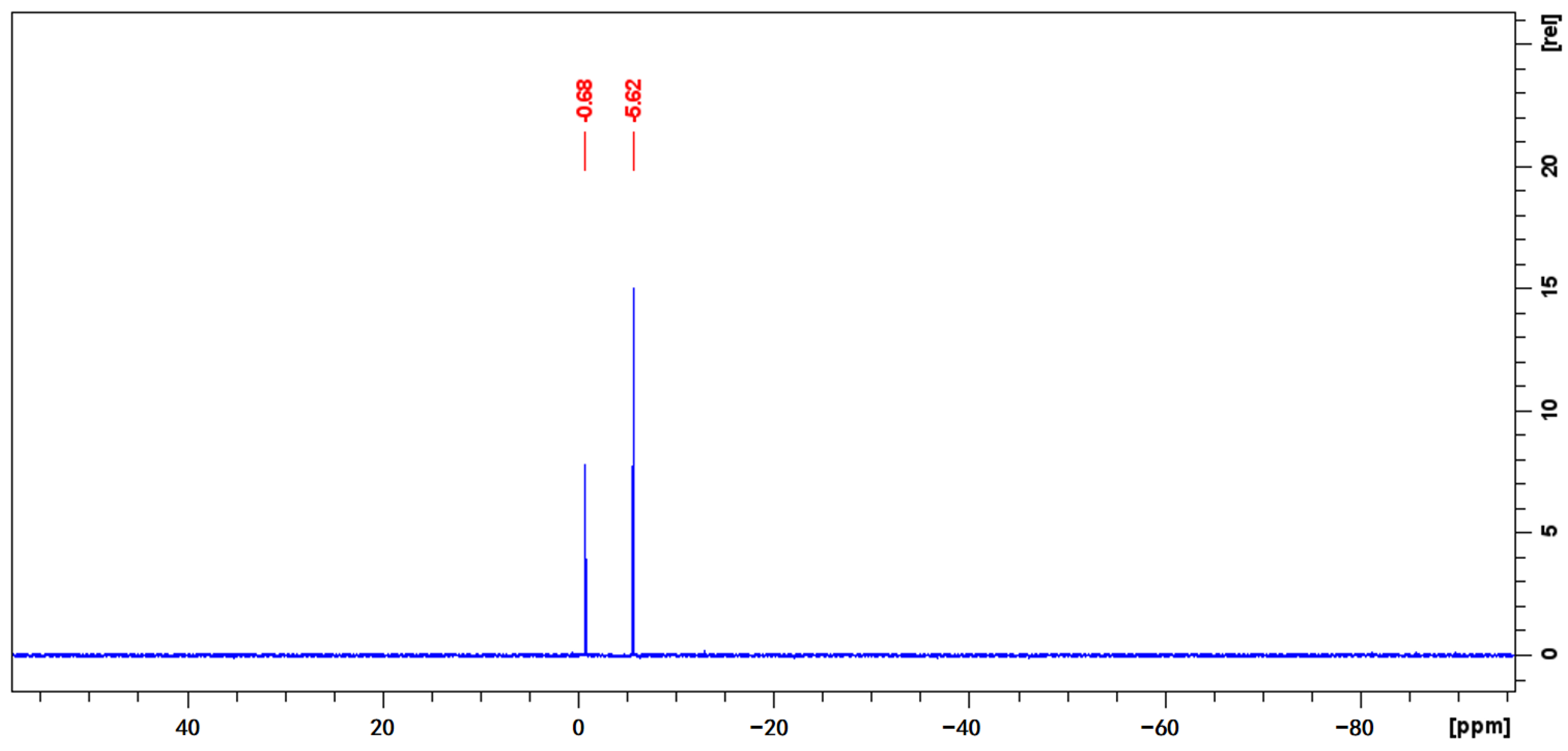
2.3.2. Quantitative Analysis by 31P-qNMR
2.4. Value Assignment
2.5. Uncertainty Evaluation
3. Materials and Methods
3.1. Chemicals and Materials
3.2. Instruments
3.3. Qualitative Characterization
3.3.1. LC-MS/MS Analysis
3.3.2. 1H-NMR and 31P-NMR Qualitative Analysis
3.4. Quantitative Analysis by MB
3.4.1. Structurally Related Impurities Determination
3.4.2. Water Determination
3.4.3. Inorganic Impurity Determination
3.4.4. Residual Organic Solvent Determination
3.5. Quantitative Analysis by qNMR
3.5.1. Quantitative Analysis by 1H-qNMR
3.5.2. Quantitative Analysis by 31P-qNMR
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Van der Veen, I.; De Boer, J. Phosphorus flame retardants: Properties, production, environmental occurrence, toxicity and analysis. Chemosphere 2012, 88, 1119–1153. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Sun, H.; Zhu, H.; Yao, Y.; Chen, H.; Ren, C.; Wu, F.; Kannan, K. Occurrence and distribution of organophosphate flame retardants (OPFRs) in soil and outdoor settled dust from a multi-waste recycling area in China. Sci. Total Environ. 2018, 625, 1056–1064. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Hao, Y.; Li, Y.; Yang, R.; Wang, P.; Zhang, G.; Zhang, Q.; Jiang, G. Occurrence and distribution of organophosphate esters in the air and soils of Ny-Ålesund and London Island, Svalbard, Arctic. Environ. Pollut. 2020, 263, 114495. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Guo, J.-Q.; Liu, L.-Y.; Sverko, E.; Zhang, Z.; Tian, C.-G.; Huo, C.-Y.; Li, H.-L.; Ma, W.-L.; Zhang, Z.-F.; et al. Seasonal variation and influence factors of organophosphate esters in air particulate matter of a northeastern Chinese test home. Sci. Total Environ. 2020, 740, 140048. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yao, Y.; Li, W.; Zhu, H.; Wang, L.; Sun, H.; Kannan, K. A nationwide survey of 19 organophosphate esters in soils from China: Spatial distribution and hazard assessment. Sci. Total Environ. 2019, 671, 528–535. [Google Scholar] [CrossRef] [PubMed]
- Yadav, I.C.; Devi, N.L.; Li, J.; Zhang, G.; Covaci, A. Concentration and spatial distribution of organophosphate esters in the soil-sediment profile of Kathmandu Valley, Nepal: Implication for risk assessment. Sci. Total Environ. 2018, 613–614, 502–512. [Google Scholar] [CrossRef] [PubMed]
- Gustavsson, J.; Wiberg, K.; Ribeli, E.; Nguyen, M.A.; Josefsson, S.; Ahrens, L. Screening of organic flame retardants in Swedish river water. Sci. Total Environ. 2018, 625, 1046–1055. [Google Scholar] [CrossRef] [PubMed]
- Kim, U.-J.; Kannan, K. Occurrence and Distribution of Organophosphate Flame Retardants/Plasticizers in Surface Waters, Tap Water, and Rainwater: Implications for Human Exposure. Environ. Sci. Technol. 2018, 52, 5625–5633. [Google Scholar] [CrossRef] [PubMed]
- Olisah, C.; Rubidge, G.; Human, L.R.D.; Adams, J.B. Organophosphate pesticides in South African eutrophic estuaries: Spatial distribution, seasonal variation, and ecological risk assessment. Environ. Pollut. 2022, 306, 119446. [Google Scholar] [CrossRef]
- Zhong, M.; Wang, T.; Qi, C.; Peng, G.; Lu, M.; Huang, J.; Blaney, L.; Yu, G. Automated online solid-phase extraction liquid chromatography tandem mass spectrometry investigation for simultaneous quantification of per- and polyfluoroalkyl substances, pharmaceuticals and personal care products, and organophosphorus flame retardants in environmental waters. J. Chromatogr. A 2019, 1602, 350–358. [Google Scholar]
- Zhang, X.; Zou, W.; Mu, L.; Chen, Y.; Ren, C.; Hu, X.; Zhou, Q. Rice ingestion is a major pathway for human exposure to organophosphate flame retardants (OPFRs) in China. J. Hazard. Mater. 2016, 318, 686–693. [Google Scholar] [CrossRef] [PubMed]
- Castro-Jiménez, J.; Ratola, N. An innovative approach for the simultaneous quantitative screening of organic plastic additives in complex matrices in marine coastal areas. Environ. Sci. Pollut. Res. 2020, 27, 11450–11457. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Rebholz, C.M.; Wong, E.; Buckley, J.P. Urinary organophosphate ester concentrations in relation to ultra-processed food consumption in the general US population. Environ. Res. 2020, 182, 109070. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-E.; Luo, X.-J.; Huang, L.-Q.; Zeng, Y.-H.; Mai, B.-X. Organophosphorus flame retardants in fish from Rivers in the Pearl River Delta, South China. Sci. Total Environ. 2019, 663, 125–132. [Google Scholar] [CrossRef]
- Chen, Z.-F.; Tang, Y.-T.; Liao, X.-L.; Jiang, J.-R.; Qi, Z.; Cai, Z. A QuEChERS-based UPLC-MS/MS method for rapid determination of organophosphate flame retardants and their metabolites in human urine. Sci. Total Environ. 2022, 826, 153989. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhou, X.; Li, X.; Li, X.; Gao, Y.; Zhang, Q. Simultaneous determination of 21 organophosphorus flame retardants in rice by gas chromatography quadrupole time-of-flight mass spectrometry. Talanta 2023, 253, 124103. [Google Scholar] [CrossRef]
- Ding, J.; Deng, T.; Xu, M.; Wang, S.; Yang, F. Residuals of organophosphate esters in foodstuffs and implication for human exposure. Environ. Pollut. 2018, 233, 986–991. [Google Scholar] [CrossRef]
- Zhao, L.; Jian, K.; Su, H.; Zhang, Y.; Li, J.; Letcher, R.J.; Su, G. Organophosphate esters (OPEs) in Chinese foodstuffs: Dietary intake estimation via a market basket method, and suspect screening using high-resolution mass spectrometry. Environ. Int. 2019, 128, 343–352. [Google Scholar] [CrossRef]
- Li, J.; Zhao, L.; Letcher, R.J.; Zhang, Y.; Jian, K.; Zhang, J.; Su, G. A review on organophosphate Ester (OPE) flame retardants and plasticizers in foodstuffs: Levels, distribution, human dietary exposure, and future directions. Environ. Int. 2019, 127, 35–51. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.; Wang, G.; Gao, S.; Wang, Z. Aryl organophosphate flame retardants induced cardiotoxicity during zebrafish embryogenesis: By disturbing expression of the transcriptional regulators. Aquat. Toxicol. 2015, 161, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Yang, J.; Jiao, H.; Li, X. Advancement in Organic Chemical Purity Measurement by Mass Balance Method. Huaxue Shiji 2020, 42, 931–939. [Google Scholar]
- Westwood, S.; Choteau, T.; Daireaux, A.; Josephs, R.D.; Wielgosz, R.I. Mass Balance Method for the SI Value Assignment of the Purity of Organic Compounds. Anal. Chem. 2013, 85, 3118–3126. [Google Scholar] [CrossRef] [PubMed]
- Gong, H.; Huang, T.; Yang, Y.; Wang, H. Purity determination and uncertainty evaluation of folic acid by mass balance method. Talanta 2012, 101, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Tu, M.; Yang, B.; Zhu, Y.; Li, X. Research Progress of Neonicotinoid Pesticides Certified Reference. Metrol. Sci. Technol. 2023, 67, 65–71. [Google Scholar]
- Lee, S.; Baek, S.-Y.; Kwon, H.-J.; Choi, K.H.; Han, J. Analytical Methods Based on the Mass Balance Approach for Purity Evaluation of Tetracycline Hydrochloride. Molecules 2023, 28, 7568. [Google Scholar] [CrossRef] [PubMed]
- Choi, K.; Myoung, S.; Seo, Y.; Ahn, S. Quantitative NMR as a Versatile Tool for the Reference Material Preparation. Magnetochemistry 2021, 7, 15. [Google Scholar] [CrossRef]
- Nishizaki, Y.; Lankin, D.C.; Chen, S.-N.; Pauli, G.F. Accurate and Precise External Calibration Enhances the Versatility of Quantitative NMR (qNMR). Anal. Chem. 2021, 93, 2733–2741. [Google Scholar] [CrossRef] [PubMed]
- Westwood, S.; Yamazaki, T.; Huang, T.; Garrido, B.; Ün, I.; Zhang, W.; Martos, G.; Stoppacher, N.; Saito, T.; Wielgosz, R. Development and validation of a suite of standards for the purity assignment of organic compounds by quantitative NMR spectroscopy. Metrologia 2019, 56, 064001. [Google Scholar] [CrossRef]
- Uchiyama, N.; Kiyota, K.; Hosoe, J.; Komatsu, T.; Sugimoto, N.; Ishizuki, K.; Koide, T.; Murabayashi, M.; Kobayashi, K.; Fujimine, Y.; et al. Quantitative 31P-NMR for Purity Determination of Sofosbuvir and Method Validation. Chem. Pharm. Bull. 2022, 70, 892–900. [Google Scholar] [CrossRef] [PubMed]
- Agrahari, V.; Meng, J.; Purohit, S.S.; Oyler, N.A.; Youan, B.-B.C. Real-Time Analysis of Tenofovir Release Kinetics Using Quantitative Phosphorus (31P) Nuclear Magnetic Resonance Spectroscopy. J. Pharm. Sci. 2017, 106, 3005–3015. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Chen, H.; Cai, N.; Zou, J.; Ju, X. Quantitative 31P-NMR spectroscopy for the determination of fosfomycin and impurity A in pharmaceutical products of fosfomycin sodium or calcium. Magn. Reson. Chem. 2015, 53, 454–459. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Wu, H. A Review on the Advancements in Quantitative Nuclear Magnetic Resonance Spectroscopy of Fluorine and Phosphorus. Metrol. Sci. Technol. 2023, 67, 9–15. [Google Scholar]
- Gard, D.R.; Burquin, J.C.; Gard, J.K. Quantitative analysis of short-chain phosphates by phosphorus-31 nuclear magnetic resonance and interlaboratory comparison with infrared and chromatographic methods. Anal. Chem. 1992, 64, 557–561. [Google Scholar] [CrossRef]
- Martino, R.; Gilard, V.; Desmoulin, F.; Malet-Martino, M. Fluorine-19 or phosphorus-31 NMR spectroscopy: A suitable analytical technique for quantitative in vitro metabolic studies of fluorinated or phosphorylated drugs. J. Pharm. Biomed. Anal. 2005, 38, 871–891. [Google Scholar] [CrossRef] [PubMed]
- Maniara, G.; Rajamoorthi, K.; Rajan, S.; Stockton, G.W. Method Performance and Validation for Quantitative Analysis by 1H and 31P NMR Spectroscopy. Applications to Analytical Standards and Agricultural Chemicals. Anal. Chem. 1998, 70, 4921–4928. [Google Scholar] [CrossRef] [PubMed]
- Saed Al Deen, T.; Brynn Hibbert, D.; Hook, J.M.; Wells, R.J. Quantitative nuclear magnetic resonance spectrometry: II. Purity of phosphorus-based agrochemicals glyphosate (N-(phosphonomethyl)-glycine) and profenofos (O-(4-bromo-2-chlorophenyl) O-ethyl S-propyl phosphorothioate) measured by 1H and 31P QNMR spectrometry. Anal. Chim. Acta 2002, 474, 125–135. [Google Scholar]
- Kato, T.; Nishimiya, M.; Kawata, A.; Kishida, K.; Suzuri, K.; Saito, M.; Fujita, K.; Igarashi, T.; Inagaki, M. Quantitative 31P NMR Method for Individual and Concomitant Determination of Phospholipid Classes in Polar Lipid Samples. J. Oleo Sci. 2018, 67, 1279–1289. [Google Scholar] [CrossRef] [PubMed]
- Spectral Database for Organic Compounds. Available online: https://sdbs.db.aist.go.jp/sdbs/cgi-bin/direct_frame_top.cgi (accessed on 23 January 2024).
- Uchiyama, N. Quantitative 31P-NMR for Purity Determination of Organophosphorus Compounds (Pharmaceuticals). Yakugaku Zasshi 2024, 144, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Uchiyama, N.; Hosoe, J.; Komatsu, T.; Sugimoto, N.; Ishizuki, K.; Koide, T.; Murabayashi, M.; Shinozaki, T.; Kobayashi, K.; Fujimine, Y.; et al. Quantitative 31P-NMR for the Purity Determination of the Organophosphorus Compound Brigatinib and Its Method Validation. Chem. Pharm. Bull. 2024, 72, 36–40. [Google Scholar] [CrossRef] [PubMed]
- ISO GUIDE 33407-2024; Guidance for the Production of Pure Organic Substance Certified Reference Materials. International Organization for Standardization: Geneva, Switzerland, 2024.
- JJF 1855–2020; Metrological Technical Specification for Purity Assessment of Certified Reference Materials-Organic Purity Certified Reference Materials. Standards Press of China: Beijing, China, 2020.
- ISO GUIDE 35-2017; Reference Materials. Guidance for Characterization and Assessment of Homogeneity and Stability. International Organization for Standardization: Geneva, Switzerland, 2017.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number of Measurements | Relative Content (mg/g) | Average Value (mg/g) | RSD (%) |
|---|---|---|---|
| 1 | 2.84 | 3.16 | 6.40 |
| 2 | 2.99 | ||
| 3 | 3.23 | ||
| 4 | 3.26 | ||
| 5 | 3.31 | ||
| 6 | 3.35 |
| Number of Measurements | MB (mg/g) | 1H-qNMR (mg/g) | 31P-qNMR (mg/g) |
|---|---|---|---|
| 1 | 994.60 | 994.7 | 992.6 |
| 2 | 994.60 | 992.6 | 993.7 |
| 3 | 994.61 | 993.6 | 994.6 |
| 4 | 994.60 | 995.7 | 994.3 |
| 5 | 994.59 | 994.8 | 992.0 |
| 6 | 994.60 | 992.7 | 994.5 |
| 7 | 994.59 | 994.4 | 992.7 |
| 8 | 994.58 | - | - |
| 9 | 994.60 | - | - |
| 10 | 994.60 | - | - |
| 11 | 994.59 | - | - |
| Mean | 994.60 | 994.1 | 993.5 |
| SD 1 | 0.008 | 1.151 | 1.048 |
| Fcalculate | 3.41 | ||
| F0.05(22,2) | 3.44 | ||
| Conclusion | Fcalculate < F0.05(22,2), the means are equal | ||
| tcalculate | 1.54 | ||
| t0.05(16) | 2.12 | ||
| Conclusion | tcalculate < t0.05(16), the SDs are equal | ||
| Components | Sources of Uncertainty | Mass Fraction (mg/g) | Standard Uncertainty (mg/g) |
|---|---|---|---|
| uGC | PGC | 998.63 | 0.21 |
| uW | XW | 3.16 | 2.10 |
| uV | XV | 0.00 | 0.00 |
| uNV | XNV | 0.87 | 0.20 |
| uMB | PMB | 994.60 | 2.10 |
| Components | Sources of Uncertainty | 1H-qNMR Mass Fraction (mg/g) | 1H-qNMR Uncertainty (mg/g) | 31P-qNMR Mass Fraction (mg/g) | 31P-qNMR Uncertainty (mg/g) |
|---|---|---|---|---|---|
| u(Ix/Istd) | Ix/Istd | 994.1 | 1.2 | 993.5 | 1.0 |
| u(Mx) | Mx | 224.2368 | 0.0043 | 224.2368 | 0.0043 |
| u(Mstd) | Mstd | 122.1230 | 0.0033 | 223.1648 | 0.0034 |
| u(mx) | mx | 3.003 | 0.002 | 3.003 | 0.002 |
| u(mstd) | mstd | 2.000 | 0.002 | 2.000 | 0.002 |
| u(Pstd) | Pstd | 999.9 | 0.2 | 999.0 | 4.0 |
| u(PTnPP-NMR) | PTnPP-NMR | 994.1 | 1.6 | 993.5 | 4.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wan, Y.; Li, K.; Li, X.; Li, X.; Chu, H.; Zhang, Q. Purity Assessment of Tripropyl Phosphate through Mass Balance and 1H and 31P Quantitative Nuclear Magnetic Resonance. Molecules 2024, 29, 1975. https://doi.org/10.3390/molecules29091975
Wan Y, Li K, Li X, Li X, Chu H, Zhang Q. Purity Assessment of Tripropyl Phosphate through Mass Balance and 1H and 31P Quantitative Nuclear Magnetic Resonance. Molecules. 2024; 29(9):1975. https://doi.org/10.3390/molecules29091975
Chicago/Turabian StyleWan, Yuebing, Kangcong Li, Xiuqin Li, Xiaomin Li, Hongtao Chu, and Qinghe Zhang. 2024. "Purity Assessment of Tripropyl Phosphate through Mass Balance and 1H and 31P Quantitative Nuclear Magnetic Resonance" Molecules 29, no. 9: 1975. https://doi.org/10.3390/molecules29091975
APA StyleWan, Y., Li, K., Li, X., Li, X., Chu, H., & Zhang, Q. (2024). Purity Assessment of Tripropyl Phosphate through Mass Balance and 1H and 31P Quantitative Nuclear Magnetic Resonance. Molecules, 29(9), 1975. https://doi.org/10.3390/molecules29091975