

Unraveling Meso-Substituent Steric Effects on the Mechanism of Hydrogen Evolution Reaction in NiII Porphyrin Hydrides Using DFT Method


Abstract

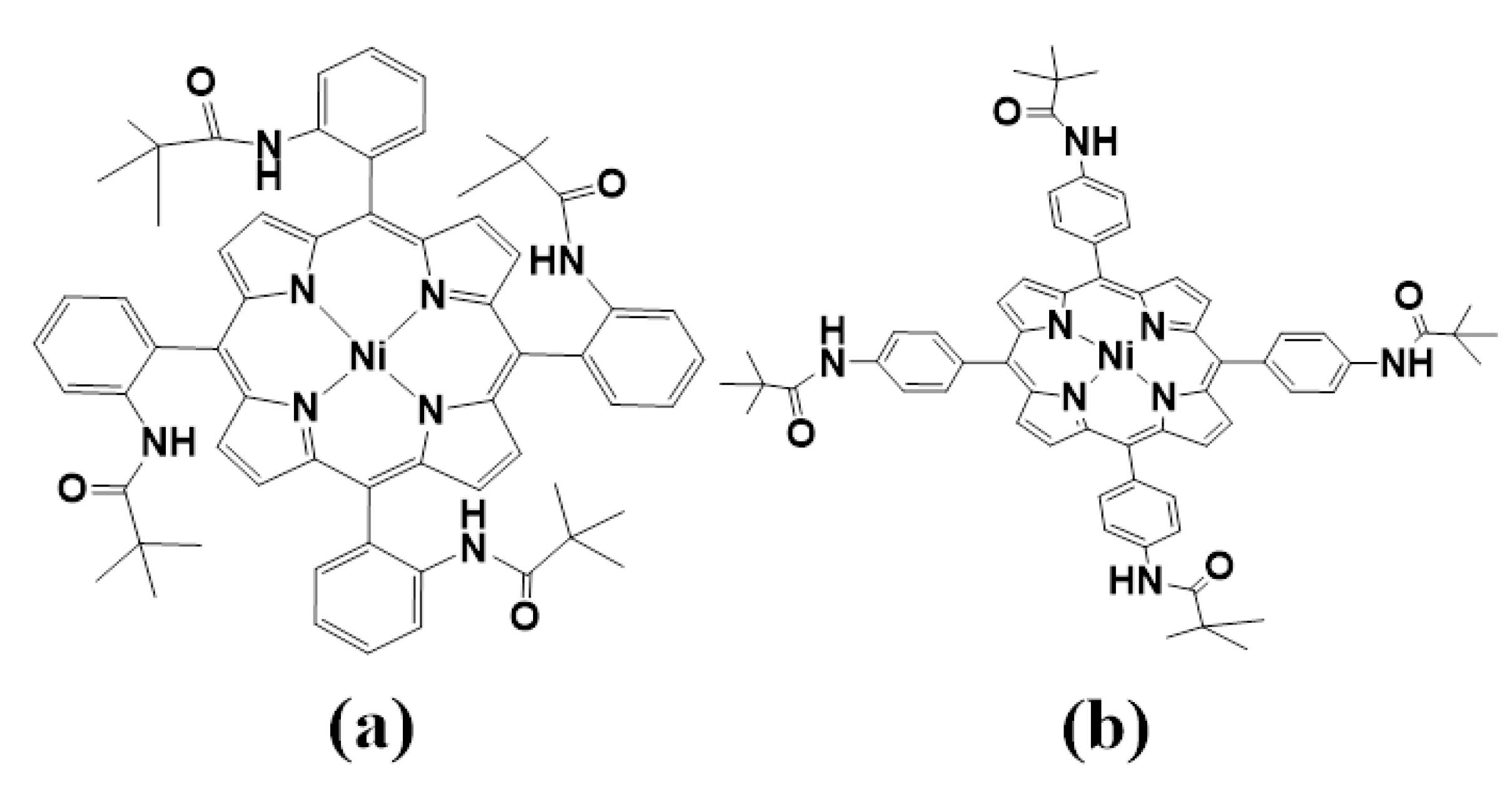
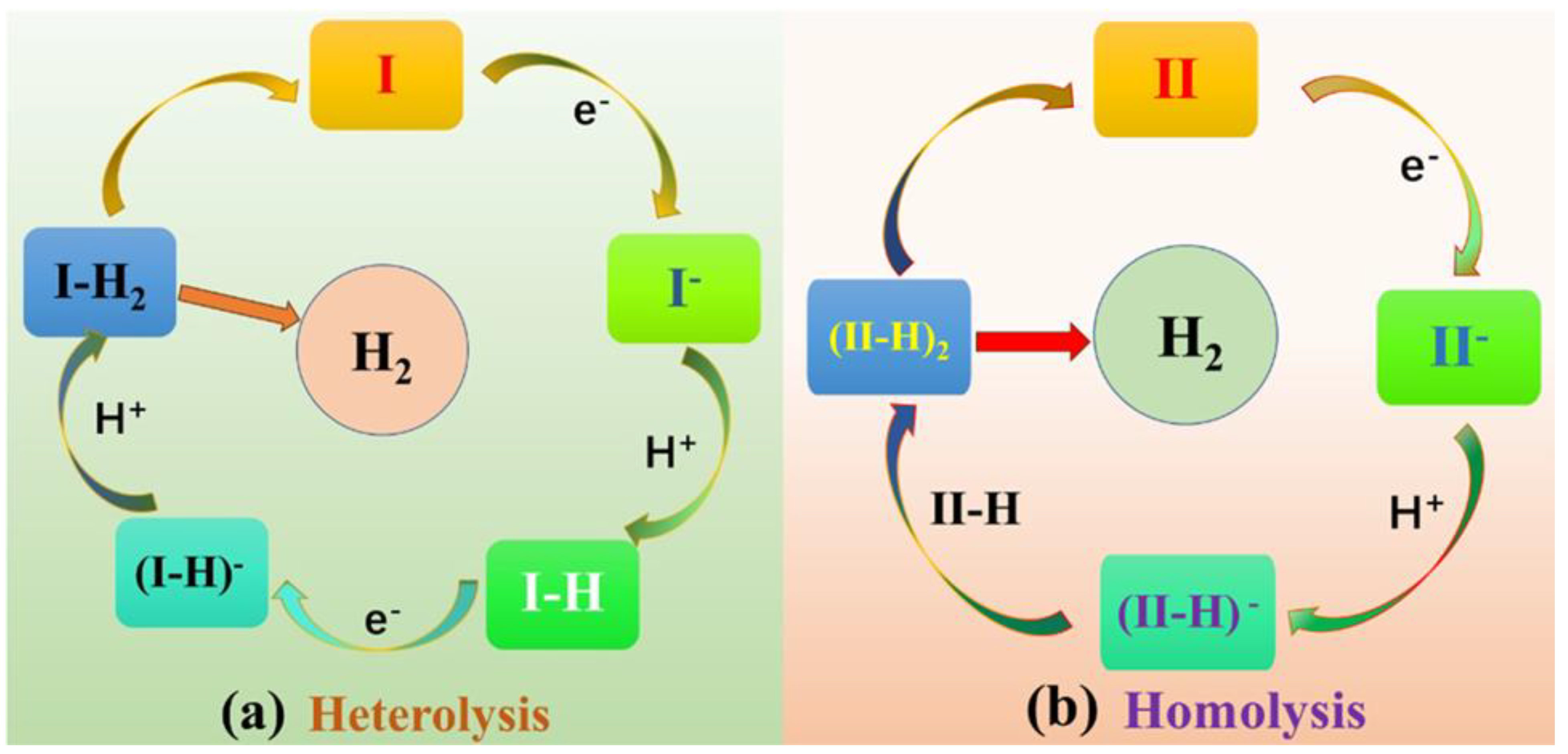
1. Introduction
2. Results and Discussion
2.1. Optimized Structure Analysis
2.2. Atomic Charge Analysis
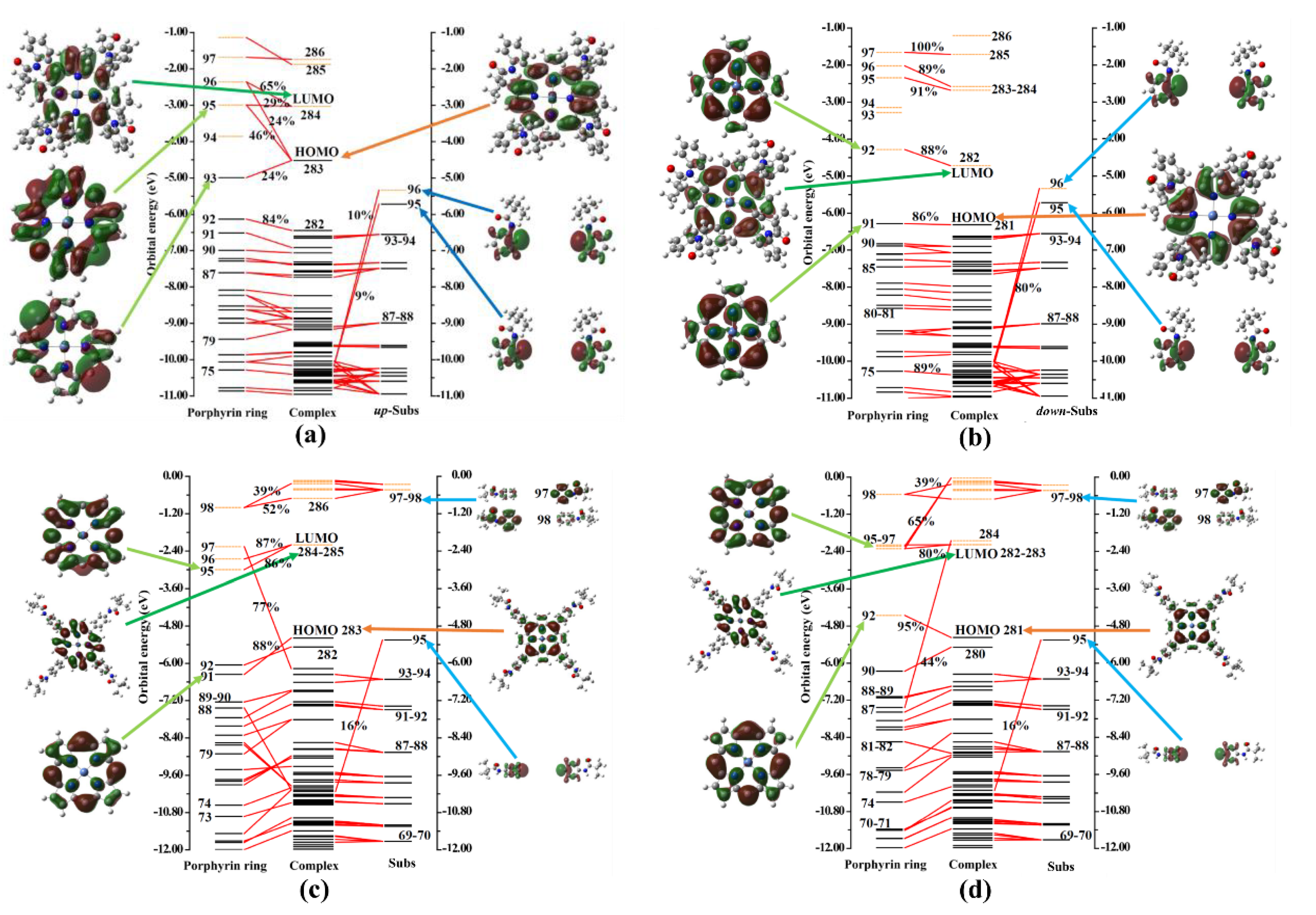
2.3. Fragment Orbital Interaction Analysis
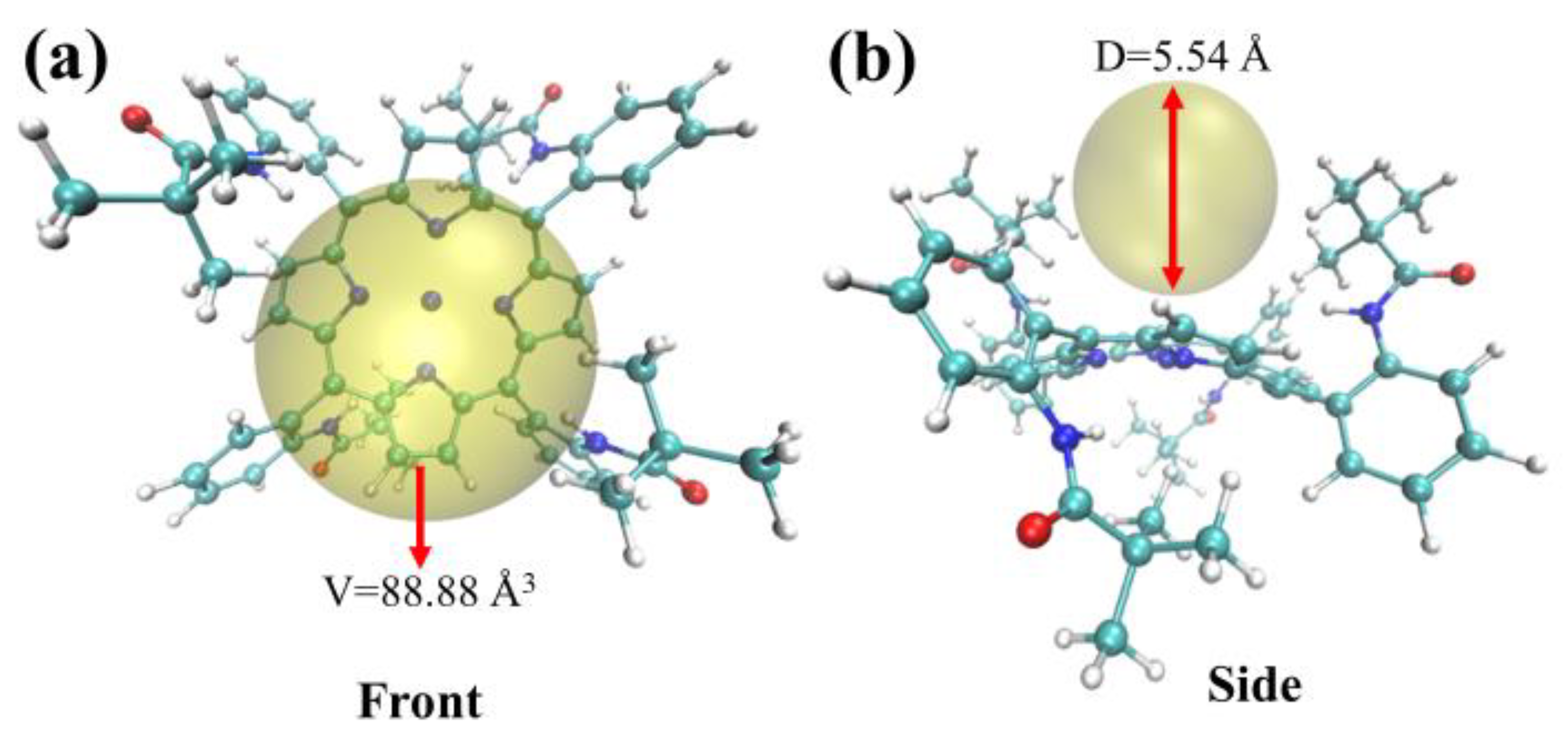
2.4. Steric Hindrance Analysis
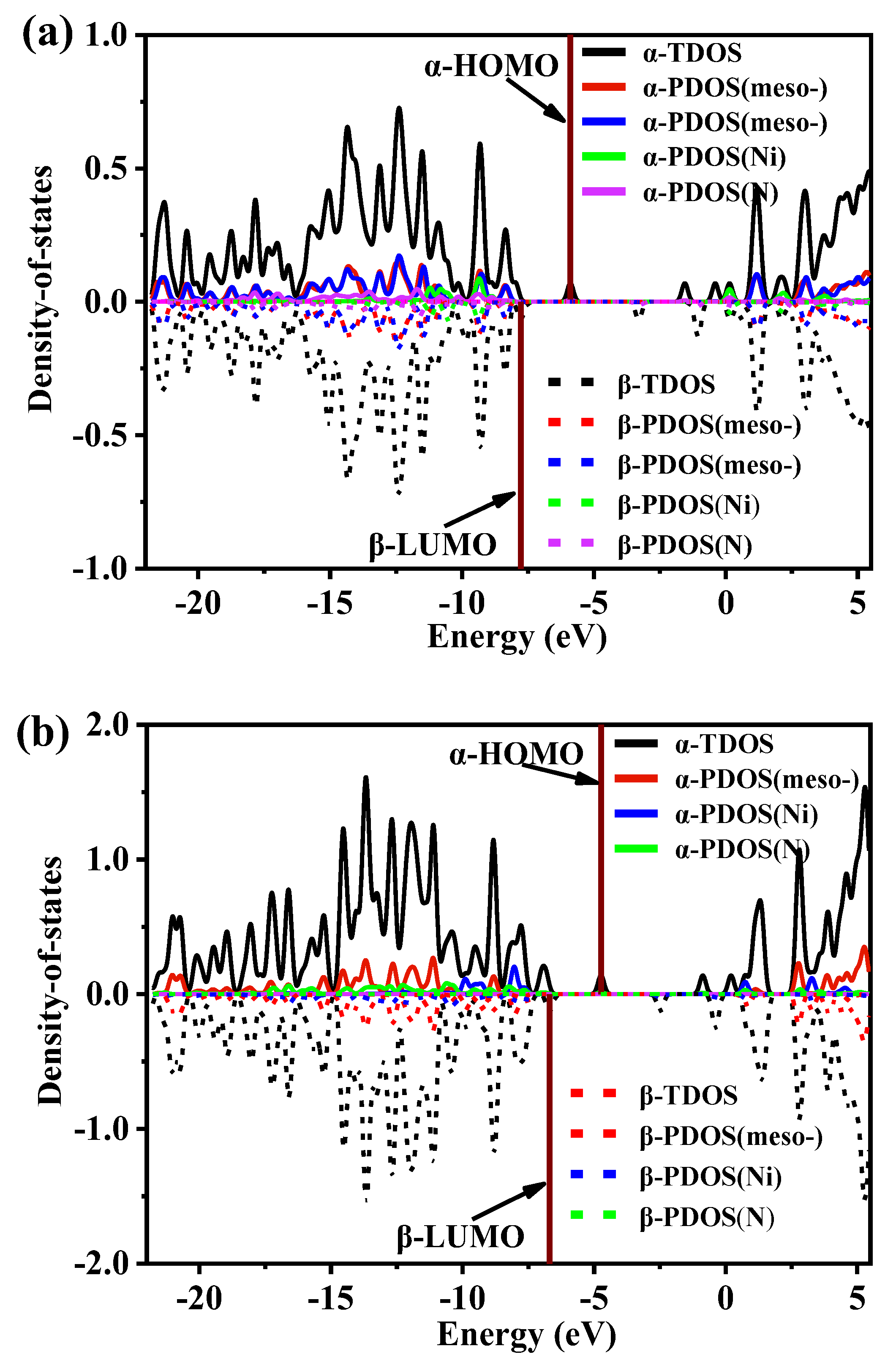
2.5. Density-of-State Analysis
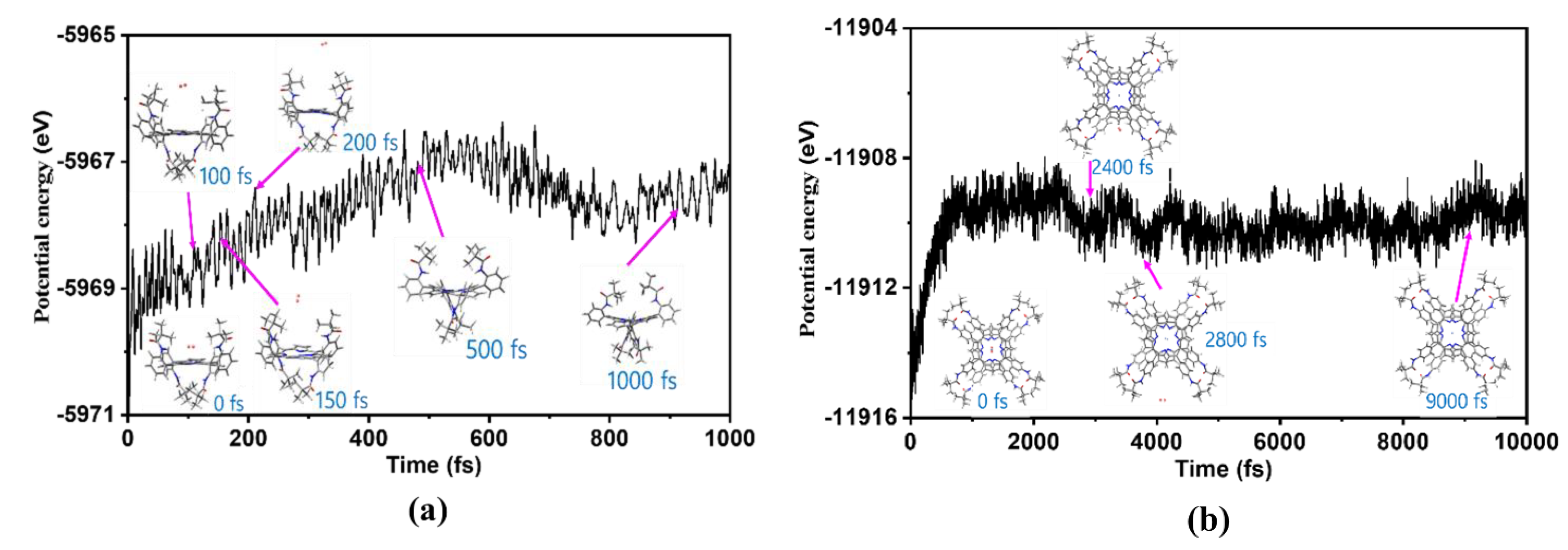
2.6. Molecules Dynamics Analysis
3. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Capurso, T.; Stefanizzi, M.; Torresi, M.; Camporeale, S.M. Perspective of the role of hydrogen in the 21st century energy transition. Energy Convers. Manag. 2022, 251, 114898. [Google Scholar] [CrossRef]
- Faye, O.; Szpunar, J.; Eduok, U. A critical review on the current technologies for the generation, storage, and transportation of hydrogen. Int. J. Hydrogen Energy 2022, 47, 13771–13802. [Google Scholar] [CrossRef]
- Ishaq, H.; Dincer, I.; Crawford, C. A review on hydrogen production and utilization: Challenges and opportunities. Int. J. Hydrogen Energy 2022, 47, 26238–26264. [Google Scholar] [CrossRef]
- Heppe, N.; Gallenkamp, C.; Paul, S.; Segura-Salas, N.; von Rhein, N.; Kaiser, B.; Jaegermann, W.; Jafari, A.; Sergueev, I.; Krewald, V.; et al. Substituent Effects in Iron Porphyrin Catalysts for the Hydrogen Evolution Reaction. Chem. Eur. J. 2023, 29, e202202465. [Google Scholar] [CrossRef]
- Qi, X.W.; Yang, G.; Guo, X.S.; Si, L.P.; Zhang, H.; Liu, H.Y. Electrocatalytic Hydrogen Evolution by Water-Soluble Cobalt (II), Copper (II) and Iron (III) meso-Tetrakis(carboxyl)porphyrin. Eur. J. Inorg. Chem. 2022, 26, e202200613. [Google Scholar] [CrossRef]
- Zhou, Y.Z.; Zhang, T.; Zhu, W.; Qin, L.; Kang, S.-Z.; Li, X. Enhanced light absorption and electron transfer in dimensionally matched carbon nitrideporphyrin nanohybrids for photocatalytic hydrogen production. Fuel 2023, 338, e127394. [Google Scholar] [CrossRef]
- Cook, B.J.; Barona, M.; Johnson, S.I.; Raugei, S.; Bullock, R.M. Weakening the N–H Bonds of NH3Ligands: Triple Hydrogen-Atom Abstraction to Form a Chromium(V) Nitride. Inorg. Chem. 2022, 61, 11165–11172. [Google Scholar] [CrossRef] [PubMed]
- Joseph, M.; Haridas, S. Recent progresses in porphyrin assisted hydrogen evolution. Int. J. Hydrogen Energy 2020, 45, 11954–11975. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, Y.; Li, T.; He, Y.Z.; Han, E.C.; Chen, Y.L.; Jiang, X.Y.; Ni, C.L.; Yang, L.M.; Liu, W. Cobalt-based metalloporphyrins as efficient electro-catalysts for hydrogen evolution from acetic acid and water. Electrocatalysis. 2023, 14, 752–762. [Google Scholar] [CrossRef]
- Zhao, W.; Peng, J.; Wang, W.; Jin, B.; Chen, T.; Liu, S.; Zhao, Q.; Huang, W. Interlayer Hydrogen-Bonded Metal Porphyrin Frameworks/MXene Hybrid Film with High Capacitance for Flexible All-Solid-State Supercapacitors. Small 2019, 15, e1901351. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Han, J.; Li, X.; Liu, G.; Xu, Y.; Peng, Y.; Nie, S.; Li, W.; Li, X.; Chen, Z.; et al. Electrocatalytic hydrogen evolution with a copper porphyrin bearing meso-(o-carborane) substituents. Chem. Commun. 2023, 59, 10777–10780. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, S.; Zhang, Y.; Li, R.; Zhao, B.; Peng, T. Hydrogen-Bond Regulation of the Microenvironment of Ni(II)-Porphyrin Bifunctional Electrocatalysts for Efficient Overall Water Splitting. Adv. Mater. 2023, 35, e2210727. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.; Li, X.; Lei, H.; Guo, K.; Lv, B.; Guo, H.; Chen, D.; Zhang, W.; Cao, R. Comparing electrocatalytic hydrogen and oxygen evolution activities of first-row transition metal complexes with similar coordination environments. J. Energy Chem. 2021, 63, 659–666. [Google Scholar] [CrossRef]
- Yuasa, M.; Nishihara, R.; Shi, C.; Anson, F.C. Comparison of Several Meso-Tetraalkyl Cobalt Porphyrins as Catalysts for the Electroreduction of Dioxygen. Polym. Adv. Technol. 2001, 12, 266–270. [Google Scholar] [CrossRef]
- Ardakani, M.M.; Rahimi, P.; Dehghani, H.; Karami, P.E.; Zare, H.R.; Karami, S. Electrocatalytic Reduction of Dioxygen on the Surface of Glassy Carbon Electrodes Modified with Cobalt Porphyrin Complexes. Electroanalysis 2007, 19, 2258–2263. [Google Scholar] [CrossRef]
- Qin, H.; Wang, Y.; Wang, B.; Duan, X.; Lei, H.; Zhang, X.; Zheng, H.; Zhang, W.; Cao, R. Cobalt porphyrins supported on carbon nanotubes as model catalysts of metal-N4/C sites for oxygen electrocatalysis. J. Energy Chem. 2021, 53, 77–81. [Google Scholar] [CrossRef]
- Sinha, S.; Aaron, M.S.; Blagojevic, J.; Warren, J.J. Electrocatalytic Dioxygen Reduction by Carbon Electrodes Noncovalently Modified with Iron Porphyrin Complexes: Enhancements from a Single Proton Relay. Chem. —A Eur. J. 2015, 21, 18072–18075. [Google Scholar] [CrossRef]
- Sinha, S.; Ghosh, M.; Warren, J.J. Changing the Selectivity of O2 Reduction Catalysis with One Ligand Heteroatom. ACS Catal. 2019, 9, 2685–2691. [Google Scholar] [CrossRef]
- Su, B.; Hatay, I.; Trojánek, A.; Samec, Z.; Khoury, T.; Gros, C.P.; Barbe, J.-M.; Daina, A.; Carrupt, P.-A.; Girault, H.H. Molecular Electrocatalysis for Oxygen Reduction by Cobalt Porphyrins Adsorbed at Liquid/Liquid Interfaces. J. Am. Chem. Soc. 2010, 132, 2655–2662. [Google Scholar] [CrossRef]
- Shi, F.C.A.C. (5,10,15,20-Tetramethylporphyrinato)cobalt(II): A Remarkably Active Catalyst for the Electroreduction of O2 to H2O. Inorg. Chem. 1998, 37, 1037–1043. [Google Scholar] [CrossRef]
- Lei, H.; Li, X.; Meng, J.; Zheng, H.; Zhang, W.; Cao, R. Structure Effects of Metal Corroles on Energy-Related Small Molecule Activation Reactions. ACS Catal. 2019, 9, 4320–4344. [Google Scholar] [CrossRef]
- Aarabi, M.; Omidyan, R.; Soorkia, S.; Grégoire, G.; Broquier, M.; Crestoni, M.-E.; de la Lande, A.; Soep, B.; Shafizadeh, N. The dramatic effect of N-methylimidazole on trans axial ligand binding to ferric heme: Experiment and theory. Phys. Chem. Chem. Phys. 2019, 21, 1750–1760. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Nakatani, N.; Fujii, H.; Hada, M. DFT insight into axial ligand effects on electronic structure and mechanistic reactivity of oxoiron(iv) porphyrin. Phys. Chem. Chem. Phys. 2020, 22, 12173–12179. [Google Scholar] [CrossRef] [PubMed]
- NElgrishi, N.; Kurtz, D.A.; Dempsey, J.L. Reaction Parameters Influencing Cobalt Hydride Formation Kinetics: Implications for Benchmarking H2-Evolution Catalysts. J. Am. Chem. Soc. 2016, 139, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Rose, M.J.; Gray, H.B.; Winkler, J.R. Hydrogen Generation Catalyzed by Fluorinated Diglyoxime–Iron Complexes at Low Overpotentials. J. Am. Chem. Soc. 2012, 134, 8310–8313. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Liang, G.; Reddy, M.R.; Long, M.; Driskill, K.; Lyons, C.; Donnadieu, B.; Bollinger, J.C.; Webster, C.E.; Zhao, X. Electronic and Steric Tuning of Catalytic H2 Evolution by Cobalt Complexes with Pentadentate Polypyridyl-Amine Ligands. J. Am. Chem. Soc. 2018, 140, 9219–9229. [Google Scholar] [CrossRef] [PubMed]
- Marinescu, S.C.; Winkler, J.R.; Gray, H.B. Molecular mechanisms of cobalt-catalyzed hydrogen evolution. Proc. Natl. Acad. Sci. USA 2012, 109, 15127–15131. [Google Scholar] [CrossRef]
- Han, Y.; Fang, H.; Jing, H.; Sun, H.; Lei, H.; Lai, W.; Cao, R. Singly versus Doubly Reduced Nickel Porphyrins for Proton Reduction: Experimental and Theoretical Evidence for a Homolytic Hydrogen-Evolution Reaction. Angew. Chem. 2016, 128, 5547–5552. [Google Scholar] [CrossRef]
- Ramakrishnan, S.; Chakraborty, S.; Brennessel, W.W.; Chidsey, C.E.D.; Jones, W.D. Rapid oxidative hydrogen evolution from a family of square-planar nickel hydride complexes. Chem. Sci. 2016, 7, 117–127. [Google Scholar] [CrossRef]
- Liberman, I.; Shimoni, R.; Ifraemov, R.; Rozenberg, I.; Singh, C.; Hod, I. Active-Site Modulation in an Fe-Porphyrin-Based Metal–Organic Framework through Ligand Axial Coordination: Accelerating Electrocatalysis and Charge-Transport Kinetics. J. Am. Chem. Soc. 2020, 142, 1933–1940. [Google Scholar] [CrossRef]
- Meng, J.; Lei, H.; Li, X.; Zhang, W.; Cao, R. The Trans Axial Ligand Effect on Oxygen Reduction. Immobilization Method May Weaken Catalyst Design for Electrocatalytic Performance. J. Phys. Chem. C 2020, 124, 16324–16331. [Google Scholar] [CrossRef]
- Samanta, S.; Das, P.K.; Chatterjee, S.; Dey, A. Effect of axial ligands on electronic structure andO2 reduction by iron porphyrin complexes: Towards a quantitative understanding of the “push effect”. J. Porphyrins Phthalocyanines 2015, 19, 92–108. [Google Scholar] [CrossRef]
- Guo, X.; Wang, N.; Li, X.; Zhang, Z.; Zhao, J.; Ren, W.; Ding, S.; Xu, G.; Li, J.; Apfel, U.; et al. Homolytic versus Heterolytic Hydrogen Evolution Reaction Steered by a Steric Effect. Angew. Chem. Int. Ed. 2020, 59, 8941–8946. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Lei, H.; Xie, L.; Wang, N.; Zhang, W.; Cao, R. Metalloporphyrins as Catalytic Models for Studying Hydrogen and Oxygen Evolution and Oxygen Reduction Reactions. Accounts Chem. Res. 2022, 55, 878–892. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Lv, X.-L.; Feng, D.; Chen, S.; Sun, J.; Song, L.; Xie, Y.; Li, J.-R.; Zhou, H.-C. Pyrazolate-Based Porphyrinic Metal-Organic Framework with Extraordinary Base-Resistance. J. Am. Chem. Soc. 2016, 138, 914–919. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Chen, F. Atomic Dipole Moment Corrected Hirshfeld Population Method. J. Theor. Comput. Chem. 2012, 11, 163–183. [Google Scholar] [CrossRef]
- Xia-Yu, Z.; Chun-Ying, R.; Tian, L.U.; Shu-Bin, L.I.U. Hirshfeld Charge as a Quantitative Measure of Electrophilicity and Nucleophilicity: Nitrogen-Containing Systems. Acta Phys.-Chim. Sin. 2014, 30, 2055–2062. [Google Scholar] [CrossRef]
- Chakravorty, E.R.D.A.S. A test of the Hirshfeld definition of atomic charges and moments. Theor. Chim. Acta 1992, 83, 319–330. [Google Scholar]
- Wiberg, K.B.; Rablen, P.R. Comparison of Atomic Charges Derived via Different Procedures. J. Comput. Chem. 1993, 14, 1504–1518. [Google Scholar] [CrossRef]
- Tian, L.; Fei-Wu, C. Comparison of Computational Methods for Atomic Charges. Acta Phys. Chim. Sin. 2012, 28, 1–18. [Google Scholar] [CrossRef]
- Dapprich, S.; Frenking, G. Investigation of Donor- Acceptor Interactions: A Charge Decomposition Analysis Using Fragment Molecular Orbitals. J. Phys. Chem. 1995, 99, 9352–9362. [Google Scholar] [CrossRef]
- Xiao, M.; Lu, T. Generalized Charge Decomposition Analysis (GCDA) Method. J. Adv. Phys. Chem. 2015, 4, 111–124. [Google Scholar] [CrossRef]
- Liu, Z.; Lu, T.; Chen, Q. An sp-hybridized all-carboatomic ring, cyclo[18]carbon: Bonding character, electron delocalization, and aromaticity. Carbon 2020, 165, 468–475. [Google Scholar] [CrossRef]
- Frisch, G.W.T.M.J.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09, versions D.01.; Gaussian, Inc.: Wallingford, UK, 2013. [Google Scholar]
- Chai, J.-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, A.D.W.; Klaus Schulten, V.M.D. Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Perdew, J.P.; Ruzsinszky, A.; Csonka, G.I.; Vydrov, O.A.; Scuseria, G.E.; Constantin, L.A.; Zhou, X.; Burke, K. Restoring the Density-Gradient Expansion for Exchange in Solids and Surfaces. Phys. Rev. Lett. 2008, 100, 13640. [Google Scholar] [CrossRef]
- Fernández, E.M.; Balbás, L.C. GGA versus van der Waals density functional results for mixed gold/mercury molecules and pure Au and Hg cluster properties. Phys. Chem. Chem. Phys. 2011, 13, 20863–20870. [Google Scholar] [CrossRef]
- Hutter, J.; Iannuzzi, M.; Schiffmann, F.; VandeVondele, J. cp2k: Atomistic simulations of condensed matter systems. WIREs Comput. Mol. Sci. 2013, 4, 15–25. [Google Scholar] [CrossRef]
- VandeVondele, J.; Krack, M.; Mohamed, F.; Parrinello, M.; Chassaing, T.; Hutter, J. Quickstep: Fast and accurate density functional calculations using a mixed Gaussian and plane waves approach. Comput. Phys. Commun. 2005, 167, 103–128. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complexes | Distance (Å) | ||
|---|---|---|---|
| Ni-N1 a | Ni-N2 b | Ni-H | |
| I I− I-H (I-H)− II II− II-H | 1.94 1.94 2.02 1.97 1.95 1.95 2.08 | 1.95 1.95 2.04 2.06 1.95 1.96 2.09 | - - 1.72 1.41 - - 1.67 |
| Complexes | ADCH Charge (a.u.) | ||||
|---|---|---|---|---|---|
| up-Sub a | down-Sub b | Ni | N1 c | N2 d | |
| I I− I-H (I-H)− I-H2 II II II-H | −0.009 −0.075 0.043 −0.003 0.016 0.012 −0.060 0.053 | 0.008 −0.080 0.030 −0.025 0.031 0.012 −0.059 0.058 | 0.135 0.087 0.233 0.138 0.251 0.397 0.087 0.266 | 0.143 −0.156 −0.135 −0.170 −0.177 −0.204 −0.165 −0.112 | −0.179 −0.157 −0.200 −0.310 −0.307 −0.204 −0.165 −0.176 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, X.; Feng, A.; Zu, Y.; Liu, P. Unraveling Meso-Substituent Steric Effects on the Mechanism of Hydrogen Evolution Reaction in NiII Porphyrin Hydrides Using DFT Method. Molecules 2024, 29, 986. https://doi.org/10.3390/molecules29050986
Li X, Feng A, Zu Y, Liu P. Unraveling Meso-Substituent Steric Effects on the Mechanism of Hydrogen Evolution Reaction in NiII Porphyrin Hydrides Using DFT Method. Molecules. 2024; 29(5):986. https://doi.org/10.3390/molecules29050986
Chicago/Turabian StyleLi, Xiaodong, Ailing Feng, Yanqing Zu, and Peitao Liu. 2024. "Unraveling Meso-Substituent Steric Effects on the Mechanism of Hydrogen Evolution Reaction in NiII Porphyrin Hydrides Using DFT Method" Molecules 29, no. 5: 986. https://doi.org/10.3390/molecules29050986
APA StyleLi, X., Feng, A., Zu, Y., & Liu, P. (2024). Unraveling Meso-Substituent Steric Effects on the Mechanism of Hydrogen Evolution Reaction in NiII Porphyrin Hydrides Using DFT Method. Molecules, 29(5), 986. https://doi.org/10.3390/molecules29050986