
Potent Apoptosis Induction by a Novel Trispecific B7-H3xCD16xTIGIT 2+1 Common Light Chain Natural Killer Cell Engager

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Anti-B7-H3 Fab: Immunization, Screening, and Characterization
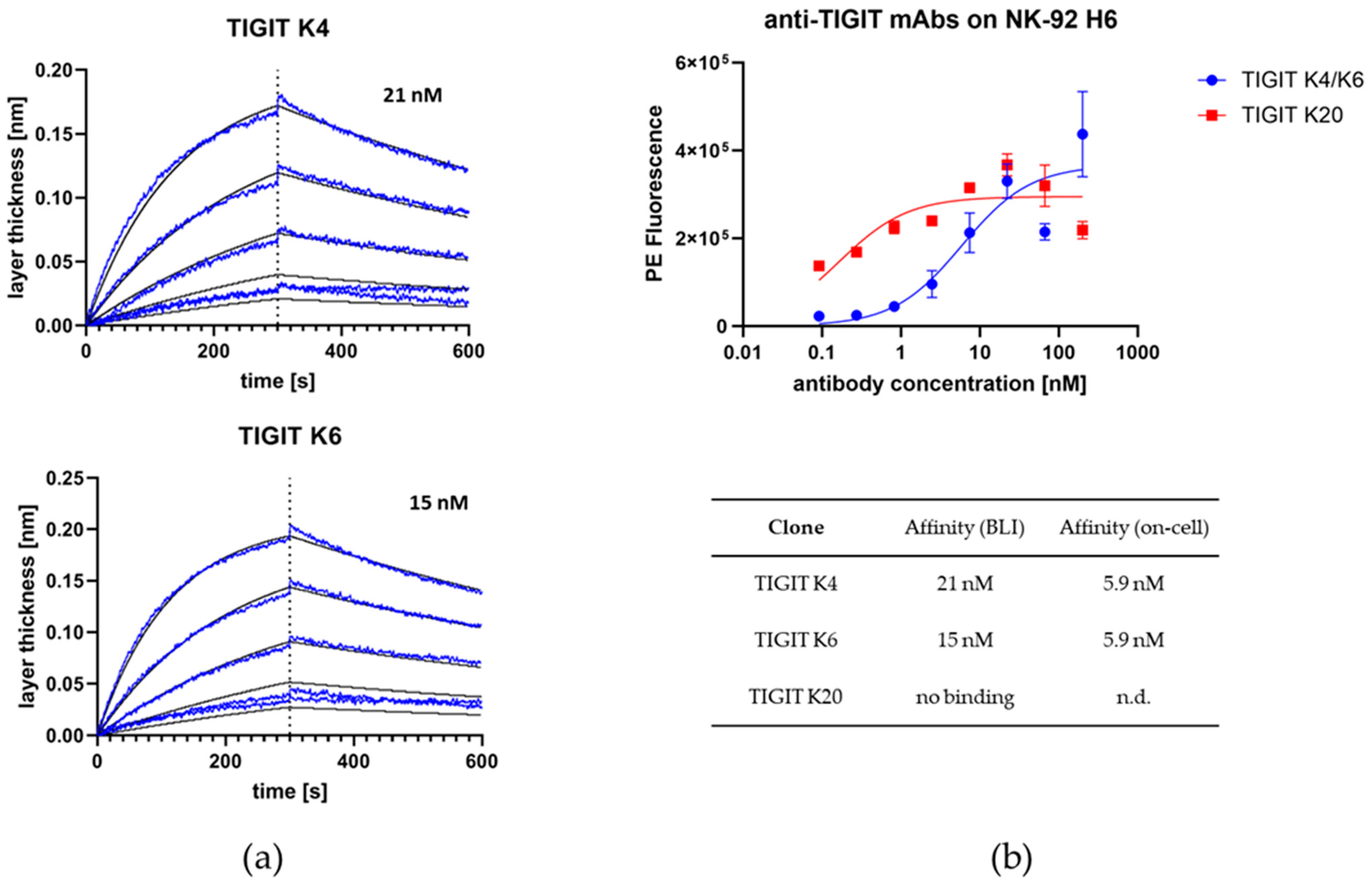
2.2. Anti-TIGIT Fab: Immunization, Screening, Characterization, and Humanization
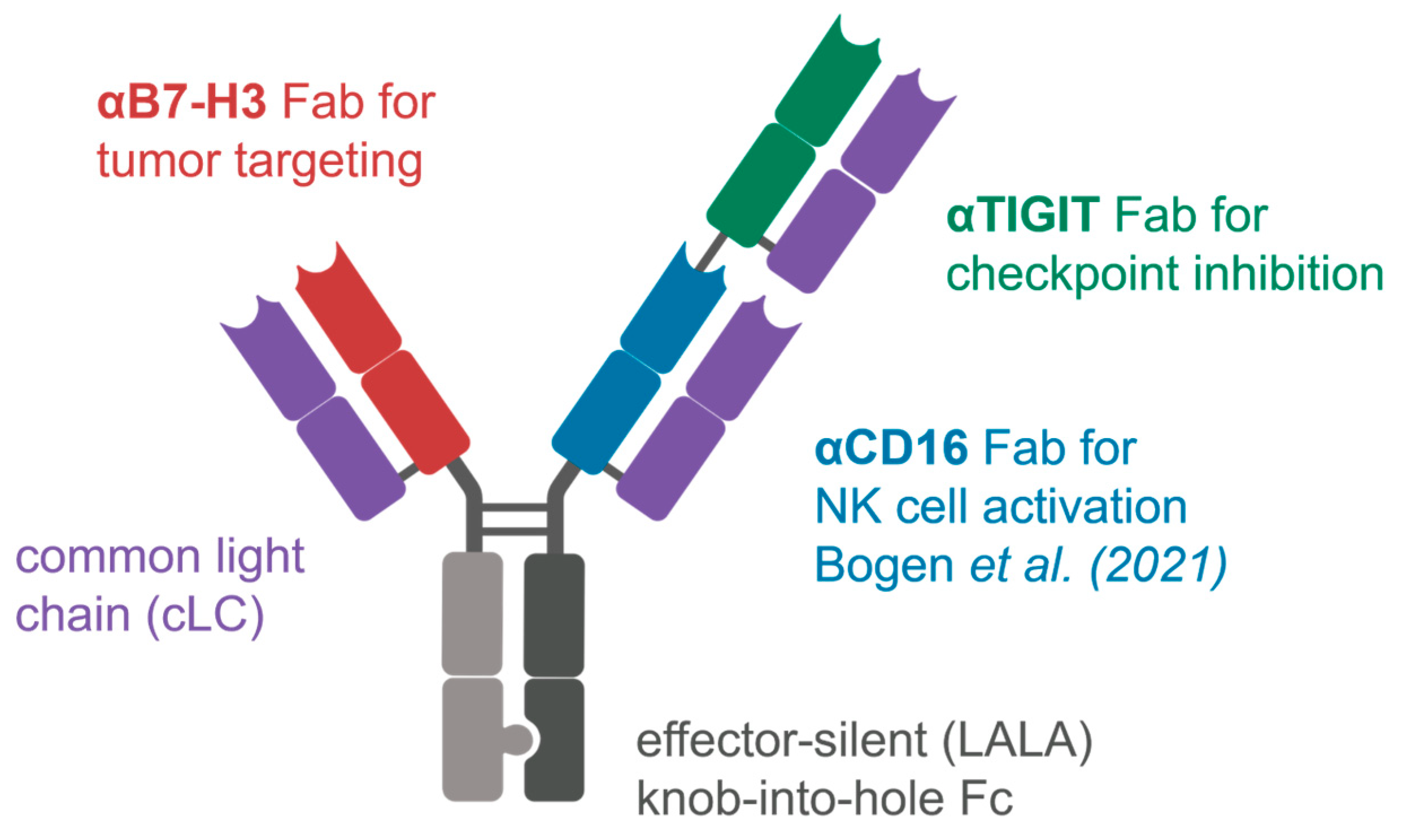
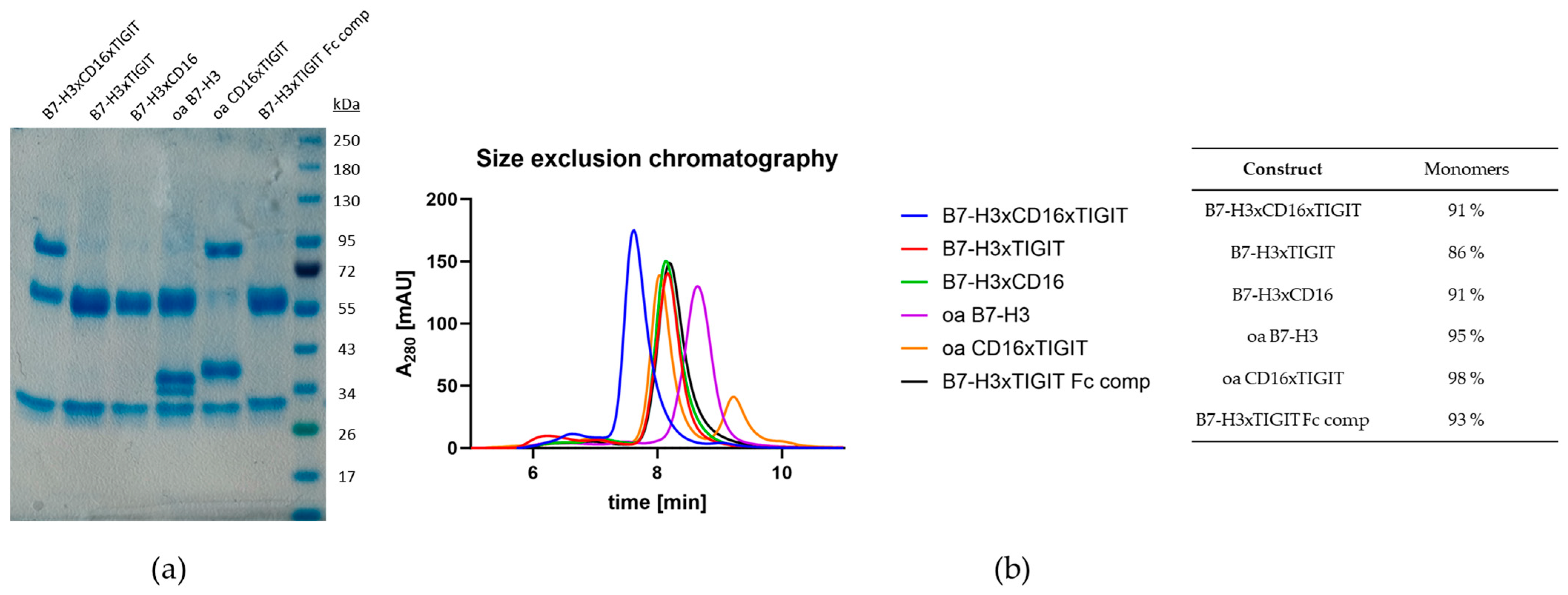
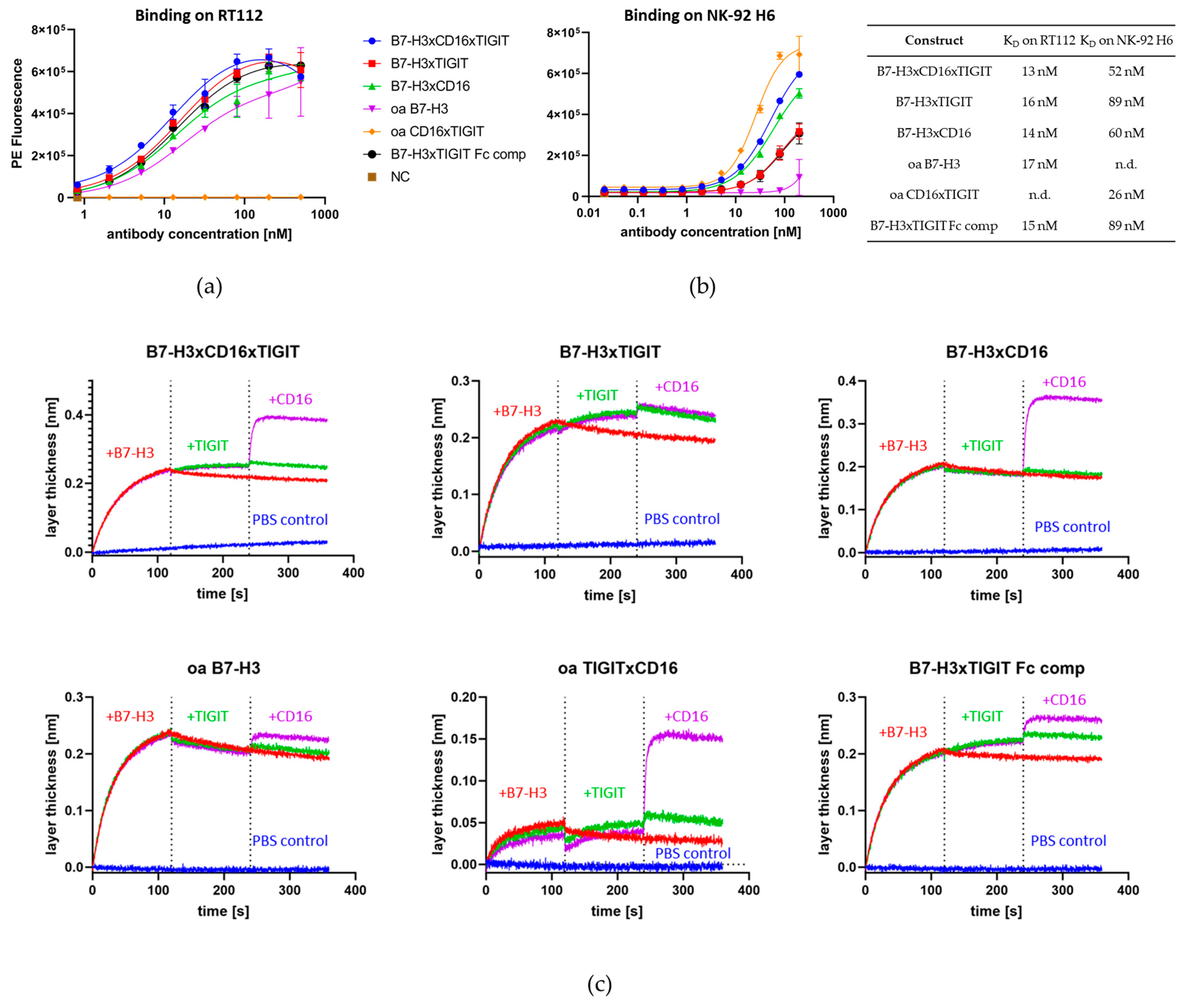
2.3. Construction and Characterization of Trispecific Dual-NK Engaging Antibody
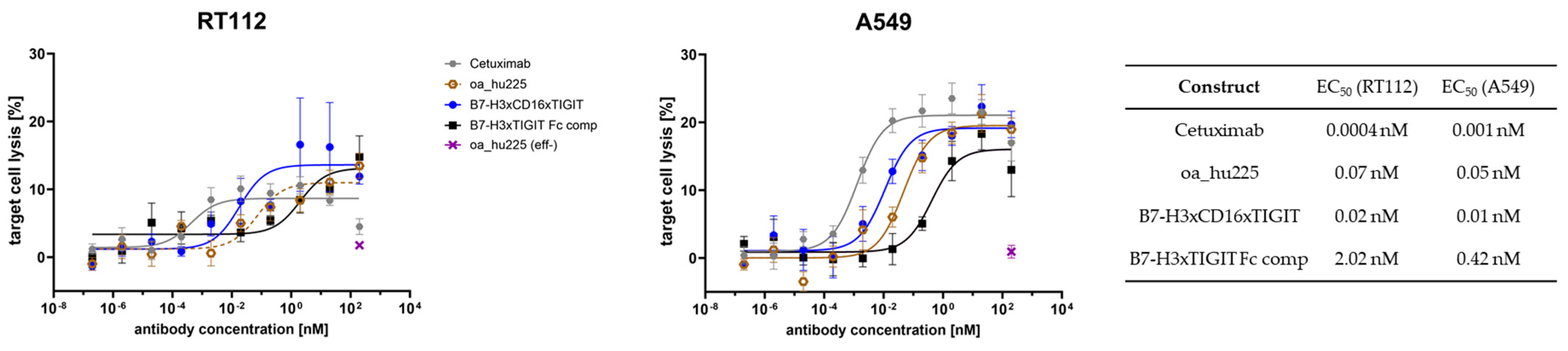
2.4. Cell-Based NK Killing Assay
2.5. PBMC Killing Assay
3. Discussion
4. Materials and Methods
4.1. Chicken Immunization
4.2. Yeast Strains and Plasmids
4.3. Yeast Library Generation
4.4. Yeast Library Screening Procedure
4.5. Reformatting, Expression, and Purification of Full-Length Antibodies
4.6. SDS-PAGE
4.7. Affinity Determination and Simultaneous Binding Assay via Biolayer Interferometry
4.8. Size Exclusion Chromatography
4.9. Cultivation of NK-92 H6 Cells
4.10. Cultivation of RT112 and A549 Cells
4.11. On-Cell Binding Affinities
4.12. NK-92 Killing Assay and EC50 Determination
4.13. PBMC Killing Assay and EC50 Determination
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Waldhauer, I.; Steinle, A. NK Cells and Cancer Immunosurveillance. Oncogene 2008, 27, 5932–5943. [Google Scholar] [CrossRef]
- Yokoyama, W.M.; Riley, J.K. NK Cells and Their Receptors. Reprod. Biomed. Online 2008, 16, 173–191. [Google Scholar] [CrossRef]
- Ramírez-Labrada, A.; Pesini, C.; Santiago, L.; Hidalgo, S.; Calvo-Pérez, A.; Oñate, C.; Andrés-Tovar, A.; Garzón-Tituaña, M.; Uranga-Murillo, I.; Arias, M.A.; et al. All About (NK Cell-Mediated) Death in Two Acts and an Unexpected Encore: Initiation, Execution and Activation of Adaptive Immunity. Front. Immunol. 2022, 13, 1–14. [Google Scholar] [CrossRef]
- Chin, D.S.; Lim, C.S.Y.; Nordin, F.; Arifin, N.; Jun, T.G. Antibody-Dependent Cell-Mediated Cytotoxicity through Natural Killer (NK) Cells: Unlocking NK Cells for Future Immunotherapy. Curr. Pharm. Biotechnol. 2021, 23, 552–578. [Google Scholar] [CrossRef]
- Demaria, O.; Gauthier, L.; Vetizou, M.; Blanchard Alvarez, A.; Vagne, C.; Habif, G.; Batista, L.; Baron, W.; Belaïd, N.; Girard-Madoux, M.; et al. Antitumor Immunity Induced by Antibody-Based Natural Killer Cell Engager Therapeutics Armed with Not-Alpha IL-2 Variant. Cell Rep. Med. 2022, 3, 100783. [Google Scholar] [CrossRef]
- Arvindam, U.S.; van Hauten, P.M.M.; Schirm, D.; Schaap, N.; Hobo, W.; Blazar, B.R.; Vallera, D.A.; Dolstra, H.; Felices, M.; Miller, J.S. A Trispecific Killer Engager Molecule against CLEC12A Effectively Induces NK-Cell Mediated Killing of AML Cells. Leukemia 2021, 35, 1586–1596. [Google Scholar] [CrossRef]
- Bogen, J.P.; Carrara, S.C.; Fiebig, D.; Grzeschik, J.; Hock, B.; Kolmar, H. Design of a Trispecific Checkpoint Inhibitor and Natural Killer Cell Engager Based on a 2 + 1 Common Light Chain Antibody Architecture. Front. Immunol. 2021, 12, 669496. [Google Scholar] [CrossRef]
- Vallera, D.A.; Felices, M.; McElmurry, R.; McCullar, V.; Zhou, X.; Schmohl, J.U.; Zhang, B.; Lenvik, A.J.; Panoskaltsis-Mortari, A.; Verneris, M.R.; et al. IL15 Trispecific Killer Engagers (TriKE) Make Natural Killer Cells Specific to CD33+ Targets While Also Inducing Persistence, in Vivo Expansion, and Enhanced Function. Clin. Cancer Res. 2016, 22, 3440–3450. [Google Scholar] [CrossRef] [PubMed]
- Beha, N.; Harder, M.; Ring, S.; Kontermann, R.E.; Muller, D. IL15-Based Trifunctional Antibody-Fusion Proteins with Costimulatory TNF-Superfamily Ligands in the Single-Chain Format for Cancer Immunotherapy. Mol. Cancer Ther. 2019, 18, 1278–1288. [Google Scholar] [CrossRef] [PubMed]
- Wingert, S.; Reusch, U.; Knackmuss, S.; Kluge, M.; Damrat, M.; Pahl, J.; Schniegler-Mattox, U.; Mueller, T.; Fucek, I.; Ellwanger, K.; et al. Preclinical Evaluation of AFM24, a Novel CD16A-Specific Innate Immune Cell Engager Targeting EGFR-Positive Tumors; Taylor & Francis: Milton Park, UK, 2021; Volume 13. [Google Scholar] [CrossRef]
- Kakiuchi-Kiyota, S.; Ross, T.; Wallweber, H.A.; Kiefer, J.R.; Schutten, M.M.; Adedeji, A.O.; Cai, H.; Hendricks, R.; Cohen, S.; Myneni, S.; et al. A BCMA/CD16A Bispecific Innate Cell Engager for the Treatment of Multiple Myeloma. Leukemia 2022, 36, 1006–1014. [Google Scholar] [CrossRef] [PubMed]
- Nikkhoi, S.K.; Li, G.; Eleya, S.; Yang, G.; Vandavasi, V.G.; Hatefi, A. Bispecific Killer Cell Engager with High Affinity and Specificity toward CD16a on NK Cells for Cancer Immunotherapy. Front. Immunol. 2023, 13, 7658. [Google Scholar] [CrossRef] [PubMed]
- Vallera, D.A.; Ferrone, S.; Kodal, B.; Hinderlie, P.; Bendzick, L.; Ettestad, B.; Hallstrom, C.; Zorko, N.A.; Rao, A.; Fujioka, N.; et al. NK-Cell-Mediated Targeting of Various Solid Tumors Using a B7-H3 Tri-Specific Killer Engager In Vitro and In Vivo. Cancers 2020, 12, 2659. [Google Scholar] [CrossRef]
- Ellwanger, K.; Reusch, U.; Fucek, I.; Wingert, S.; Ross, T.; Müller, T.; Schniegler-Mattox, U.; Haneke, T.; Rajkovic, E.; Koch, J.; et al. Redirected Optimized Cell Killing (ROCK®): A Highly Versatile Multispecific Fit-for-Purpose Antibody Platform for Engaging Innate Immunity; Taylor & Francis: Milton Park, UK, 2019; Volume 11, pp. 899–918. [Google Scholar] [CrossRef]
- Gauthier, L.; Morel, A.; Anceriz, N.; Rossi, B.; Blanchard-Alvarez, A.; Grondin, G.; Trichard, S.; Cesari, C.; Sapet, M.; Bosco, F.; et al. Multifunctional Natural Killer Cell Engagers Targeting NKp46 Trigger Protective Tumor Immunity. Cell 2019, 177, 1701–1713.e16. [Google Scholar] [CrossRef]
- Van Hall, T.; André, P.; Horowitz, A.; Ruan, D.F.; Borst, L.; Zerbib, R.; Narni-Mancinelli, E.; Van Der Burg, S.H.; Vivier, E. Monalizumab: Inhibiting the Novel Immune Checkpoint NKG2A. J. Immunother. Cancer 2019, 7, 263. [Google Scholar] [CrossRef]
- Kanchan, R.K.; Doss, D.; Khan, P.; Nasser, M.W.; Mahapatra, S. To Kill a Cancer: Targeting the Immune Inhibitory Checkpoint Molecule, B7-H3. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2022, 1877, 188783. [Google Scholar] [CrossRef]
- Dong, P.; Xiong, Y.; Yue, J.; Hanley, S.J.B.; Watari, H. B7H3 As a Promoter of Metastasis and Promising Therapeutic Target. Front. Oncol. 2018, 8, 264. [Google Scholar] [CrossRef] [PubMed]
- Flem-Karlsen, K.; Fodstad, Ø.; Tan, M.; Nunes-Xavier, C.E. B7-H3 in Cancer—Beyond Immune Regulation. Trends Cancer 2018, 4, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Picarda, E.; Ohaegbulam, K.C.; Zang, X. Molecular Pathways: Targeting B7-H3 (CD276) for Human Cancer Immunotherapy. Clin. Cancer Res. 2016, 22, 3425–3431. [Google Scholar] [CrossRef]
- Yang, S.; Wei, W.; Zhao, Q. B7-H3, a Checkpoint Molecule, as a Target for Cancer Immunotherapy. Int. J. Biol. Sci. 2020, 16, 1767–1773. [Google Scholar] [CrossRef]
- Capuano, C.; Pighi, C.; Battella, S.; De Federicis, D.; Galandrini, R.; Palmieri, G. Harnessing Cd16-Mediated Nk Cell Functions to Enhance Therapeutic Efficacy of Tumor-Targeting Mabs. Cancers 2021, 13, 2500. [Google Scholar] [CrossRef]
- Srpan, K.; Ambrose, A.; Karampatzakis, A.; Saeed, M.; Cartwright, A.N.R.; Guldevall, K.; De Matos, G.D.S.C.; Önfelt, B.; Davis, D.M. Shedding of CD16 Disassembles the NK Cell Immune Synapse and Boosts Serial Engagement of Target Cells. J. Cell Biol. 2018, 217, 3267–3283. [Google Scholar] [CrossRef] [PubMed]
- Romee, R.; Foley, B.; Lenvik, T.; Wang, Y.; Zhang, B.; Ankarlo, D.; Luo, X.; Cooley, S.; Verneris, M.; Walcheck, B.; et al. NK Cell CD16 Surface Expression and Function Is Regulated by a Disintegrin and Metalloprotease-17 (ADAM17). Blood 2013, 121, 3599–3608. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Correa, B.; Valhondo, I.; Hassouneh, F.; Lopez-Sejas, N.; Pera, A.; Bergua, J.M.; Arcos, M.J.; Bañas, H.; Casas-Avilés, I.; Durán, E.; et al. DNAM-1 and the TIGIT/PVRIG/TACTILE Axis: Novel Immune Checkpoints for Natural Killer Cell-Based Cancer Immunotherapy. Cancers 2019, 11, 877. [Google Scholar] [CrossRef] [PubMed]
- Stanietsky, N.; Simic, H.; Arapovic, J.; Toporik, A.; Levy, O.; Novik, A.; Levine, Z.; Beiman, M.; Dassa, L.; Achdout, H.; et al. The Interaction of TIGIT with PVR and PVRL2 Inhibits Human NK Cell Cytotoxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 17858–17863. [Google Scholar] [CrossRef]
- Chauvin, J.-M.; Zarour, H.M. TIGIT in Cancer Immunotherapy. J. Immunother. Cancer 2020, 8, e000957. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Xue, L.; Ding, X.; Zhang, J.; Jiang, L.; Liu, S.; Hou, H.; Jiang, B.; Cheng, L.; Zhu, Q.; et al. An Fc-Competent Anti-Human TIGIT Blocking Antibody Ociperlimab (BGB-A1217) Elicits Strong Immune Responses and Potent Anti-Tumor Efficacy in Pre-Clinical Models. Front. Immunol. 2022, 13, 828319. [Google Scholar] [CrossRef]
- Mu, S.; Liang, Z.; Wang, Y.; Chu, W.; Chen, Y.-L.; Wang, Q.; Wang, G.; Wang, C. PD-L1/TIGIT Bispecific Antibody Showed Survival Advantage in Animal Model. Clin. Transl. Med. 2022, 12, e754. [Google Scholar] [CrossRef]
- Rousseau, A.; Parisi, C.; Barlesi, F. Anti-TIGIT Therapies for Solid Tumors: A Systematic Review. ESMO Open 2023, 8, 101184. [Google Scholar] [CrossRef]
- Bogen, J.P.; Grzeschik, J.; Krah, S.; Zielonka, S.; Kolmar, H. Rapid Generation of Chicken Immune Libraries for Yeast Surface Display. Methods Mol. Biol. 2020, 2070, 289–302. [Google Scholar] [CrossRef]
- Bogen, J.P.; Storka, J.; Yanakieva, D.; Fiebig, D.; Grzeschik, J.; Hock, B.; Kolmar, H. Isolation of Common Light Chain Antibodies from Immunized Chickens Using Yeast Biopanning and Fluorescence-Activated Cell Sorting. Biotechnol. J. 2021, 16, 2000240. [Google Scholar] [CrossRef]
- Romera-Cárdenas, G.; Thomas, L.M.; Lopez-Cobo, S.; García-Cuesta, E.M.; Long, E.O.; Reyburn, H.T. Ionomycin Treatment Renders NK Cells Hyporesponsive. PLoS ONE 2016, 11, e0150998. [Google Scholar] [CrossRef]
- Gantke, T.; Weichel, M.; Herbrecht, C.; Reusch, U.; Ellwanger, K.; Fucek, I.; Eser, M.; Müller, T.; Griep, R.; Molkenthin, V.; et al. Trispecific Antibodies for CD16A-Directed NK Cell Engagement and Dual-Targeting of Tumor Cells. Protein Eng. Des. Sel. 2017, 30, 673–684. [Google Scholar] [CrossRef]
- Schubert, I.; Kellner, C.; Stein, C.; Kügler, M.; Schwenkert, M.; Saul, D.; Mentz, K.; Singer, H.; Stockmeyer, B.; Hillen, W.; et al. A Single-Chain Triplebody with Specificity for CD19 and CD33 Mediates Effective Lysis of Mixed Lineage Leukemia Cells by Dual Targeting; Taylor & Francis: Milton Park, UK, 2011; Volume 3, pp. 21–30. [Google Scholar] [CrossRef]
- Schmohl, J.U.; Felices, M.; Taras, E.; Miller, J.S.; Vallera, D.A. Enhanced ADCC and NK Cell Activation of an Anticarcinoma Bispecific Antibody by Genetic Insertion of a Modified IL-15 Cross-Linker. Mol. Ther. 2016, 24, 1312. [Google Scholar] [CrossRef]
- Tapia-Galisteo, A.; Compte, M.; Álvarez-Vallina, L.; Sanz, L. When Three Is Not a Crowd: Trispecific Antibodies for Enhanced Cancer Immunotherapy. Theranostics 2023, 13, 1028–1041. [Google Scholar] [CrossRef]
- Coënon, L.; Villalba, M. From CD16a Biology to Antibody-Dependent Cell-Mediated Cytotoxicity Improvement. Front. Immunol. 2022, 13, 913215. [Google Scholar] [CrossRef] [PubMed]
- Gauthier, L.; Virone-Oddos, A.; Beninga, J.; Rossi, B.; Nicolazzi, C.; Amara, C.; Blanchard-Alvarez, A.; Gourdin, N.; Courta, J.; Basset, A.; et al. Control of Acute Myeloid Leukemia by a Trifunctional NKp46-CD16a-NK Cell Engager Targeting CD123. Nat. Biotechnol. 2023, 41, 1296–1306. [Google Scholar] [CrossRef] [PubMed]
- Cancer Cell-Expressed B7-H3 Regulates the Differentiation of Tumor-Associated Macrophages in Human Colorectal Carcinoma. Available online: https://www.spandidos-publications.com/10.3892/ol.2017.6935 (accessed on 27 February 2024).
- Liu, X.; Zhelev, D.; Adams, C.; Chen, C.; Mellors, J.W.; Dimitrov, D.S. Effective Killing of Cells Expressing CD276 (B7-H3) by a Bispecific T Cell Engager Based on a New Fully Human Antibody. Transl. Oncol. 2021, 14, 101232. [Google Scholar] [CrossRef]
- Feng, Y.; Xie, K.; Yin, Y.; Li, B.; Pi, C.; Xu, X.; Huang, T.; Zhang, J.; Wang, B.; Gu, H.; et al. A Novel Anti-B7-H3 × Anti-CD3 Bispecific Antibody with Potent Antitumor Activity. Life 2022, 12, 157. [Google Scholar] [CrossRef]
- Liu, J.; Yang, S.; Cao, B.; Zhou, G.; Zhang, F.; Wang, Y.; Wang, R.; Zhu, L.; Meng, Y.; Hu, C.; et al. Targeting B7-H3 via Chimeric Antigen Receptor T Cells and Bispecific Killer Cell Engagers Augments Antitumor Response of Cytotoxic Lymphocytes. J. Hematol. Oncol. 2021, 14, 21. [Google Scholar] [CrossRef] [PubMed]
- Vivier, E.; Rebuffet, L.; Narni-Mancinelli, E.; Cornen, S.; Igarashi, R.Y.; Fantin, V.R. Natural Killer Cell Therapies. Nature 2024, 626, 727–736. [Google Scholar] [CrossRef]
- Yang, R.; Huang, S.; Huang, C.; Fay, N.S.; Wang, Y.; Putrevu, S.; Wright, K.; Zaman, M.S.; Cai, W.; Huang, B.; et al. Fc-Competent Multispecific PDL-1/TIGIT/LAG-3 Antibodies Potentiate Superior Anti-Tumor T Cell Response. Sci. Rep. 2023, 13, 9865. [Google Scholar] [CrossRef]
- Datta-Mannan, A.; Brown, R.; Key, S.; Cain, P.; Feng, Y. Pharmacokinetic Developability and Disposition Profiles of Bispecific Antibodies: A Case Study with Two Molecules. Antibodies 2022, 11, 2. [Google Scholar] [CrossRef]
- Felices, M.; Lenvik, T.R.; Davis, Z.B.; Miller, J.S.; Vallera, D.A. Generation of BiKEs and TriKEs to Improve NK Cell-Mediated Targeting of Tumor Cells. In Methods in Molecular Biology; Springer: Berlin, Germany, 2016; pp. 333–346. [Google Scholar]
- Waldmann, T.A.; Strober, W. Metabolism of Immunoglobulins. Prog. Immunol. 1971, 13, 891–903. [Google Scholar] [CrossRef]
- Bogen, J.P.; Elter, A.; Grzeschik, J.; Hock, B.; Kolmar, H. Humanization of Chicken-Derived Antibodies by Yeast Surface Display. Methods Mol. Biol. 2022, 2491, 335–360. [Google Scholar] [CrossRef] [PubMed]
- Benatuil, L.; Perez, J.M.; Belk, J.; Hsieh, C.M. An Improved Yeast Transformation Method for the Generation of Very Large Human Antibody Libraries. Protein Eng. Des. Sel. 2010, 23, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Dyballa, N.; Metzger, S. Fast and Sensitive Colloidal Coomassie G-250 Staining for Proteins in Polyacrylamide Gels. J. Vis. Exp. 2009, 30, 2–5. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ulitzka, M.; Harwardt, J.; Lipinski, B.; Tran, H.; Hock, B.; Kolmar, H. Potent Apoptosis Induction by a Novel Trispecific B7-H3xCD16xTIGIT 2+1 Common Light Chain Natural Killer Cell Engager. Molecules 2024, 29, 1140. https://doi.org/10.3390/molecules29051140
Ulitzka M, Harwardt J, Lipinski B, Tran H, Hock B, Kolmar H. Potent Apoptosis Induction by a Novel Trispecific B7-H3xCD16xTIGIT 2+1 Common Light Chain Natural Killer Cell Engager. Molecules. 2024; 29(5):1140. https://doi.org/10.3390/molecules29051140
Chicago/Turabian StyleUlitzka, Michael, Julia Harwardt, Britta Lipinski, Hue Tran, Björn Hock, and Harald Kolmar. 2024. "Potent Apoptosis Induction by a Novel Trispecific B7-H3xCD16xTIGIT 2+1 Common Light Chain Natural Killer Cell Engager" Molecules 29, no. 5: 1140. https://doi.org/10.3390/molecules29051140
APA StyleUlitzka, M., Harwardt, J., Lipinski, B., Tran, H., Hock, B., & Kolmar, H. (2024). Potent Apoptosis Induction by a Novel Trispecific B7-H3xCD16xTIGIT 2+1 Common Light Chain Natural Killer Cell Engager. Molecules, 29(5), 1140. https://doi.org/10.3390/molecules29051140