Abstract
A novel, efficient and environmentally friendly solvent-free and catalyst-free approach for the synthesis of structurally diverse gem-difluorinated and polyfluoroarylated derivatives with readily available nucleophilic and electrophilic fluorine-containing reaction partners, difluoroenoxysilane and pentafluorobenzaldehyde, is described. This neat protocol is induced by the direct hydrogen-bond interactions between fluorinated and non-fluorinated reactants without the use of heavy metal catalysts or volatile organic solvents and with no need for column chromatographic separation for most cases.
1. Introduction
The last decade has witnessed growing interest in developing new solvent- and catalyst-free organic transformations among researchers both in academia and industry [1,2,3]. This is not only because of the increased demand for sustainable and environmentally benign synthetic approaches in green synthesis, but also because solvent and catalyst-free synthesis could tremendously accelerate the reaction rates, minimize the side reactions caused by the solvents and avoid the use of expensive, metallic and corrosive catalysts. Furthermore, when reactions are conducted under solvent-free conditions, solvation phenomena will not need to be considered, which may provide an opportunity for chemists to realize the reactions that could not be achieved in conventional solvents before. Therefore, developing novel reactions without requiring the usage of solvents and catalysts is an important subject in green chemistry [4,5,6].
The widespread applications of fluorine-containing organic molecules and their derivatives in pharmaceuticals, agrochemicals, polymers, dyes, and optical functional materials have raised demand greatly in recent years [7,8]. Up to now, a series of synthetic methods for accessing new fluorine-containing compounds have been developed [9,10,11]. Among the existing organofluorine compounds, gem-difluoromethylene and perfluorinated arene-containing molecules have long been recognized as fascinating targets for organic synthesis due to their special structures and important properties (Figure 1) [12,13,14].
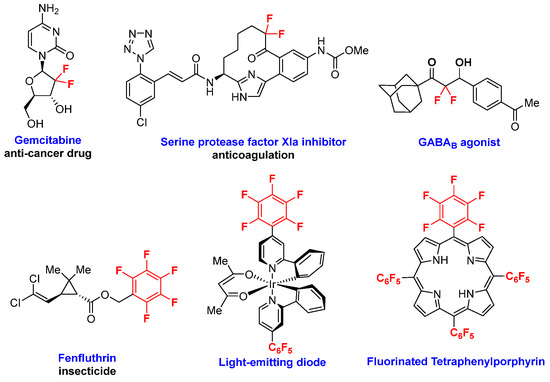
Figure 1.
Representative functional molecules containing gem-difluoromethylene and perfluorinated arene scaffolds.
Accordingly, two general approaches have been developed for the construction of such compounds [12,15]. The first one is the nucleophilic difluoroalkylation and perfluoroarylation of electrophilic acceptors, and the second strategy is based on the addition of different nucleophiles to difluoromethylene- and perfluorinated arene-containing reaction partners. Due to the low reactivities of most fluorine-containing reactants, catalysts, promoters or additives are essential for enabling the reactions to proceed smoothly (Scheme 1a) [16,17,18,19]. Although significant progress has been made, these methodologies usually show some drawbacks such as requiring expensive metal Lewis acid catalysts, strong acid catalysts, volatile organic solvents, relatively high temperatures and longer reaction periods. To further activate fluorine-containing reactants under mild conditions, a new activation model for nucleophilic difluoroenoxysilane and electrophilic difluoroacetaldehyde ethyl hemiacetal has been developed by our group, using 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) as the organocatalyst or promoter in various cases due to its high H-bond donating ability, mild acidity and strong cation stabilization (Scheme 1b) [20,21]. In those processes, fluorine-containing reactants were activated via strong hydrogen bonding and the competing side reactions were significantly suppressed. In addition, it should be pointed out that although quite a number of neat reactions have been developed to generate structurally diverse molecules over the past decades, reports on the efficient construction of fluorine-containing organic molecules without solvent or catalyst are fairly limited [22,23,24,25]. Thus, considering the formation of hydrogen bonds between the reactants and HFIP, we were interested in determining whether the non-fluorinated reactants could be activated by fluorinated reactants directly through the intermolecular hydrogen-bond interactions without the participation of additional catalysts (Scheme 1c). In this context, we report a novel solvent- and catalyst-free method for the efficient synthesis of gem-difluorinated and polyfluoroarylated compounds with nucleophilic and electrophilic fluorine-containing reaction partners. To the best of our knowledge, this is the first report on the direct reaction with nucleophilic difluoroenoxysilane and electrophilic pentafluorobenzaldehyde in neat conditions without a solvent and catalyst.
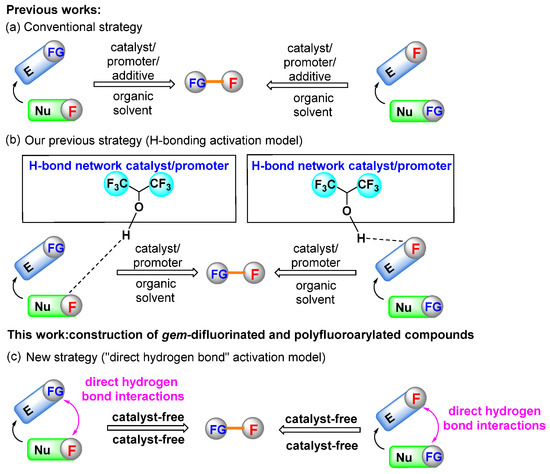
Scheme 1.
Synthetic strategies for the construction of organofluorine compounds.
2. Results and Discussion
To find out the feasibility of the direct synthesis of gem-difluorinated compounds using nucleophilic difluoroenoxysilanes under solvent- and catalyst-free conditions, the reaction of glyoxal monohydrate 1a with difluoroenoxysilane 2a was chosen as a model reaction. To our delight, the reaction under neat conditions using 2.0 equiv. of 2a at room temperature (Table 1, entry 1) led to the direct formation of gem-difluorinated 2-hydroxy-1,4-dicarbonyl adduct 3a in 91% yield, smoothly, after only 4 h. The reaction time was greatly reduced in comparison with our previous experiment results, using HFIP as the catalyst and DCM as solvent [26]. Then, a reduced amount of difluoroenoxysilane 2a (1.0 equiv, entry 2) was investigated to obtain a higher reaction utilization. The desired product, 3a, was obtained in 89% yield. A reaction at a higher temperature (60 °C) by reducing the reaction duration was not helpful due to the partial decomposition of the both reactants (entry 3). These studies have shown that considerable rate acceleration is formed under neat conditions, compared to the reported results in organic solvents with different catalysts. Further, the reactions in organic solvents and water (entries 4–9) delivered the gem-difluorinated products in rather low yields. On the basis of these investigations, reactions under neat conditions using 1.0 equiv. of difluoroenoxysilane were chosen as optimized conditions for direct nucleophilic addition reactions at room temperature. More importantly, this solvent-free procedure with minimal usage of difluoroenoxysilanes makes the separation and purification of the final products even easier using only simple filtration with an H2O work-up (Figure 2 shows the main reaction process; see the Supplementary Materials for details). Finally, for comparison, several Lewis acid and Brønsted acid catalysts such as Sc(OTf)3, PhB(OH)2 and PTSA were tested in DCM with 10 mol % amount and the desired product was obtained in 13–42% yield, which showed the superiority of the developed neat reactions in the absence of catalysts.

Table 1.
Optimization of the reaction conditions a.

Figure 2.
The main reaction process for the synthesis of compound 3a.
The scope of the solvent- and catalyst-free difluoroalkylation reaction was then explored with a range of glyoxal monohydrates 1, including aromatic (1a–1o), heteroaromatic (1p–1q), aliphatic derivatives (1u), and difluoroenoxysilane derivatives 2, as outlined in Scheme 2. The results indicate that the neat reaction is highly efficient (2–6 h of reaction time) for all series of reactants, proceeding at room temperature to afford gem-difluorinated 2-hydroxy-1,4-dicarbonyl products 3 in moderate to good yields and with high purity, without the need for chromatographic purification in most cases. It was found that electron-withdrawing groups which substituted glyoxal monohydrates (1i–1j and 1l–1m) afforded the desired products in higher yield than the substrates with electron-donating groups (1b–1h). And this neat reaction was found to be sensitive to the steric effect. Ortho-substituted phenyl glyoxal monohydrate gave a much lower yield (3d, 70% yield) compared with the para- and meta-substituted substrates (3b, 84%; 3c, 81%). Finally, a gram-scale synthesis of 3a was conducted under the neat conditions without the need for chromatographic purification to further illustrate the practicability of this solvent- and catalyst-free reaction (5.0 mmol scale, 1.17 g, 81% yield). It should be noted that substrates 2 with electron-donating groups (Me, MeO) were also tested under the neat condition; only trace products were detected, with most difluoroenoxysilanes recovered.
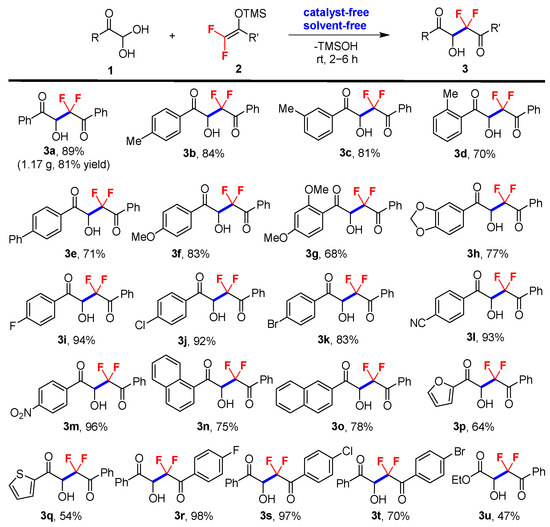
Scheme 2.
Standard reaction conditions: glyoxal monohydrates 1 (1.0 mmol) and difluoroenoxysilanes 2 (1.0 mmol) at room temperature. The yields are isolated yields.
Having developed an efficient procedure for the preparation of gem-difluorinated 2-hydroxy-1,4-dicarbonyl compounds, we next turned our attention to investigating the synthesis of polyfluoroarylated compounds with pentafluorobenzaldehyde through the nucleophilic attack pathway. As depicted in Scheme 3, pentafluorobenzene-derived bis(tetrahydroquinolinyl)methane 6a, bis(pyrrolyl)methanes 6b and bis(indolyl)methanes 6c–6f were obtained in moderate yields within 0.5–6 h. Meanwhile, for N,N-dimethylaniline 4g without free NH moiety in the molecular skeleton, the reaction rate was greatly slowed down. Both the bis(aryl)methane product 6g (48% yield) and the direct hydroxyarylation product 6g’ (13% yield) were produced despite the reaction time extending to even six times longer than before (36 h). It should be noted that benzothiazole-bearing trifluoromethyl ketone hydrate was also a viable substrate for this neat reaction, although only 41% yield was achieved for product 6h. And it should be noted that a relatively higher temperature (80 °C) was required for the reaction of pentafluorobenzaldehyde with these different reactants in order to improve the reaction yields (see the Supplementary Materials for complete details of reaction optimization).
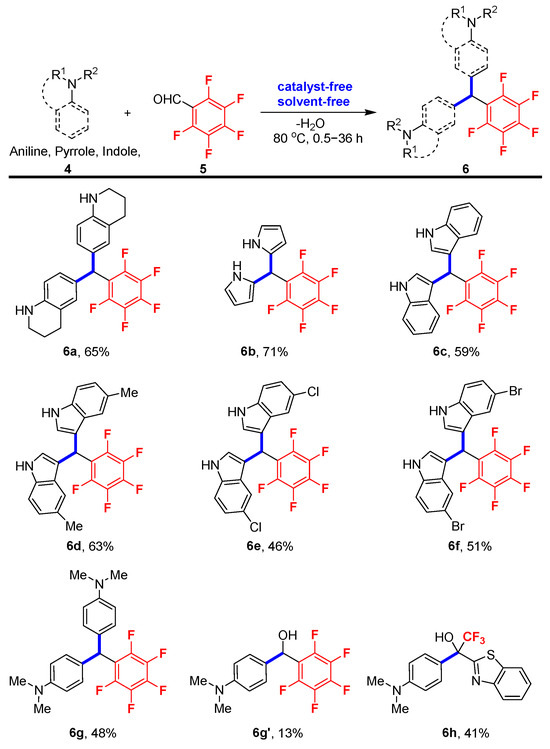
Scheme 3.
Standard reaction conditions: aniline, pyrrole or indole 4 (1.0 mmol) and pentafluorobenzaldehyde 5 (0.5 mmol) at 80 °C. The yields are isolated yields.
Several control experiments were carried out to better understand the mechanisms of solvent- and catalyst-free synthesis of gem-difluorinated and polyfluoroarylated molecules (Scheme 4). First, the role of the fluorine atom with the reaction partners was investigated with non-fluorinated analogues 7 and 8, and no reactions occurred after 24 h (Scheme 4a–c). These results proved the indispensable role of the fluorine atom in reactants to smoothly promote this neat reaction, and the installation of fluorine atoms in reactants would have a dramatic impact on reactivity. Next, the reaction with benzaldehyde 8 still could not proceed under the same condition (Scheme 4d). A similar result was obtained with anhydrous phenylglyoxal 1a′, indicating that the hydrate form of reactants plays a key role in promoting solvent- and catalyst-free synthesis of gem-difluorinated compounds with nucleophilic difluoroenoxysilanes (Scheme 4e). Interestingly, it was found that the reaction could proceed smoothly to afford 3a with the addition of 1.0 equiv. H2O, albeit in a lower yield (38%). The reaction conducted under standard conditions without the addition of H2O gave 3a in fairly low yield (6%). Taken together, these preliminary results clearly revealed the important role of the hydrate of glyoxal to promote the neat reaction.
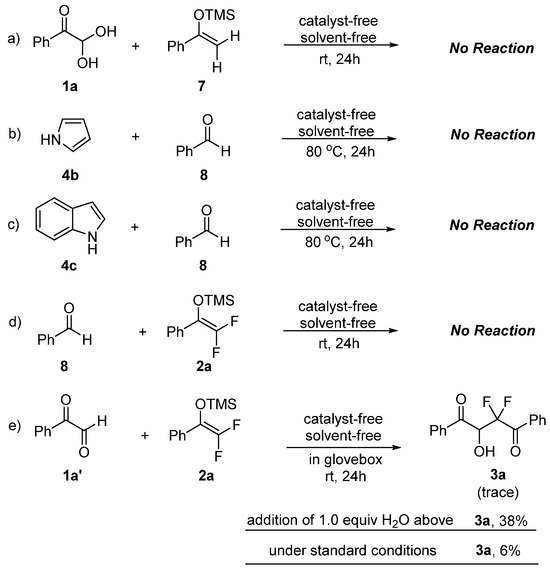
Scheme 4.
Control experiments.
Based on the current control experiments and previous studies [27,28,29], a possible mechanism for the solvent- and catalyst-free synthesis of gem-difluorinated 2-hydroxy-1,4-dicarbonyl products 3 is illustrated in Scheme 5. The glyoxal monohydrates are in equilibrium with their aldehyde form [30], which may drive the occurrence of the neat reaction. Arylglyoxals and difluoroenoxysilanes could be activated through the simultaneous hydrogen-bonding interaction and C–F···H–O interaction between the substrates [31,32]. The water produced from glyoxal monohydrates may help to activate the reaction partners. Then, the additional intermediate A could be formed through an intramolecular-like nucleophilic attack of difluoroenoxysilane 2a on arylglyoxal 1a′. Finally, the protonation of the generated intermediate A would provide the desired gem-difluorinated product 3a smoothly. The synthesis of polyfluoroarylated compounds with pentafluorobenzaldehyde may be promoted through the C–F···H–N interaction between fluorinated and non-fluorinated reactants in a similar pathway.
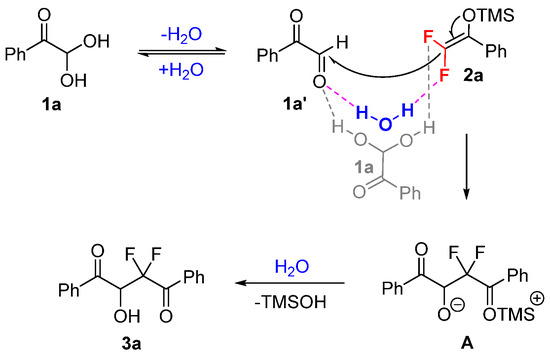
Scheme 5.
Plausible mechanism.
3. Materials and Methods
3.1. Materials
All reagents were obtained from commercial sources and used directly without further purification unless otherwise noted. 1H, 13C and 19F were recorded on a Bruker AV 400 MHz instrument at 400 MHz (1H NMR) and 100 MHz (13C NMR), as well as 376 MHz (19F NMR). Chemical shifts were reported in ppm downfield from internal Me4Si and external CCl3F, respectively.
3.2. General Procedure for the Synthesis of gem-Difluorinated 2-Hydroxy-1,4-dicarbonyl Products
To a 5.0 mL vial was added glyoxal monohydrates 1 (1.0 mmol) and difluoroenoxysilanes 2 (1.0 mmol, 1.0 equiv.). The resulting mixture was stirred at room temperature until the completion of the reaction (monitored by TLC, approximately 2–6 h). Then, water (1.0 mL) was added to the liquefied mixture, followed by vigorous stirring for about 0.5 hour until precipitate was generated. The precipitate was directly filtered, washed with saturated sodium bicarbonate (3 × 2.0 mL) and dried under vacuum to afford the desired pure products 3.
3.3. General Procedure for the Synthesis of Polyfluoroarylated Compounds
To a 5.0 mL oven-dried Schlenk flask was added aniline, pyrrole or indole 4 (1.0 mmol) and pentafluorobenzaldehyde 5 (0.5 mmol) under an argon atmosphere. The resulting mixture was stirred at 80 °C until the completion of the reaction (monitored by TLC, approximately 0.5–36 h). Then, the reaction mixture was separated by column chromatography (petroleum ether/ethyl acetate) to produce the pure polyfluoroarylated products.
4. Conclusions
In summary, we have successfully developed a facile and environmentally friendly method for the synthesis of a series of gem-difluorinated and polyfluoroarylated compounds using nucleophilic difluoroenoxysilane and electrophilic pentafluorobenzaldehyde under neat conditions. Readily available reactants, a broad substrate scope, solvent- and catalyst-free conditions and a simple work-up procedure without column chromatographic separation are the prominent features of this green and concise protocol.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules29030697/s1, S2. General information, S2–S9. General procedure for the synthesis of gem-difluorinated 2-hydroxy-1,4-dicarbonyl products, S9. Optimization of the reaction conditions for the synthesis of polyfluoroarylated compounds, S10–S14. General procedure for the synthesis of polyfluoroarylated compounds, S14–S59. NMR spectra of the related compounds, [33,34,35,36].
Author Contributions
Informed consent was obtained from all subjects involved in the study. Writing—review and editing, J.L.; investigation and methodology, L.L. and J.L.; data curation, L.L. and J.L.; funding acquisition, L.L.; conceptualization, J.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Natural Science Foundation of Zhejiang Province (LQ20B020005), and a horizontal research project “70301603” from Tianjin University of Technology with Hailin International Culture Media Co., Ltd.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are contained within the article and Supplementary Materials.
Acknowledgments
We express gratitude to Lei Ren and Hua Xie.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Sarkar, A.; Santra, S.; Kundu, S.K.; Hajra, A.; Zyryanov, G.V.; Chupakhin, O.N.; Charushin, V.N.; Majee, A. A decade update on solvent and catalyst-free neat organic reactions: A step forward towards sustainability. Green Chem. 2016, 18, 4475–4525. [Google Scholar] [CrossRef]
- Cao, Z.; Zhu, Q.; Lin, Y.W.; He, W.M. The concept of dual roles design in clean organic preparation. Chin. Chem. Lett. 2019, 30, 2132–2138. [Google Scholar] [CrossRef]
- Baruah, B.; Deb, M.L. Catalyst-free and additive-free reactions enabling C−C bond formation: A journey towards a sustainablefuture. Org. Biomol. Chem. 2021, 19, 1191–1229. [Google Scholar] [CrossRef]
- Lu, B.; Xie, Z.; Lu, J.; Liu, J.; Cui, S.; Ma, Y.; Hu, X.; Liu, Y.; Zhong, K. Highly atom-economic, catalyst-free, and solvent-free synthesis of phthalazinones. ACS Sustain. Chem. Eng. 2019, 7, 134–138. [Google Scholar] [CrossRef]
- Liao, T.M.; Ma, W.J.; Gao, Y.N.; Bian, M.; Jiang, M.; Liu, J.T.; Chen, H.Y.; Liu, Z.J. Facile synthesis of (polyfluoro)alkanesulfinyl 4-isoxazolines: A stepwise solvent- and catalyst-free approach or a one-pot process in water. Green Chem. 2023, 25, 5233–5239. [Google Scholar] [CrossRef]
- Li, M.Y.; Li, J.T.; Gu, A.; Nong, X.M.; Zhai, S.Y.; Yue, Z.Y.; Feng, C.G.; Liu, Y.B.; Lin, G.Q. Solvent-free and catalyst-free direct alkylation of alkenes. Green Chem. 2023, 25, 7073–7078. [Google Scholar] [CrossRef]
- Gillis, E.P.; Eastman, K.J.; Hill, M.D.; Donnelly, D.J.; Meanwell, N.A. Applications of fluorine in medicinal chemistry. J. Med. Chem. 2015, 58, 8315–8359. [Google Scholar] [CrossRef] [PubMed]
- Ni, C.; Hu, J. The unique fluorine effects in organic reactions: Recent facts and insights into fluoroalkylations. Chem. Soc. Rev. 2016, 45, 5441–5454. [Google Scholar] [CrossRef] [PubMed]
- Belhomme, M.-C.; Besset, T.; Poisson, T.; Pannecoucke, X. Recent progress toward the introduction of functionalized difluoromethylated building blocks onto C(sp2) and C(sp) centers. Chem.-Eur. J. 2015, 21, 12836–12865. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Xiao, Y.-L.; Zhang, X. Transition-metal (Cu, Pd, Ni)-catalyzed difluoroalkylation via cross-coupling with difluoroalkyl halides. Acc. Chem. Res. 2018, 51, 2264–2278. [Google Scholar] [CrossRef] [PubMed]
- Xiao, H.; Zhang, Z.; Fang, Y.; Zhu, L.; Li, C. Radical Trifluoromethylation. Chem. Soc. Rev. 2021, 50, 6308–6319. [Google Scholar] [CrossRef]
- Pattison, G. Methods for the synthesis of α,α-difluoroketones. Eur. J. Org. Chem. 2018, 2018, 3520–3540. [Google Scholar] [CrossRef]
- Liu, C.; Zhang, B. Facile access to fluoroaromatic molecules by transition-metal-free C–F bond cleavage of polyfluoroarenes: An efficient, green, and sustainable protocol. Chem. Rec. 2016, 16, 667–687. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Zhang, M.; Tang, X.; Piper, J.L.; Peng, Z.H.; Ma, J.A.; Wu, J.; Zhang, F.G. Photoinduced defluorinative branch-selective olefination of multifluoro (hetero)arenes. Org. Lett. 2023, 25, 883–888. [Google Scholar] [CrossRef]
- Moseev, T.D.; Varaksin, M.V.; Gorlov, D.A.; Charushin, V.N.; Chupakhin, O.N. Recent advances in the functionalization of polyfluoro(aza)aromatics via C-C coupling strategies. Org. Biomol. Chem. 2021, 19, 4429–4459. [Google Scholar] [CrossRef] [PubMed]
- Decostanzi, M.; Campagne, J.M.; Leclerc, E. Fluorinated enol ethers: Their synthesis and reactivity. Org. Biomol. Chem. 2015, 13, 7351–7380. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.S.; Yu, J.S.; Zhou, J. Catalytic selective mono- and difluoroalkylation using fluorinated silyl enol ethers. Chem. Commun. 2019, 55, 13638–13648. [Google Scholar] [CrossRef]
- Du, G.F.; Xing, F.; Gu, C.Z.; Dai, B.; He, L. N-heterocyclic carbenecatalysed pentafluorophenylation of aldehydes. RSC Adv. 2015, 5, 35513–35517. [Google Scholar] [CrossRef]
- Liu, Z.Q.; Kole, G.K.; Budiman, Y.P.; Tian, Y.M.; Friedrich, A.; Luo, X.L.; Westcott, S.A.; Radius, U.; Marder, T.B. Transition metal catalyst-free, base-promoted 1,2-additions of polyfluorophenylboronates to aldehydes and ketones. Angew. Chem. Int. Ed. 2021, 60, 16529–16538. [Google Scholar] [CrossRef]
- Li, J.; Xi, W.; Zhong, R.; Yang, J.; Wang, L.; Ding, H.; Wang, Z. HFIP-catalyzed direct dehydroxydifluoroalkylation of benzylic and allylic alcohols with difluoroenoxysilanes. Chem. Commun. 2021, 57, 1050–1053. [Google Scholar] [CrossRef]
- Wu, H.; Hong, P.; Xi, W.; Li, J. Divergent synthesis of gem-difluorinated oxa-spirocyclohexadienones by one-pot sequential reactions of p-hydroxybenzyl alcohols with difluoroenoxysilanes. Org. Lett. 2022, 24, 2488–2493. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Liu, L.; Zhang, H.; Wu, Y.; Wang, D.; Chen, Y. Rapid and convenient synthesis of aryl- and heteroaryl-α-hydroxy-α-trifluoromethyl acetate via Friedel–Crafts alkylation under solvent- and catalyst-free conditions. Tetrahedron Lett. 2006, 47, 2511–2514. [Google Scholar] [CrossRef]
- Stavber, G.; Stavber, M.; Stavber, S. Solvent-free fluorination of organic compounds using N–F reagents. Tetrahedron Lett. 2007, 48, 2671–2673. [Google Scholar] [CrossRef]
- Yang, X.; Chen, Z.; Cai, Y.; Huang, Y.; Shibata, N. An aza-Michael addition protocol to fluoroalkylated β-amino acid derivatives and enantiopure trifluoromethylated N-heterocycles. Green Chem. 2014, 16, 4530–4534. [Google Scholar] [CrossRef]
- Matador, E.; Monge, D.; Fernández, R.; Lassaletta, J.M. Solvent-free synthesis of quaternary α-hydroxy α-trifluoromethyl diazenes: The key step of a nucleophilic formylation strategy. Green Chem. 2016, 18, 4042–4050. [Google Scholar] [CrossRef]
- Yang, J.; Liu, S.; Hong, P.; Li, J.; Wang, Z.; Ren, J. Synthesis of 2,2-difluoro-3-hydroxy-1,4-diketones via an HFIP-catalyzed Mukaiyama Aldol reaction of glyoxal monohydrates with difluoroenoxysilanes. J. Org. Chem. 2022, 87, 1144–1153. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.S.; Liu, Y.L.; Tang, J.; Wang, X.; Zhou, J. Highly efficient “on water” catalyst-free nucleophilic addition reactions using difluoroenoxysilanes: Dramatic fluorine effects. Angew. Chem. Int. Ed. 2014, 53, 9512–9516. [Google Scholar] [CrossRef]
- Wang, P.; Tao, W.J.; Sun, X.L.; Liao, S.; Tang, Y. A highly efficient and enantioselective intramolecular Cannizzaro reaction under TOX/Cu(II) catalysis. J. Am. Chem. Soc. 2013, 135, 16849–16852. [Google Scholar] [CrossRef]
- Rong, M.Y.; Li, J.S.; Zhou, Y.; Zhang, F.G.; Ma, J.A. Catalytic enantioselective synthesis of difluoromethylated tetrasubstituted stereocenters in isoindolones enabled by a multiple-fluorine system. Org. Lett. 2020, 22, 9010–9015. [Google Scholar] [CrossRef]
- Zhao, P.; Zhou, Y.; Yu, X.X.; Huang, C.; Wu, Y.D.; Yin, G.D.; Wu, A.X. Iodine-Promoted Multicomponent Synthesis of 2,4-Diamino-1,3,5-triazines. Org. Lett. 2020, 22, 8528–8532. [Google Scholar] [CrossRef]
- Hao, Y.J.; Yu, J.S.; Zhou, Y.; Wang, X.; Zhou, J. Influence of C−F···H−X interactions on organic reactions. Acta Chim. Sin. 2018, 76, 925–939. [Google Scholar] [CrossRef]
- Zhang, Q.X.; Li, Y.; Wang, J.; Yang, C.; Liu, C.J.; Li, X.; Cheng, J.P. B(C6F5)3/Chiral Phosphoric Acid Catalyzed Ketimine–Ene Reaction of 2-Aryl-3H-indol-3-ones and α-Methylstyrenes. Angew. Chem. Int. Ed. 2020, 59, 4550–4556. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Panda, G. Linearization of carbohydrate derived polycyclic frameworks. RSC Adv. 2014, 4, 31892. [Google Scholar] [CrossRef]
- Jagt, D.L.V.; Han, L.-P.B.; Lehman, C.H. Effects of substituents on the rates of disproportionation of substituted phenylglyoxals in alkaline. J. Org. Chem. 1972, 37, 4100. [Google Scholar] [CrossRef][Green Version]
- Tafelska-Kaczmarek, A.; Prewysz-Kwinto, A.; Skowerski, K.; Pietrasiak, K.; Kozakiewicz, A.; Zaidlewicz, M. Asymmetric synthesis of β-amino alcohols by the transfer hydrogenation of α-keto imines, Tetrahedron. Asymmetry 2010, 21, 2244. [Google Scholar] [CrossRef]
- Amii, H.; Kobayashi, T.; Hatamoto, Y.; Uneyama, K. Mg0-promoted selective C–F bond cleavage of trifluoromethyl ketones: A convenient method for the synthesis of 2,2-difluoro enol silanes. Chem. Commun. 1999, 1323–1324. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).