

Comparative Investigation of Ultrafast Excited-State Electron Transfer in Both Polyfluorene-Graphene Carboxylate and Polyfluorene-DCB Interfaces

Abstract
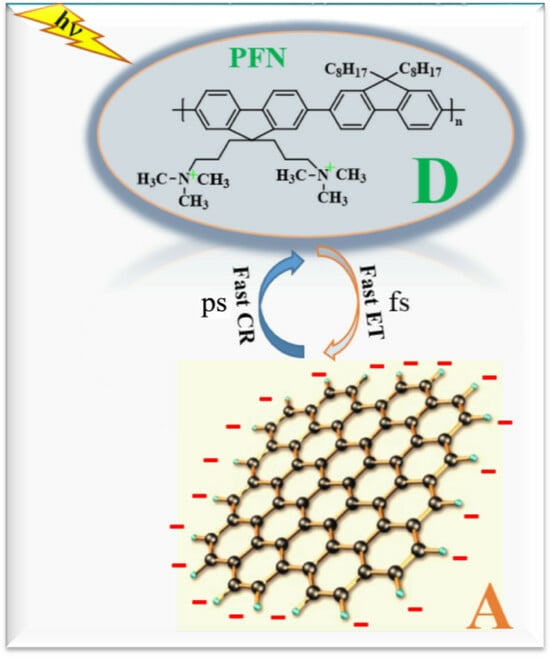
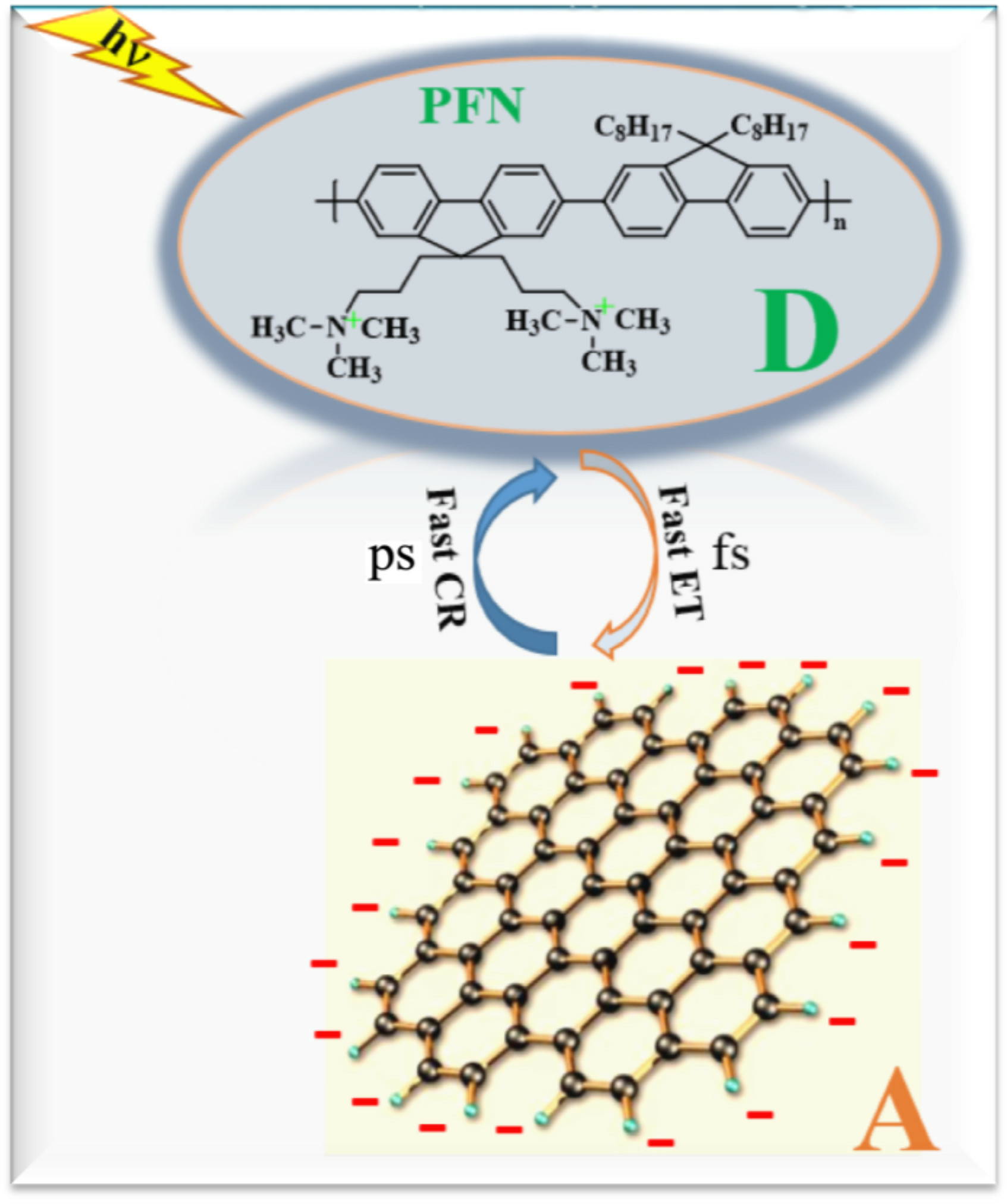{kind=link}
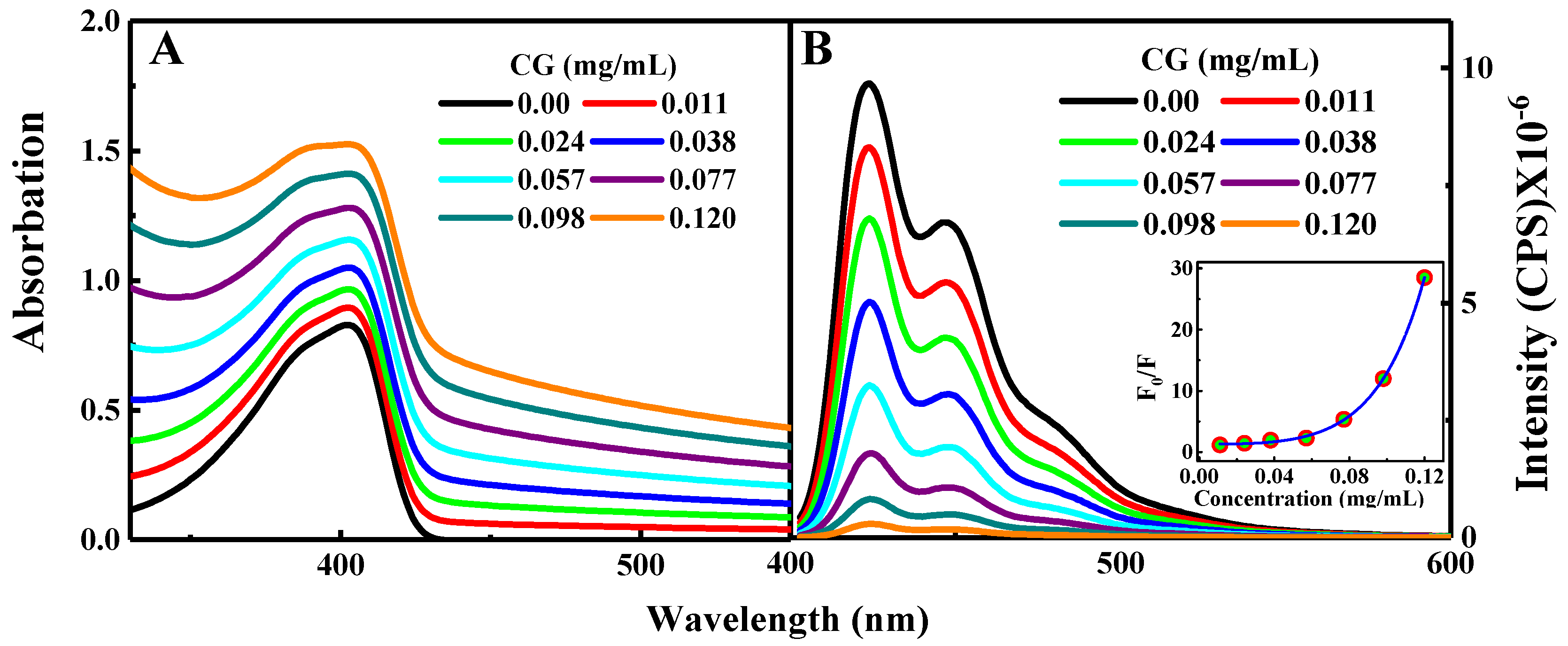{kind=link}
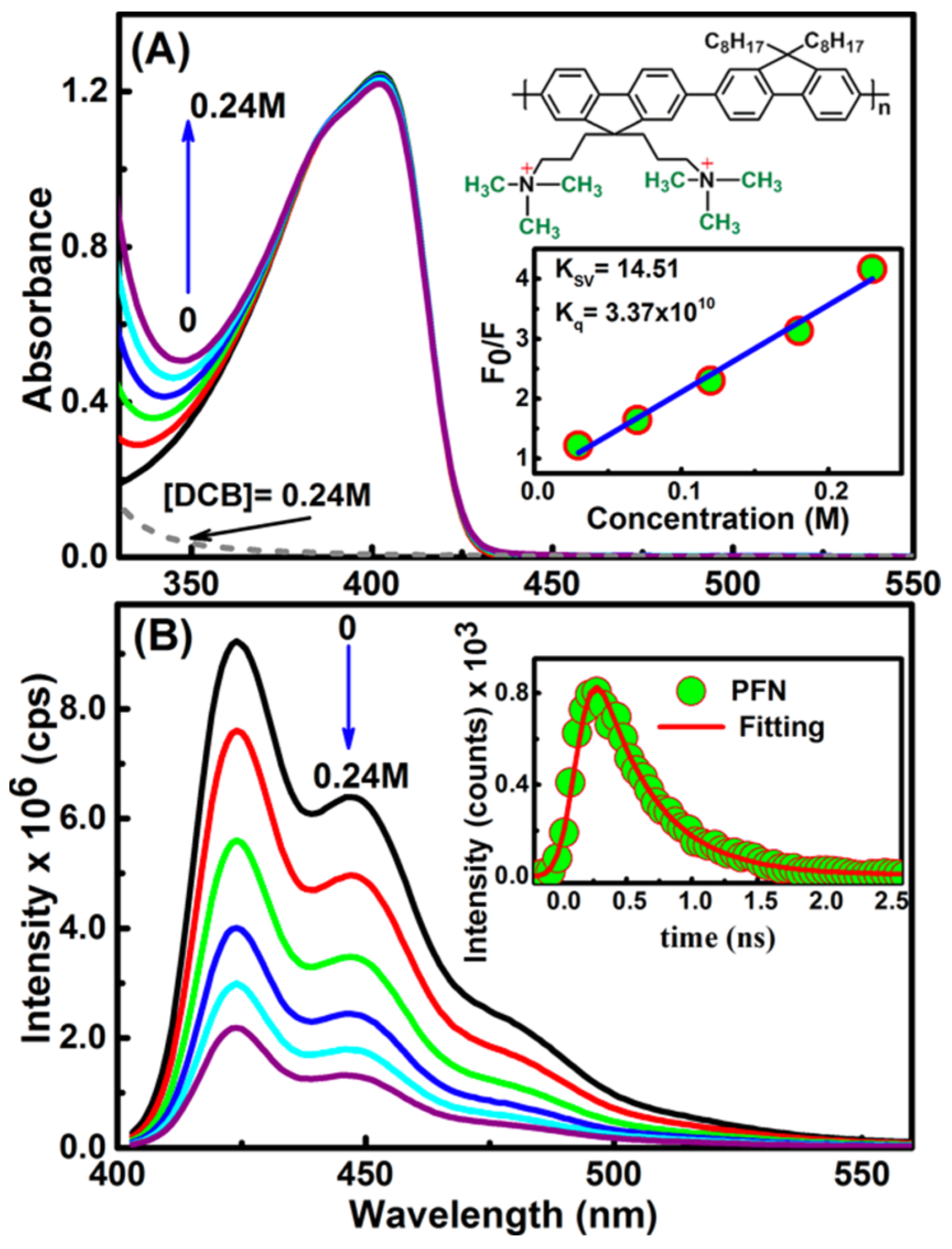{kind=link}
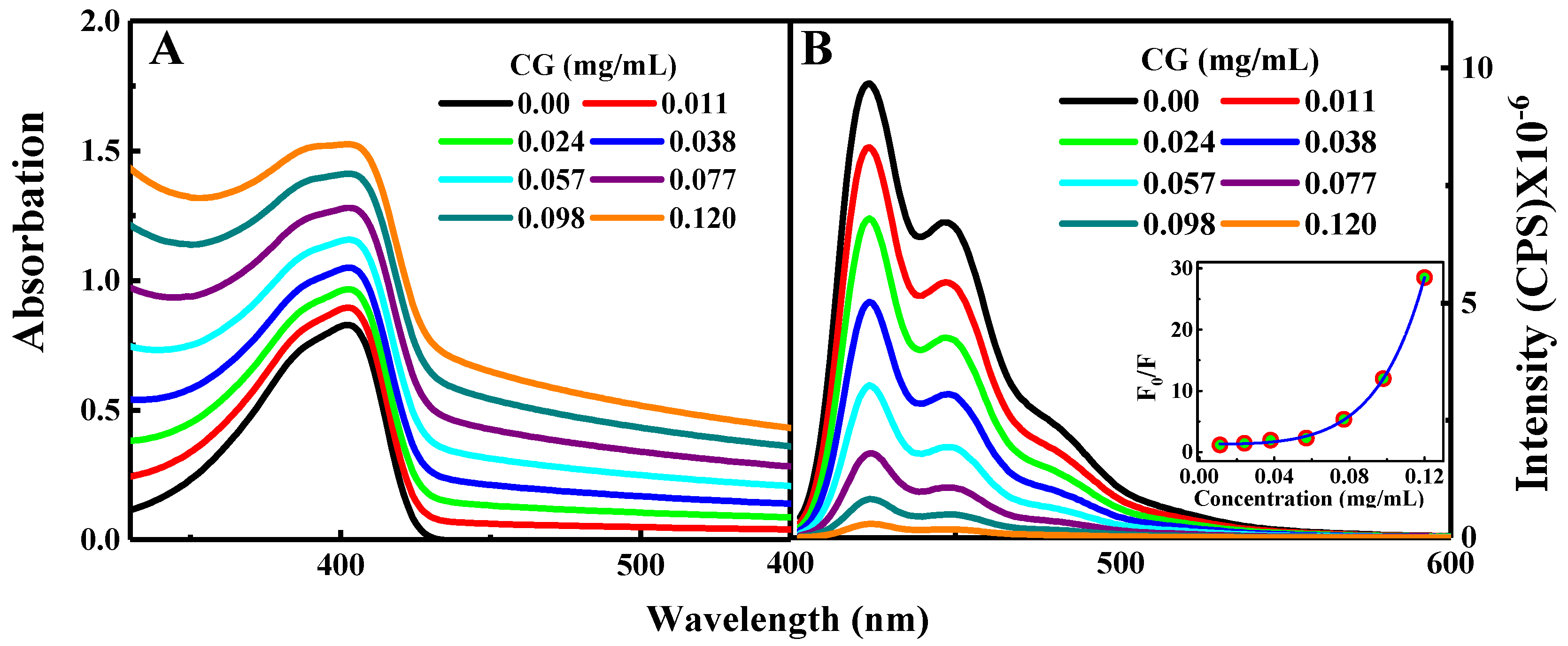
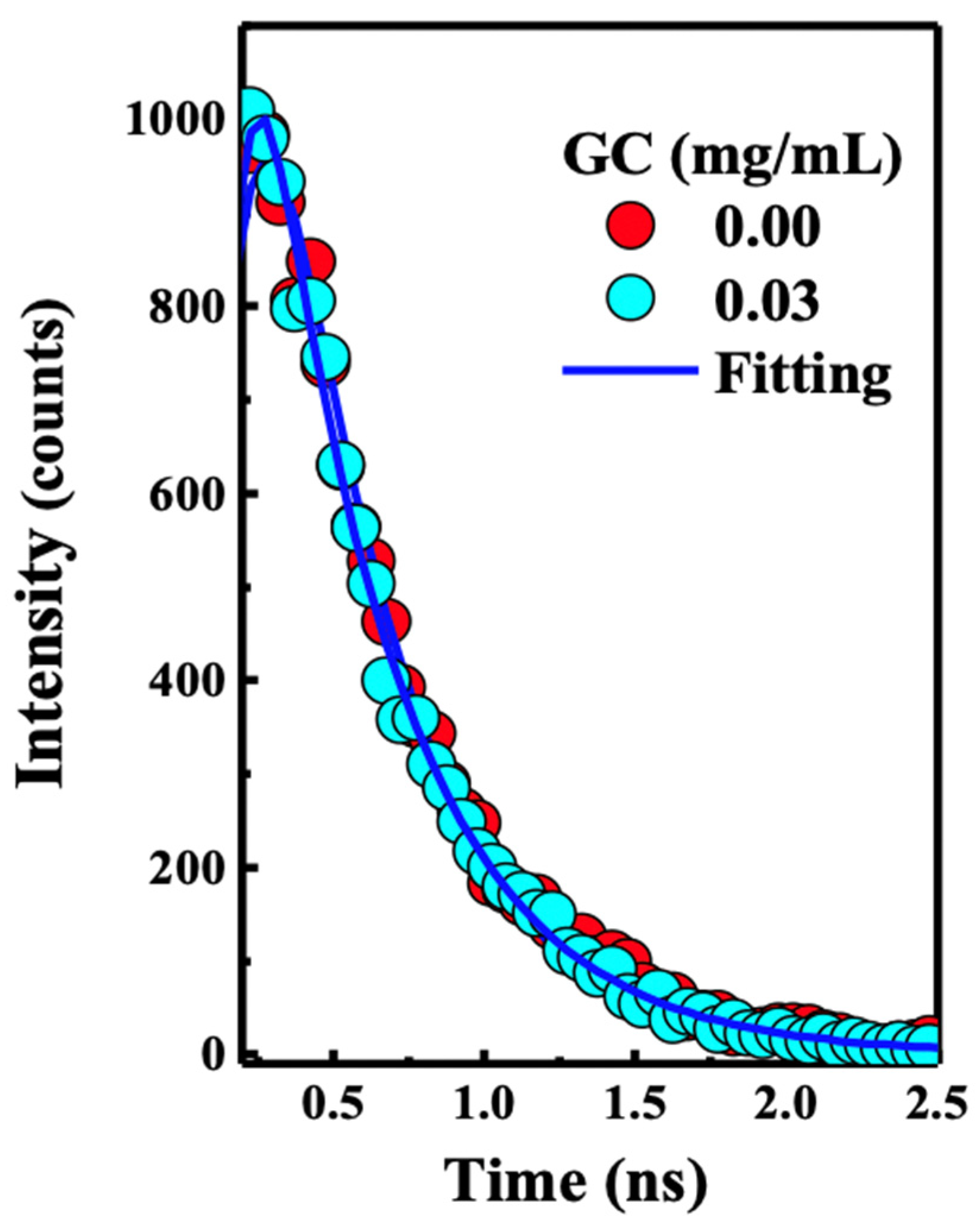{kind=link}
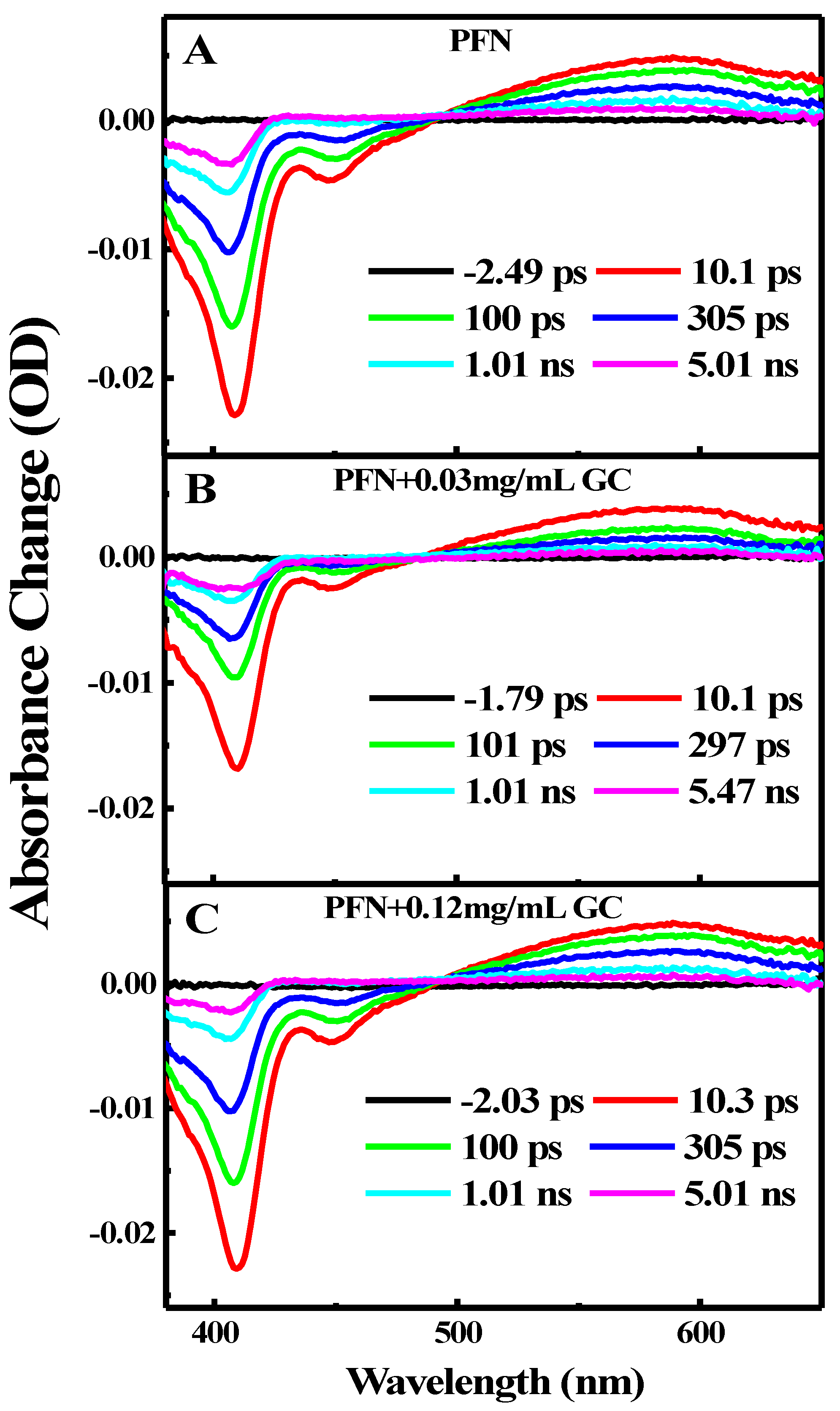{kind=link}
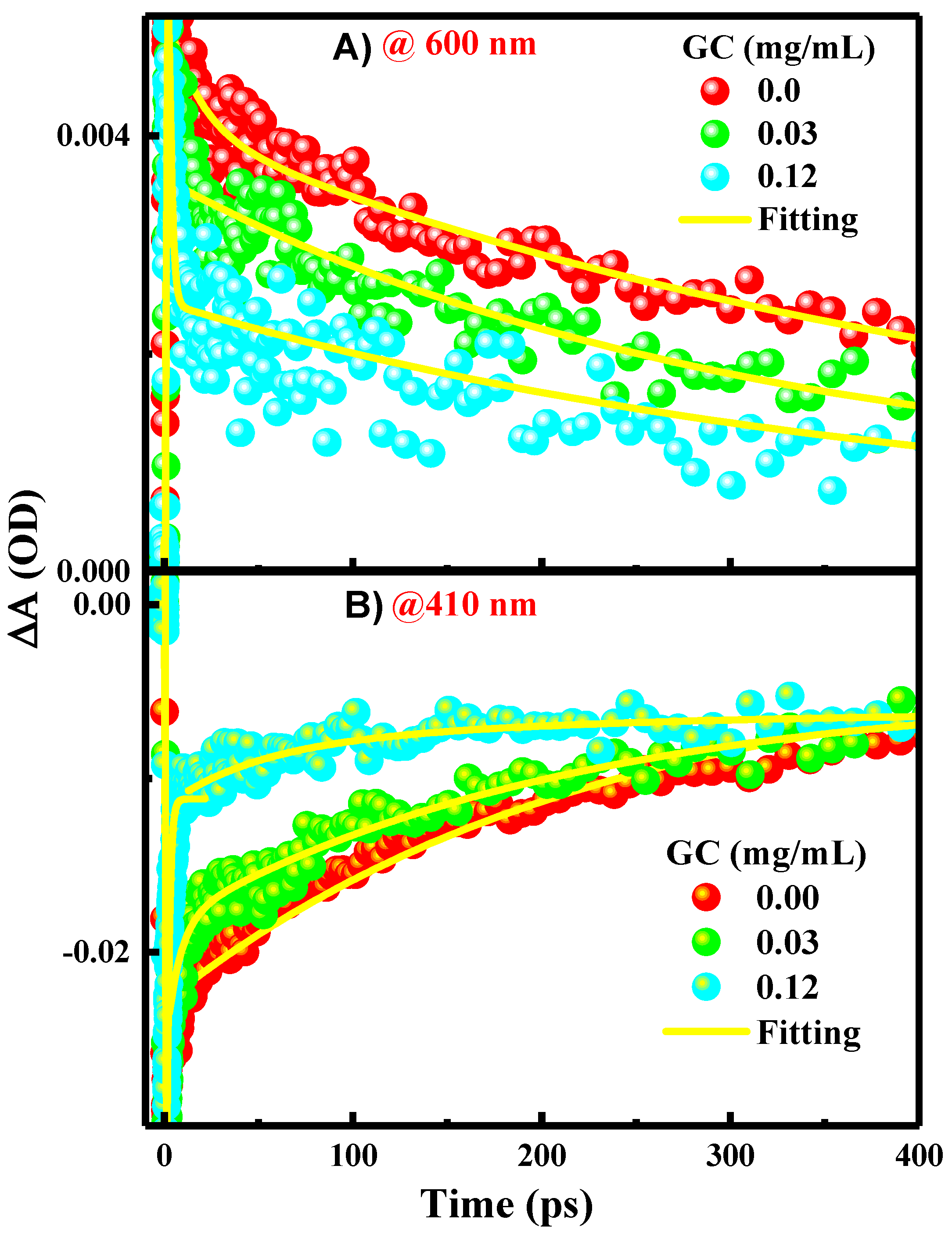{kind=link}
1. Introduction
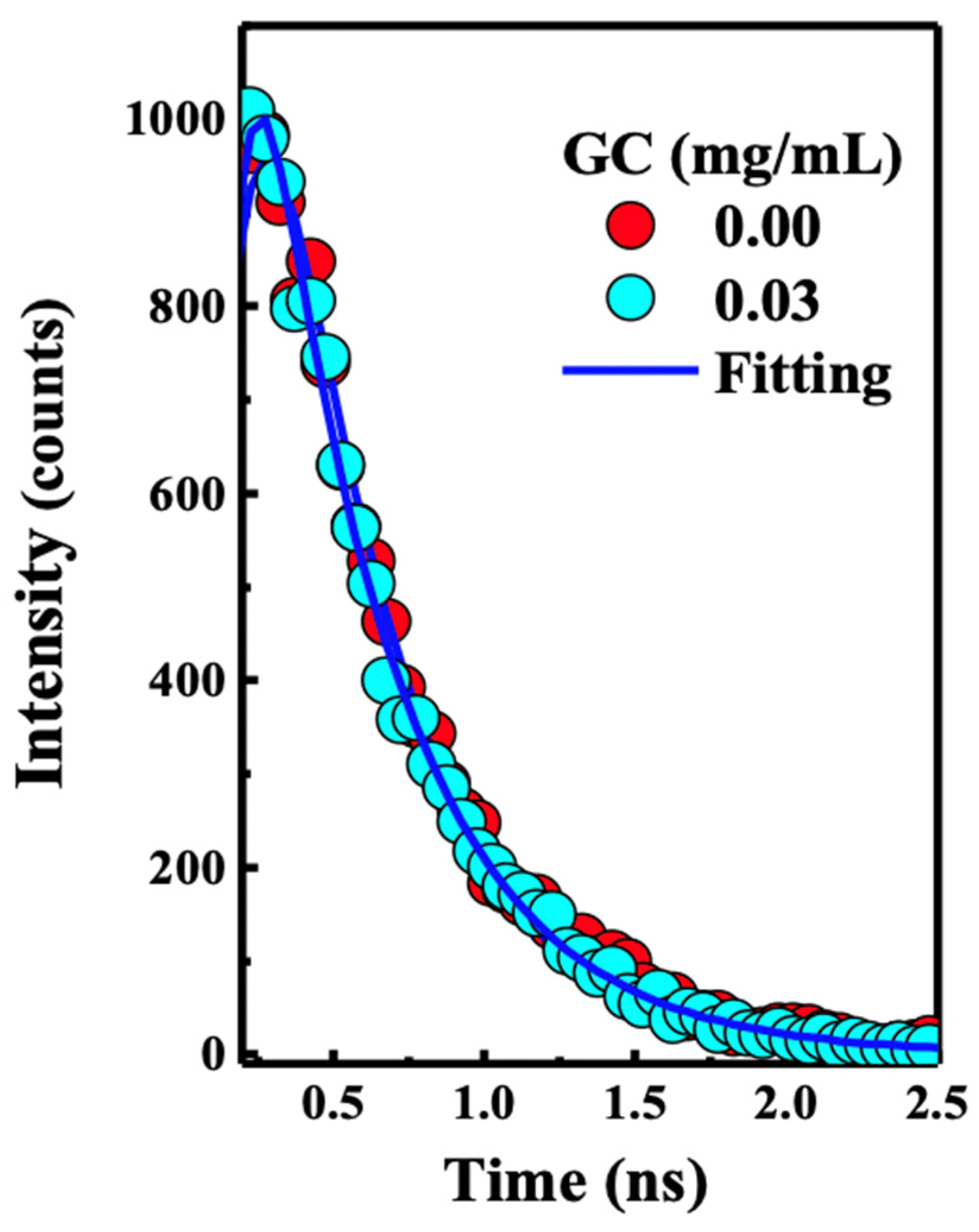
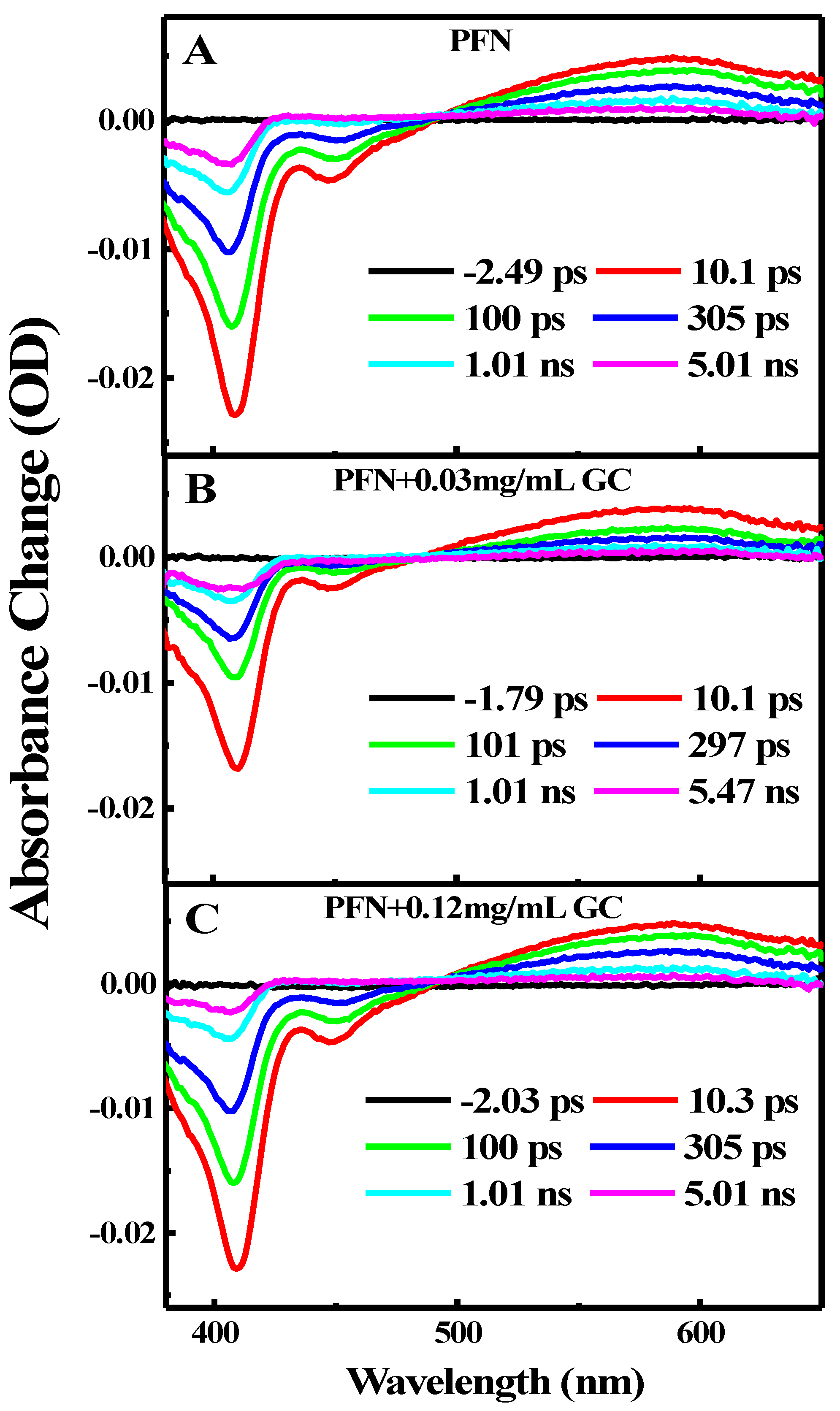
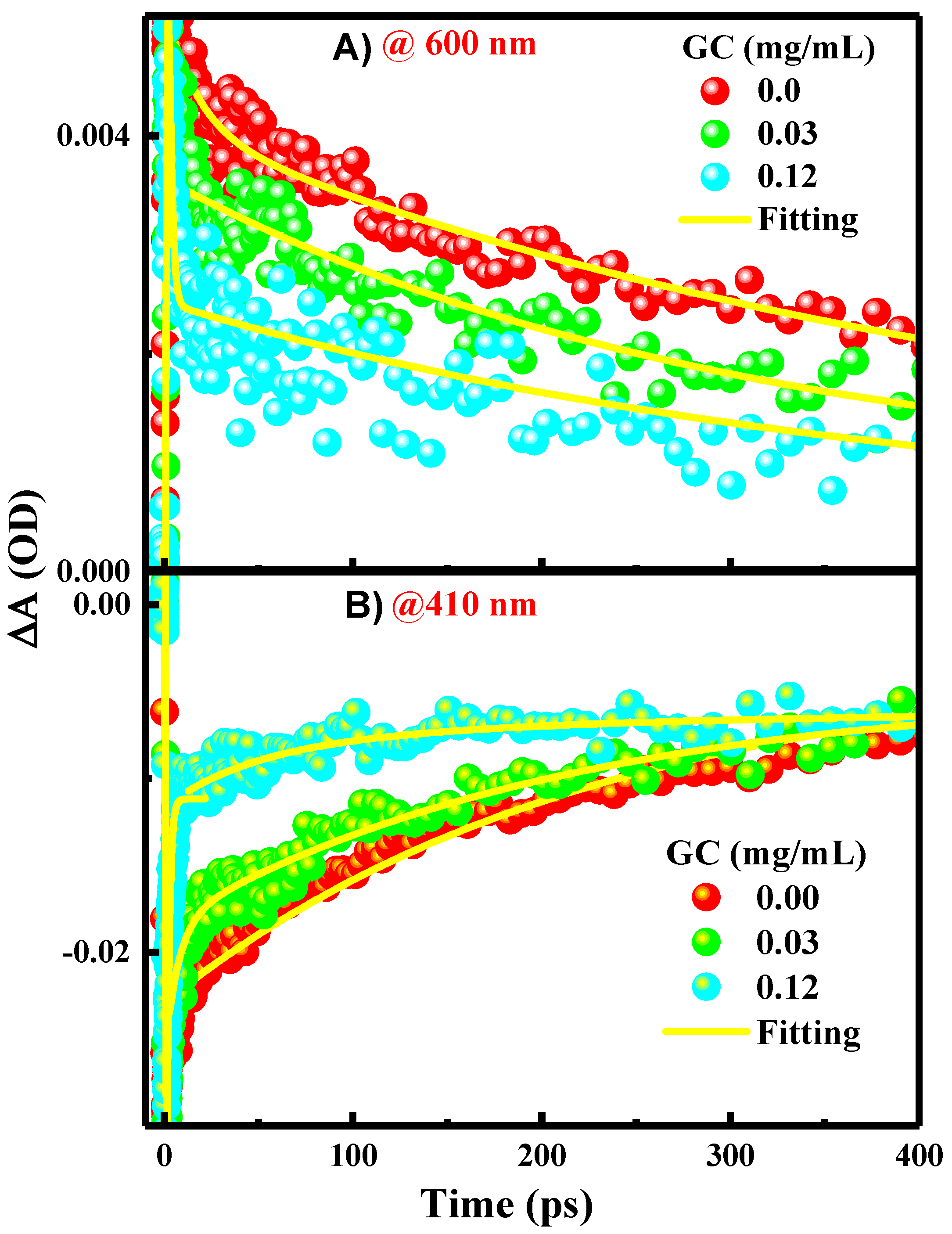
2. Results and Discussion
3. Experimental Section
3.1. Materials
3.2. Steady-State Measurements
3.3. TCSPC Setup
3.4. Femtosecond Broadband Transient Absorption (TA) Spectroscopy
4. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, C.; Shao, L.; Chen, S.; Hu, Z.; Cai, H.; Huang, F. Recent Progress in π-Conjugated Polymers for Organic Photovoltaics: Solar Cells and Photodetectors. Prog. Poly. Sci. 2023, 143, 101711–101715. [Google Scholar] [CrossRef]
- Alsam, A.A.; Aly, S.M.; Usman, A.; Parida, R.M.; Del Gobbo, S.; Alarousu, E.; Mohammed, O.F. Bimolecular Excited-State Electron Transfer with Surprisingly Long-Lived Radical Ions. J. Phys. Chem. C 2015, 119, 21896–21903. [Google Scholar] [CrossRef]
- Xie, Y.; Chen, L.; Li, H.; Yi, Y. Polymer and Hybrid Optical Devices Manipulated by the Thermo-Optic Effect. Polymer 2023, 15, 3721. [Google Scholar] [CrossRef]
- He, W.; Duan, J.; Liu, H.; Qian, C.; Zhu, M.; Zhang, W.; Liao, Y. Conjugated Microporous Polymers for Advanced Chemical Sensing Applications. Prog. Polym. Sci. 2024, 148, 101770. [Google Scholar] [CrossRef]
- Xing, X.J.; Liu, X.G.; He, Y.; Lin, Y.; Zhang, C.L.; Tang, H.W.; Pang, D.W. Amplified Fluorescent Sensing of DNA Using Graphene Oxide and a Conjugated Cationic Polymer. Biomacromolecules 2013, 14, 117–127. [Google Scholar] [CrossRef]
- Ramanaviciene, A.; Plikusiene, I. Polymers in Sensor and Biosensor Design. Polymer 2021, 13, 917. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Alsam, A.A.; Wang, S.; Aly, S.M.; Pan, Z.; Mohammed, F.O.; Schanze, S.K. Effect of Conjugation Length on Photoinduced Charge Transfer in π-Conjugated Oligomer-Acceptor Dyads. J. Phys. Chem. A 2017, 121, 4891–4901. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Liang, S.; Karuthedath, S.; Xiao, C.; Wang, J.; Tan, W.L.; Li, R.; Li, H.; Hou, L.; Tang, Z.; et al. Random Double-Cable Conjugated Polymers with Controlled Acceptor Contents for Single-Component Organic Solar Cells. J. Mater. Chem. A 2023, 11, 12236–12244. [Google Scholar] [CrossRef]
- Al-Azzawi, A.G.S.; Aziz, S.B.; Dannoun, E.M.A.; Iraqi, A.; Nofal, M.M.; Murad, A.R.; Hussein, A.M. A Mini Review on the Development of Conjugated Polymers: Steps towards the Commercialization of Organic Solar Cells. Polymer 2023, 15, 158–164. [Google Scholar]
- Kang, R.; Oh, S.H.; Kim, D.Y. Influence of the Ionic Functionalities of Polyfluorene Derivatives as a Cathode Interfacial Layer on Inverted Polymer Solar Cells. ACS Appl. Mater. Interfaces 2014, 6, 6227–6236. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.; Huang, G.; Huang, H.; Wang, Y.; Gu, X.; Wang, X.; Qiu, L. Achieving High Performance Stretchable Conjugated Polymers via Donor Structure Engineering. Macromol. Rapid Commun. 2023, 44, 2300169. [Google Scholar] [CrossRef]
- Sutjianto, J.G.; Yoo, S.H.; Westerman, C.R.; Jackson, T.N.; Wilker, J.J.; Gomez, E.D. Blends of Conjugated and Adhesive Polymers for Sticky Organic Thin-Film Transistors. Adv. Electron. Mater. 2023, 9, 2300422. [Google Scholar] [CrossRef]
- Imai, T.; Sakamaki, D.; Aoyagi, S.; Amaya, T. Intramolecular Electron Transfer in Multi-Redox Systems Based on Cyclic [3]Spirobifluorenylene Compound. Chem. Eur. J. 2023, 29, 202302670. [Google Scholar] [CrossRef]
- Yi, C.; Song, B.; Tian, W.; Cui, X.; Qi, Q.; Jiang, W.; Qi, Z.; Sun, Y. Fluorescent Sensor of Fluorene Derivatives Having Phosphonic Acid as a Fluorogenic Ionophore: Synthesis and Static Quenched Properties for Fe(III). Tetrahedron Lett. 2014, 55, 5119–5123. [Google Scholar] [CrossRef]
- Duarte, A.; Pu, K.-Y.; Liu, B.; Bazan, G.C. Recent Advances in Conjugated Polyelectrolytes for Emerging Optoelectronic Applications. Chem. Mater. 2011, 23, 501–515. [Google Scholar] [CrossRef]
- Alsam, A.A.; Adhikari, A.; Parida, M.R.; Aly, S.M.; Bakr, O.M.; Mohammed, O.F. Bane of Hydrogen-Bond Formation on the Photoinduced Charge-Transfer Process in Donor-Acceptor Systems. J. Phys. Chem. C 2017, 121, 7837–7843. [Google Scholar] [CrossRef]
- Lee, K.; Kim, H.J.; Kim, J. Design Principle of Conjugated Polyelectrolytes to Make Them Water-Soluble and Highly Emissive. Adv. Funct. Mater. 2012, 22, 1076–1086. [Google Scholar] [CrossRef]
- Guo, Y.; Sun, J.; Tang, Y.; Jia, X.; Nie, Y.; Geng, Z.; Wang, C.; Zhang, J.; Tan, X.; Zhong, D.; et al. Efficient Interfacial Electron Transfer Induced by Hollow-Structured ZnIn2S4 for Extending Hot Electron Lifetimes. Energy Environ. Sci. 2023, 16, 3462–3473. [Google Scholar] [CrossRef]
- Alsam, A.A.; Aly, S.M.; Parida, R.M.; Alarousu, E.; Cao, Z.; Cavallo, L.; Mohammed, F.O. Real-Time Observation of Intersystem Crossing Induced by Charge Recombination during Bimolecular Electron Transfer Reactions. Dye Pigm. 2017, 136, 881–886. [Google Scholar] [CrossRef]
- Aly, S.M.; Parida, M.R.; Alarousu, E.; Mohammed, O.F. Ultrafast Electron Injection at the Cationic Porphyrin-Graphene Interface Assisted by Molecular Flattening. Chem. Commun. 2014, 50, 10452–10455. [Google Scholar] [CrossRef]
- Kim, I.; Kyhm, K.; Kang, M.; Woo, H.Y. Ultrafast Combined Dynamics of Förster Resonance Energy Transfer and Transient Quenching in Cationic Polyfluorene/Fluorescein-Labelled Single-Stranded DNA Complex. J. Lumin. 2014, 149, 185–189. [Google Scholar] [CrossRef]
- Alrais, L.; Almaksoud, W.; Abou-Hamad, E.; Werghi, B.; Bendjeriou-Sedjerari, A.; Hedhili, M.N.; Basset, J.-M. A Strategy For High Ethylene Polymerization Performance Using Titanium Single-Site Catalysts. Chem. Commun. 2023, 59, 12503–12506. [Google Scholar] [CrossRef]
- Clarke, T.M.; Durrant, J.R. Charge Photogeneration in Organic Solar Cells. Chem. Rev. 2010, 110, 6736–6767. [Google Scholar] [CrossRef]
- Zhao, M.-Y.; Tang, Y.-F.; Han, G.-Z. Recent Advances in the Synthesis of Aromatic Azo Compounds. Molecules 2023, 28, 6741. [Google Scholar] [CrossRef]
- Razali, N.A.; Jamain, Z. Synthesis, Chemical Identification and Biological Application of Azo-Based Molecules Containing Different Terminal Group: A review. J. Mole. Struc. 2023, 1284, 135329. [Google Scholar] [CrossRef]
- Rosspeintner, A.; Angulo, G.; Vauthey, E. Bimolecular Photoinduced Electron Transfer Beyond the Diffusion Limit: The Rehm−Weller Experiment Revisited with Femtosecond Time Resolution. J. Am. Chem. Soc. 2014, 136, 2026–2032. [Google Scholar] [CrossRef] [PubMed]
- Ge, G.; Zhang, C.; Li, X. Multi-Electron Transfer Electrode Materials for High-Energy-Density Flow Batteries. Next Energy 2023, 1, 100043. [Google Scholar] [CrossRef]
- Srinivasan, M.V.; Ito, M.; Kumar, P.; Abhirami, K.; Tsuda, N.; Yamada, J.; Shin, P.-K.; Ochiai, S. Performance Evaluation of an Organic Thin-Film Solar Cell of PTB7:PC71BM with an Alcohol-Soluble Polyelectrolyte Interlayer Prepared Using the Spray-Coating Method. Ind. Eng. Chem. Res. 2015, 54, 181–187. [Google Scholar] [CrossRef]
- Magne, T.M.; de Oliveira Vieira, T.; Alencar, L.M.R.; Junior, F.F.M.; Gemini-Piperni, S.; Carneiro, S.V.; Fechine, L.M.U.D.; Freire, R.M.; Golokhvast, K.; Metrangolo, P.; et al. Graphene and its Derivatives: Understanding the Main Chemical and Medicinal Chemistry Roles for Biomedical applications. J. Nanostruct Chem. 2022, 12, 693–727. [Google Scholar] [CrossRef] [PubMed]
- Kahveci, Z.; Martinez-Tomé, M.J.; Mallavia, R.; Mateo, C.R. Use of the Conjugated Polyelectrolyte Poly{[9,9-bis(6′-N,N,N-trimethylammonium)hexyl]fluorene-phenylene} bromide (HTMA-PFP) as a Fluorescent Membrane Marker. Biomacromolecules 2013, 14, 1990–1998. [Google Scholar] [CrossRef]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy, 3rd ed.; Springer Science+Business Media, LLC: Singapore, 2006. [Google Scholar]
- Beckwith, J.S.; Aster, A.; Vauthey, E. The Excited-State Dynamics of the Radical Anions of Cyanoanthracenes. Phys. Chem. Chem. Phys. 2022, 24, 568–575. [Google Scholar] [CrossRef]
- Zacharioudaki, D.E.; Fitilis, I.; Kotti, M. Review of Fluorescence Spectroscopy in Environmental Quality Applications. Molecules 2022, 27, 4801. [Google Scholar] [CrossRef] [PubMed]
- Ageeva, A.A.; Babenko, S.V.; Magin, I.M.; Plyusnin, V.F.; Kuznetsova, P.S.; Stepanov, A.A.; Vasilevsky, S.F.; Polyakov, N.E.; Doktorov, A.B.; Leshina, T.V. Stereoselectivity of Electron and Energy Transfer in the Quenching of (S/R)-Ketoprofen-(S)-Tryptophan Dyad Excited State. Int. J. Mol. Sci. 2020, 21, 5370. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.; Chen, T.W.; Yan, L.; You, W. Facile Synthesis of Key Building Blocks of D18 Series Conjugated Polymers for High-Performance Polymer Solar Cells. ACS Appl. Poly. Mater. 2023, 5, 1937–1944. [Google Scholar] [CrossRef]
- Tvrdy, K.; Frantsuzov, P.A.; Kamat, P.V. Photoinduced Electron Transfer from Semiconductor Quantum Dots to Metal Oxide Nanoparticles. Proc. Natl. Acad. Sci. USA 2010, 108, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; He, Y.; Almalki, M.; Yin, J.; Shekhah, O.; Jia, J.; Gutierrez-Arzaluz, L.; Cheng, Y.; Alkhazragi, O.; Maka, V.K.; et al. Engineering Metal-Organic Frameworks with Tunable Colors for High-Performance Wireless Communication. J. Am. Chem. Soc. 2023, 145, 15435–15442. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alsam, A.A. Comparative Investigation of Ultrafast Excited-State Electron Transfer in Both Polyfluorene-Graphene Carboxylate and Polyfluorene-DCB Interfaces. Molecules 2024, 29, 634. https://doi.org/10.3390/molecules29030634
Alsam AA. Comparative Investigation of Ultrafast Excited-State Electron Transfer in Both Polyfluorene-Graphene Carboxylate and Polyfluorene-DCB Interfaces. Molecules. 2024; 29(3):634. https://doi.org/10.3390/molecules29030634
Chicago/Turabian StyleAlsam, Amani A. 2024. "Comparative Investigation of Ultrafast Excited-State Electron Transfer in Both Polyfluorene-Graphene Carboxylate and Polyfluorene-DCB Interfaces" Molecules 29, no. 3: 634. https://doi.org/10.3390/molecules29030634
APA StyleAlsam, A. A. (2024). Comparative Investigation of Ultrafast Excited-State Electron Transfer in Both Polyfluorene-Graphene Carboxylate and Polyfluorene-DCB Interfaces. Molecules, 29(3), 634. https://doi.org/10.3390/molecules29030634