1. Introduction
The nozzle is a critical component of the orbital control engine, exerting a significant influence on a spacecraft’s capability for precise velocity adjustments, orbital corrections, and attitude maintenance. It plays a pivotal role in ensuring the stable operation of the spacecraft. Given the extreme operational conditions it endures, the nozzle material must exhibit exceptional comprehensive properties, particularly high-temperature strength and oxidation resistance [
1,
2]. Refractory metals such as tungsten (W), molybdenum (Mo), tantalum (Ta), iridium (Ir), and rhenium (Re) have become primary materials for nozzle construction in thrust-controlled engines due to their high melting points and superior performance in high-temperature environments [
3]. However, the rapid advancement of aerospace technology has led to increased demands for spacecraft in terms of flight distance, speed, and responsiveness. Traditional refractory metals are no longer adequate to meet these new requirements. Consequently, the advancement of a new generation of ultra-high-temperature materials with exceptional performance is crucial in the field of aerospace materials.
Rhenium (Re) is a rare metal known for its high melting point (3340 °C), high strength, creep resistance, and chemical stability. Ultramet, a U.S.-based company, utilized chemical vapor deposition (CVD) to prepare rhenium-based nozzle materials with service temperatures as high as 2200 °C. This innovation was successfully applied to the Hughes 601HP satellite, setting a new standard for nozzle material performance [
4,
5]. However, despite Re’s exceptional creep resistance and strength at high temperatures, it faces challenges, such as a high cost (USD 5163/kg), a high density (21.02 g/cm
3), and complex preparation processes [
6,
7]. In contrast, tantalum and tantalum alloys possess high melting points, excellent strength, good processability, and lower density and cost, making them ideal structural materials for environments ranging from 1600 to 1800 °C [
8]. For instance, alloys such as T-111, T-222, and ASTAR-811C, developed by NASA-affiliated research institutions, are widely used in alkali-metal corrosion-resistant pipelines, containers for radioactive materials, and rocket fuel containers [
9].
Based on the superior properties of rhenium (Re) and tantalum (Ta), we designed a new Ta/Re layered composite material. This material leverages the cost-effectiveness and lightweight characteristics of tantalum, as well as the high strength of rhenium, and shows potential for applications in aerospace engine nozzle materials. During the research process, it was observed that the Ta/Re material, when exposed to high temperatures, exhibited interdiffusion of Ta and Re atoms at the Ta/Re interface, forming Ta-Re and Re-Ta solid solutions on either side of the interface, thereby resulting in alterations in the material’s high-temperature properties [
10]. Consequently, investigating the diffusion behavior within the Ta/Re interface and studying the physical and chemical properties of the resulting solid solutions are crucial for enhancing the overall performance of Ta/Re layered composite materials [
11,
12]. However, since Ta (atomic number 73) and Re (atomic number 75) differ by only two atomic numbers and exist as a solid solution, conventional testing methods, such as transmission, struggle to distinguish their forms and mechanisms of action. Therefore, molecular dynamics simulations of the related interface reaction processes are required.
After over fifty years of development, molecular dynamics simulation (MD) has evolved into a robust tool for studying the diffusion behavior of both pure fluids and mixtures [
13,
14,
15]. These simulations typically involve thousands of atoms, enabling detailed exploration of atomic-scale processes and offering insights into experimental phenomena at the microscopic level [
16]. Consequently, MD serves as a valuable complement to experimental methods and theoretical models, providing a comprehensive understanding of diffusion mechanisms in various systems. Currently, molecular dynamics simulations are extensively employed in fields such as chemistry, biology, and materials science. Jamali et al. [
17] conducted a thorough investigation into the finite size effects of MS diffusivities in binary mixtures. They performed EMD simulations of 200 LJ and nine molecular mixtures, showing that, similar to self-diffusivities, MS-diffusivities scale linearly with system size. Wei et al. [
18] and Luo et al. [
19] investigated the interface diffusion behavior of Mo-Ti and Fe-W interfaces using MD simulations, examining the effects of diffusion time, temperature, and interface orientation on diffusion. Notably, the potential function plays a pivotal role in molecular dynamics simulations. This mathematical function defines the interactions and energy exchanges among atoms, setting the force field and energy landscape within the simulated system. The choice of an appropriate potential function significantly affects the accuracy and reliability of simulation results. Unfortunately, the existing potential functions do not cover the Ta-Re system, making the construction of an appropriate potential function crucial for simulating its molecular dynamics processes and elucidating the interface diffusion mechanisms.
There are numerous types of potential function fitting methodologies. Among them, machine learning potentials have garnered significant attention in recent years due to their remarkable accuracy and ability to capture complex atomic interactions. Their flexibility in fitting large datasets and reproducing quantum-level precision makes them a powerful tool in materials modeling [
20,
21]. However, it is worth noting that machine learning potentials often lack strong physical interpretability, and their generalization ability across different systems may be limited. In contrast, traditional force field fitting methods offer a balance between accuracy and interpretability, providing well-defined physical parameters and ensuring broader applicability across diverse systems [
22,
23]. Therefore, employing the force field approach to construct the Ta-Re potential is a feasible solution.
To develop the Ta-Re embedded potential functions, a force-matching approach was employed. In this study, 90 different structures were considered, including fcc, bcc, sc, hcp, non-equilibrium fcc, low-index surfaces of bcc, fcc, sc, and hcp, and cluster types. Additionally, 9045 force measurements, 90 energy values, and 540 stress values were obtained through first-principles calculations. These data were then fitted using the force-matching method, and the Ta-Re potential function was derived using the Potfit software package. Finally, the accuracy of the potential functions was validated through both simulations and experimental methods. This study provides theoretical insight into the high-temperature strengthening and toughening mechanisms of Ta/Re layered composites and aids in controlling their material properties.
4. Fitting Method and Experimental Details
4.1. EAM Potentials
The embedded atom method (EAM) [
30] is a computational model primarily used for analyzing atomic interactions within metal systems, focusing on the structure and interplay of atoms. This method operates on the principle of simulating atomic interactions by characterizing the embedding energy associated with electrons around each individual atom. The total energy of an alloy system can be expressed concisely in a generalized form.
Among them, Ei represents the total potential energy of the system, which is the linear sum of the individual potential energies of all embedded atoms in the system. Furthermore, i and j, respectively, represent embedded atoms and adjacent atoms within the system. The types of embedded atoms and adjacent atoms are represented by α and β, respectively.
In Equation (4), the initial component denotes the embedding energy of the
atom, illustrating the interaction between the atom
and the charge density of its background electrons. A reference is provided for the initial formulation of the embedding function
[
31]:
The formulation is derived from the more general equation proposed by Rose et al. [
32]. Real values are taken for the parameters
and
, while
represents the equilibrium density. Due to numerical instability with our optimization algorithms in the original form, we substitute
with
and set
= 1. Consequently, Equation (5) simplifies to the following expression.
The equation involves three free parameters, , , and . In our proposed model, we utilize the simplified form presented in Equation (6).
In Equation (4),
represents the charge density of the host. In this calculation, the electron density contributions from atoms within a cutoff radius
rcut are summed linearly, excluding the embedding atom itself.
It is important to note that this charge density is purely empirical and does not correspond to the actual physical density. Subsequently, the electron transfer function
can be expressed [
33].
We can see from the transfer function that this oscillation type is characterized by four free parameters, a, a1, j, and β. Among them, a1 represents the amplitude of this type of oscillation, while h is used to smooth the potential. It is worth noting that a, a1, and β also correspond to the wave vector, initial phase, and attenuation factor of the wave function, respectively.
As shown in Equation (4), the second term represents the pair potential between atoms
and
separated by a distance
rij. For our test calculations, to verify the accuracy of the model, we selected the Empirical Oscillating Pair Potential (EOPP) [
34], the Classical Morse potential (CM) [
35], and the Lennard Jones (LJ) potential [
36] for testing and calculation. Our findings indicate that the EOPP demonstrates significant flexibility in describing atomic interactions within the Ta-Re system. Consequently, we adopt the function of the pair potentials with oscillations, expressed as follows:
In the provided formulation, short-range repulsion is governed by the parameters
c1 and
η1, while
c2 and
η2 are the key to maintaining oscillation with
k as the characteristic frequency. It is worth noting that this oscillation, based on empirical definitions, has been widely applied in the study of ternary alloy systems and other complex metal alloy systems [
37,
38]. In Equations (8) and (9), the cutoff function Ψ(x) is defined [
37].
where Ψ(x) ≡ 0 for x ≥ 0 and Ψ(
x) is defined for
x < 0. This function ensures that at the distance cutoff
, the potential function and its derivatives up to the second order smoothly approach zero. Both
and
act as global parameters with uniform values across all functions. The cutoff radius
in our model is uniformly set to 6.0 Å throughout the fitting process.
All potential functions are employed in their analytical forms. The atomic interactions in a binary tantalum/rhenium (Ta-Re) alloy system are characterized by a total of seven analytical functions. These include three pair potential functions (, , and ), two electron transfer functions ( and ), and two embedding functions ( and ). There are a total of 17 free parameters included in this potential function. Among them, the electron transfer function (TF) has four free parameters, the electron potential function (PPF) has six free parameters, the embedding function (EF) has six free parameters, and, finally, there is a global parameter h as a whole. This potential function serves as the foundation for the analysis. The above parameters can be obtained by fitting the potential energy surfaces of different structures of the studied Ta Re alloy system.
4.2. Target Systems for Fitting
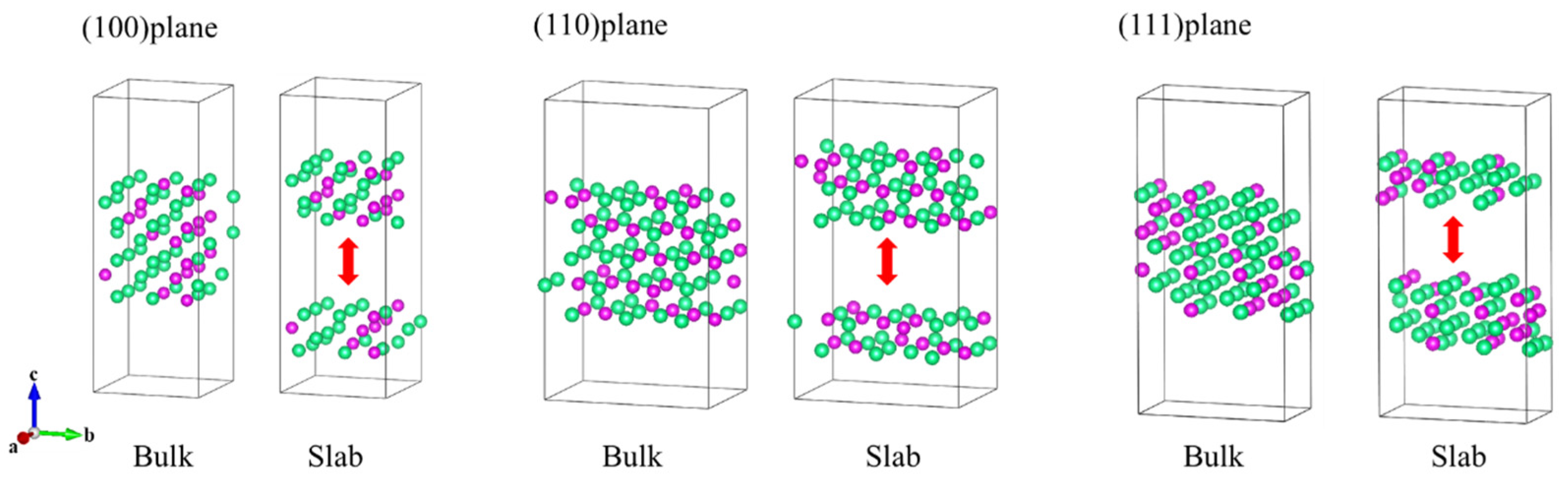
During the fitting of the EAM potential function for Ta-Re alloys, a large number of reference configurations were thoroughly evaluated to enhance the transferability of the EAM potential function. This careful evaluation ensures that the model accurately represents the alloy’s behavior under various conditions. The configurations include clusters, face-centered cubic (fcc), body-centered cubic (bcc), simple cubic (sc), hexagonal crystals, and non-equilibrium structures, as shown in
Figure 8,
Figure 9 and
Figure 10 and
Supplementary Figures S2–S4. To ensure precision, we performed full relaxation of most reference configurations using first-principles calculations, with a maximum force of less than 0.01 eVÅ
−1 per atom.
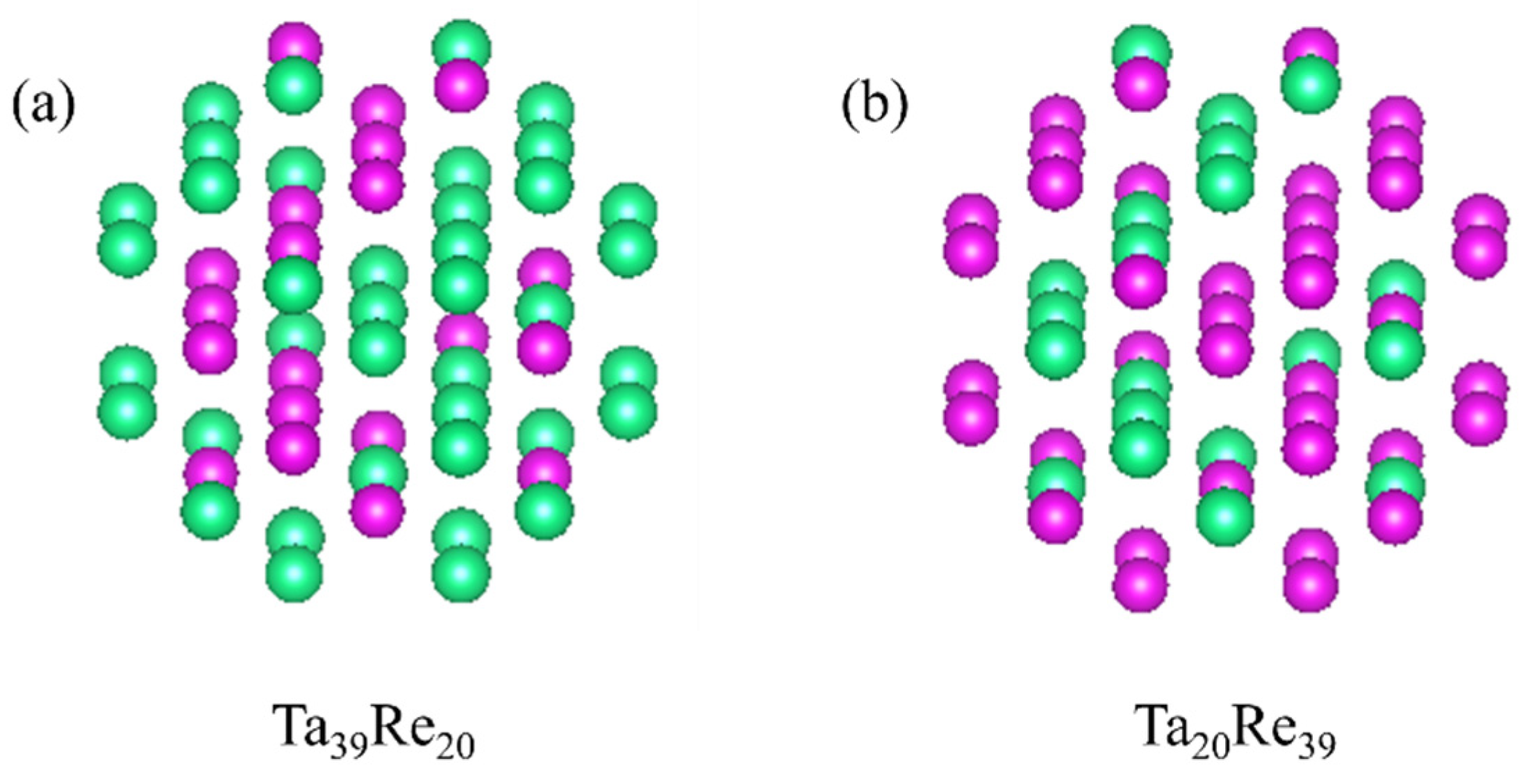
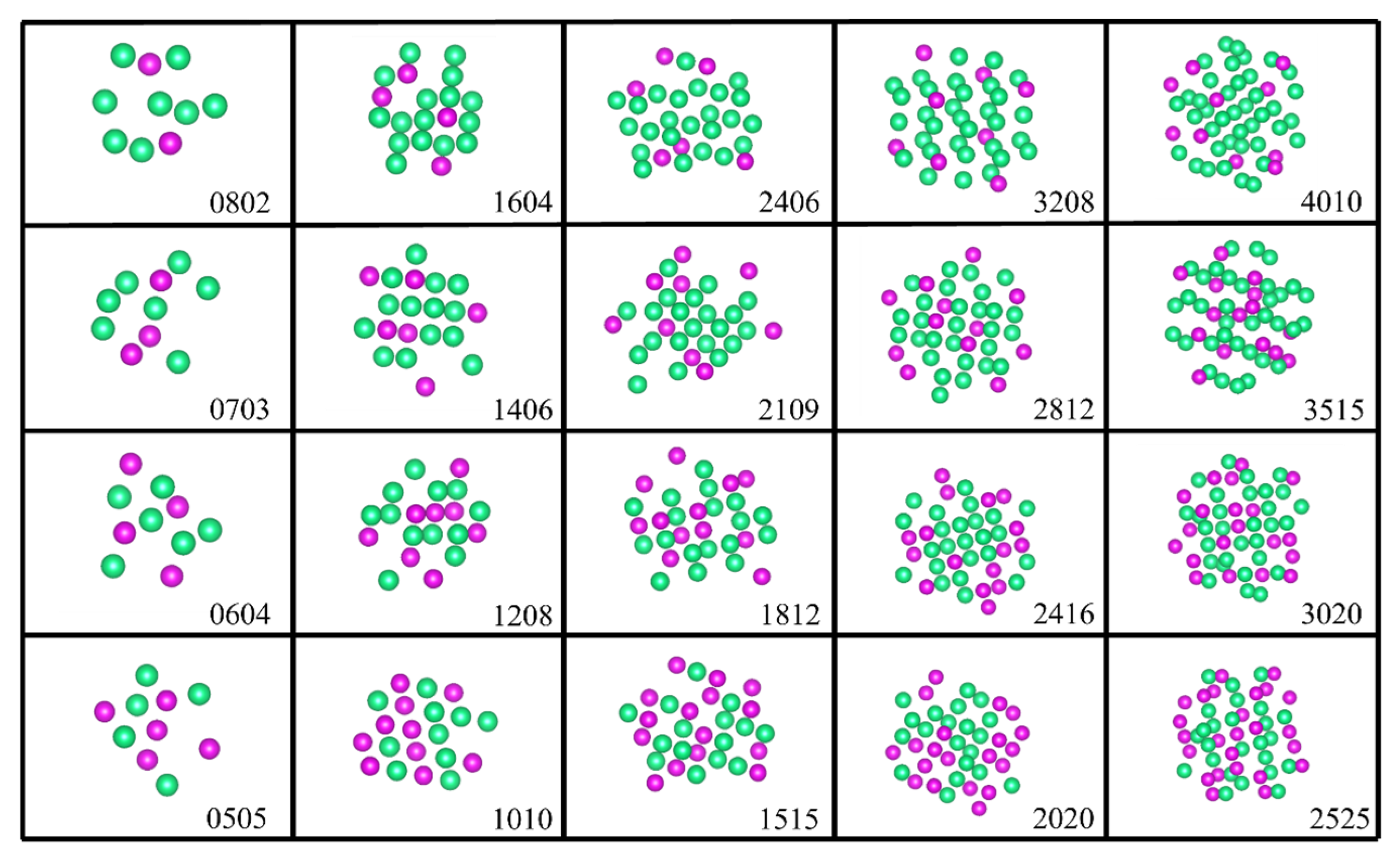
The clusters exhibit a substantial specific surface area and unique electronic properties. Their intricate electronic and structural characteristics are influenced by several factors, including size and composition. In our existing database, we have curated five clusters of varying sizes, comprising 10, 20, 30, 40, and 50 atoms, respectively. Moreover, each cluster size encompasses different proportions of Re atoms, specifically 20%, 30%, 40%, and 50%. The reference structure is depicted in
Figure 8.
To enhance the generalization ability of the potential function, it is essential to consider both equilibrium and non-equilibrium structures, particularly in temperature simulations of complex systems that involve local perturbations, segregation, clustering, and precipitation.
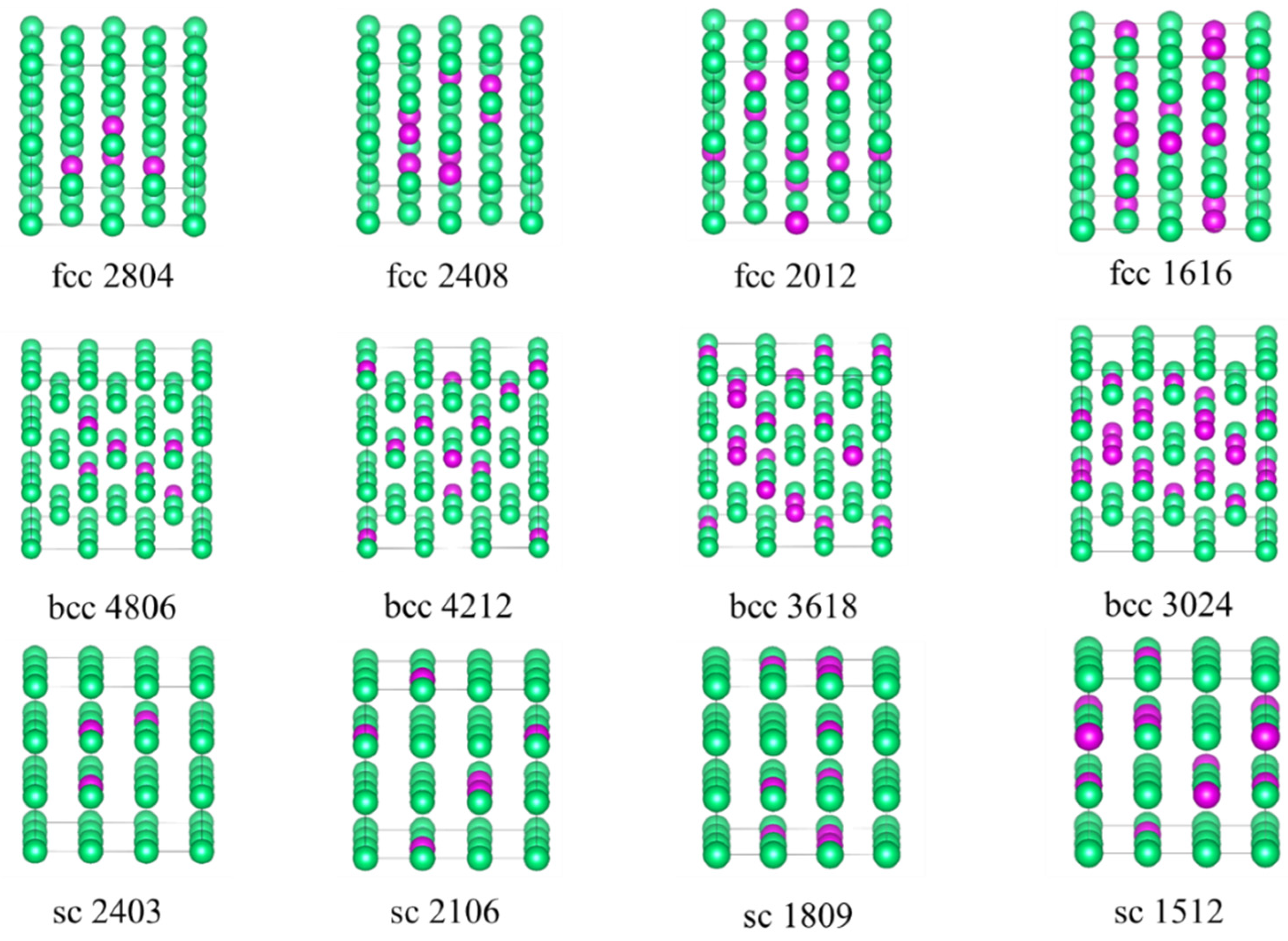
For equilibrium structures, we constructed a 32-atom 2 × 2 × 2 supercell to represent the periodic face-centered cubic (fcc) phase and selected seven Ta
32-xRe
x alloy structures with varying compositions (x = 4, 8, 12, 16, 20, 24, 28). Similarly, for the body-centered cubic (bcc) phase, a 3 × 3 × 3 supercell containing 54 atoms was constructed, and we identified eight Ta
54-xRe
x alloy structures (x = 6, 12, 18, 24, 30, 36, 42, 48). For the simple cubic (sc) phase, a 3 × 3 × 3 supercell containing 27 atoms was employed, and eight Ta
27-xRe
x alloy structures (x = 3, 6, 9, 12, 15, 18, 21, 24) were selected. Partial reference structures for the face-centered cubic (fcc), body-centered cubic (bcc), and simple cubic (sc) crystals are illustrated in
Figure 9. The added non-equilibrium structure is shown in
Table 5.
The reference database includes 90 structures, comprising 20 cluster structures, 7 face-centered cubic (fcc), 8 body-centered cubic (bcc), 8 simple cubic (sc), and 47 non-equilibrium structures. These configurations were carefully selected to provide comprehensive coverage of the Ta-Re alloy system. The database contains 9045 force measurements, 90 cohesive energies, and 540 stress values, all derived from first-principles calculations performed using the Projector Augmented Wave (PAW) method [
39,
40] within the Vienna Ab Initio Simulation Package (VASP) code [
41,
42]. The Generalized Gradient Approximation (GGA) was applied for the exchange–correlation functional, specifically using the PW91 functional [
43]. In the electronic structure calculations, the valence electrons were treated as Ta: 5d
36s
2 and Re: 5p
65d
56s
2. According to the cutoff energy and k-point convergence tests based on the Monkhorst–Pack method shown in
Figure 11, a set of plane-wave basis vectors with a cutoff energy of 650 eV is used to expand the electronic wavefunction, and the k-point grid is set to 7 × 7 × 7, which meets the accuracy requirements. Since the Ta-Re system is non-magnetic, spin polarization effects were not considered in any of the calculations. Additionally, a 15 Å vacuum region was incorporated along the x, y, and z directions to prevent unwanted interactions between clusters during the simulations.
4.3. Fitting Procedure
In this paper, we have fitted the EAM potential function for Ta-Re alloys using the force-matching method [
44] and the Potfit package [
45,
46] developed by Brommer et al. During the fitting process, the results from first-principles calculations were used as a crucial reference for refining the free parameters of the potential function. Additionally, the ranges of the electron density function
ρβ(
r) and the electron potential function
Φαβ(
r) were defined between the minimum distance
rmin and the cutoff distance
rc. The electron density function determines the effective range of the embedding function
Fα(
n). The minimum interatomic distance in a given configuration corresponds to the original value of
rmin, while the cutoff distance of 6.0 Å encompasses interactions between atoms and their fourth-nearest neighbors in the equilibrium system.
To achieve accurate parameter estimation, we employ two distinct optimization algorithms to fit our analytical function. Both algorithms are specifically designed to minimize the sum of squares, expressed as follows:
In this context, f0, E0, and S0 represent the forces, cohesive energies, and stresses calculated using first-principles methods, respectively. Similarly, f, E, and S represent the corresponding values derived from the current EAM potential function. The terms εj, εk, and εl are negligible and have no physical significance.
To optimize the parameters effectively, we first use the simulated annealing algorithm [
47], a global optimization technique aimed at minimizing energy states. Subsequently, we apply a deterministic method, similar to conjugate gradients [
48], to further refine the optimization process and achieve a more accurate local minimum.
Portability is critical when evaluating the practical application of the EAM potential function, and good portability relies on selecting appropriate reference structures. Enhancing transferability requires a larger number of reference configurations, but this introduces the risk of additional errors. Therefore, it is essential to balance the quantity of reference data with the accuracy of the model. In Equation (11),
and
are the weighting factors for cohesive energy and stress, respectively, and they play a significant role in the accuracy of force, energy, and stress fitting. Based on a series of test calculations, we determined that
should be set to 20,000 and
to 8000. The
Φαβ(
r) with PPF, the
ρβ(
r) with TF, and the
Fα(
n) with EF for each element serve as the target functions in the fitting process. In the Ta-Re alloy system,
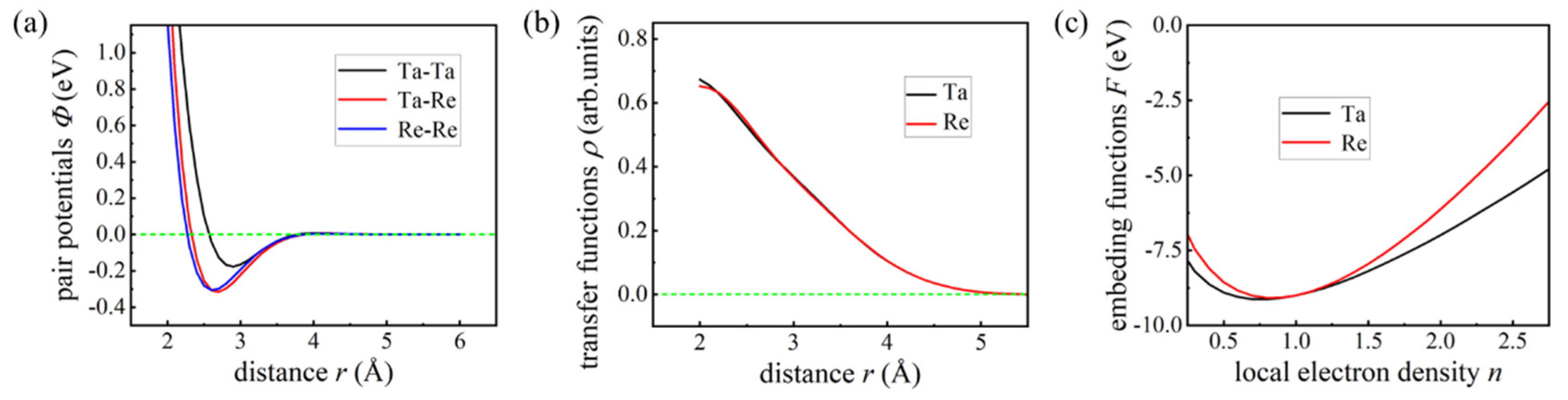
Figure 12 illustrates the fitted functions of our EAM potential model, with panels (a), (b), and (c) representing the PPF, TF, and EF, respectively. Notably, both the PPF and TF approach zero at atomic separations exceeding 6.0 Å, which led us to select a cutoff distance of 6.0 Å to terminate interactions between atoms in the Ta-Re alloy system. A comprehensive summary of all parameters obtained from the fitting process for the current EAM potential model is presented in
Table 6.
4.4. Experimental Procedure
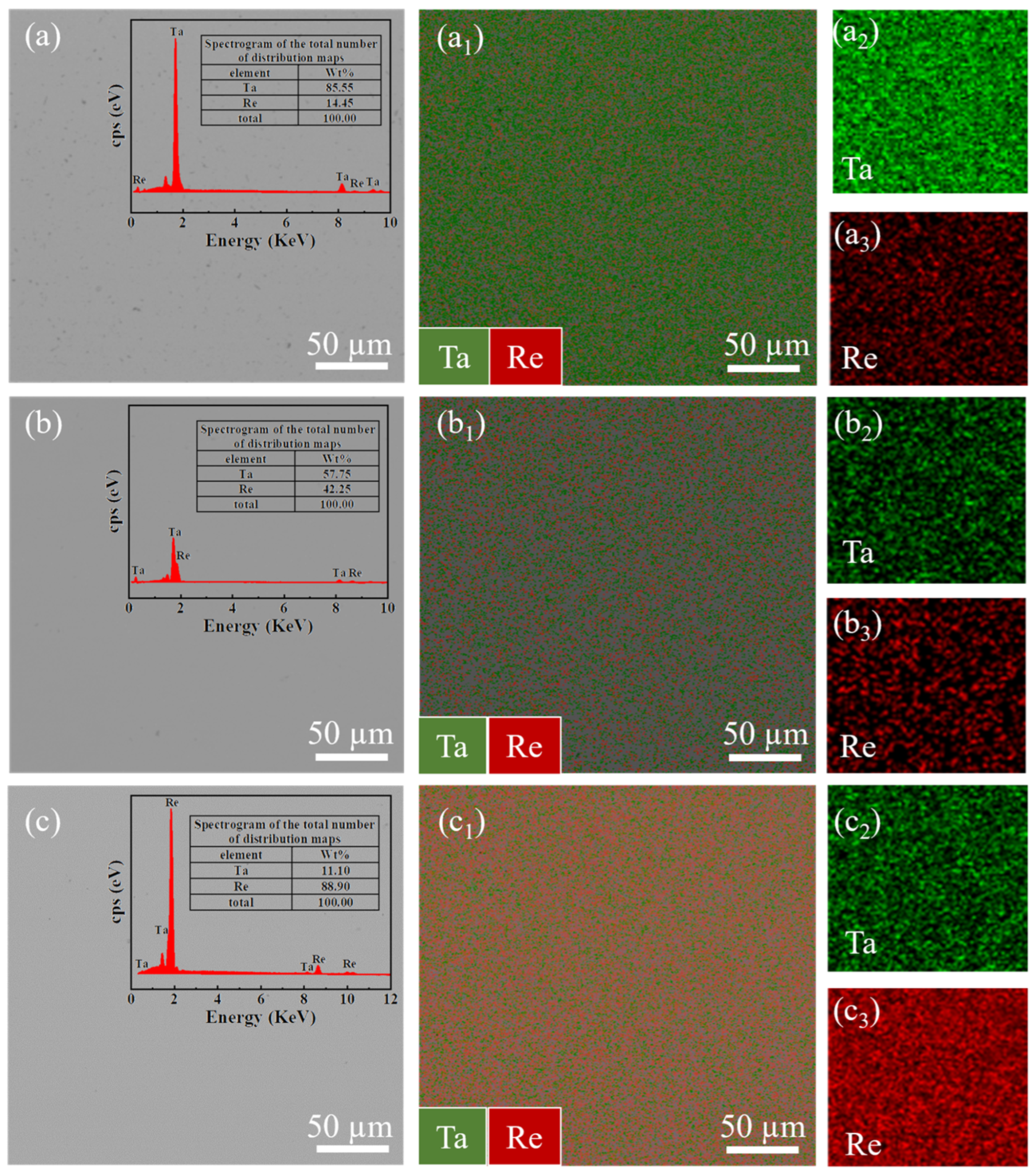
The Ta-Re binary alloy specimens were produced using a tungsten electrode arc furnace, with raw materials consisting of 99.99 wt% pure Re and Ta particles. The Re particles were obtained from Advanced Technology and Materials Co., Ltd. (Beijing, China), while the Ta particles were sourced from Ningxia Orient Tantalum Industry Co., Ltd. (Ningxia, China). To ensure uniformity, the Ta-Re alloy samples underwent five consecutive melting cycles (the elemental compositions of the Ta-Re alloys are shown in
Table 7). Electromagnetic stirring was applied during the melting process, and the vacuum level in the tungsten electrode arc furnace was maintained at no less than 5 × 10
−3 Pa.
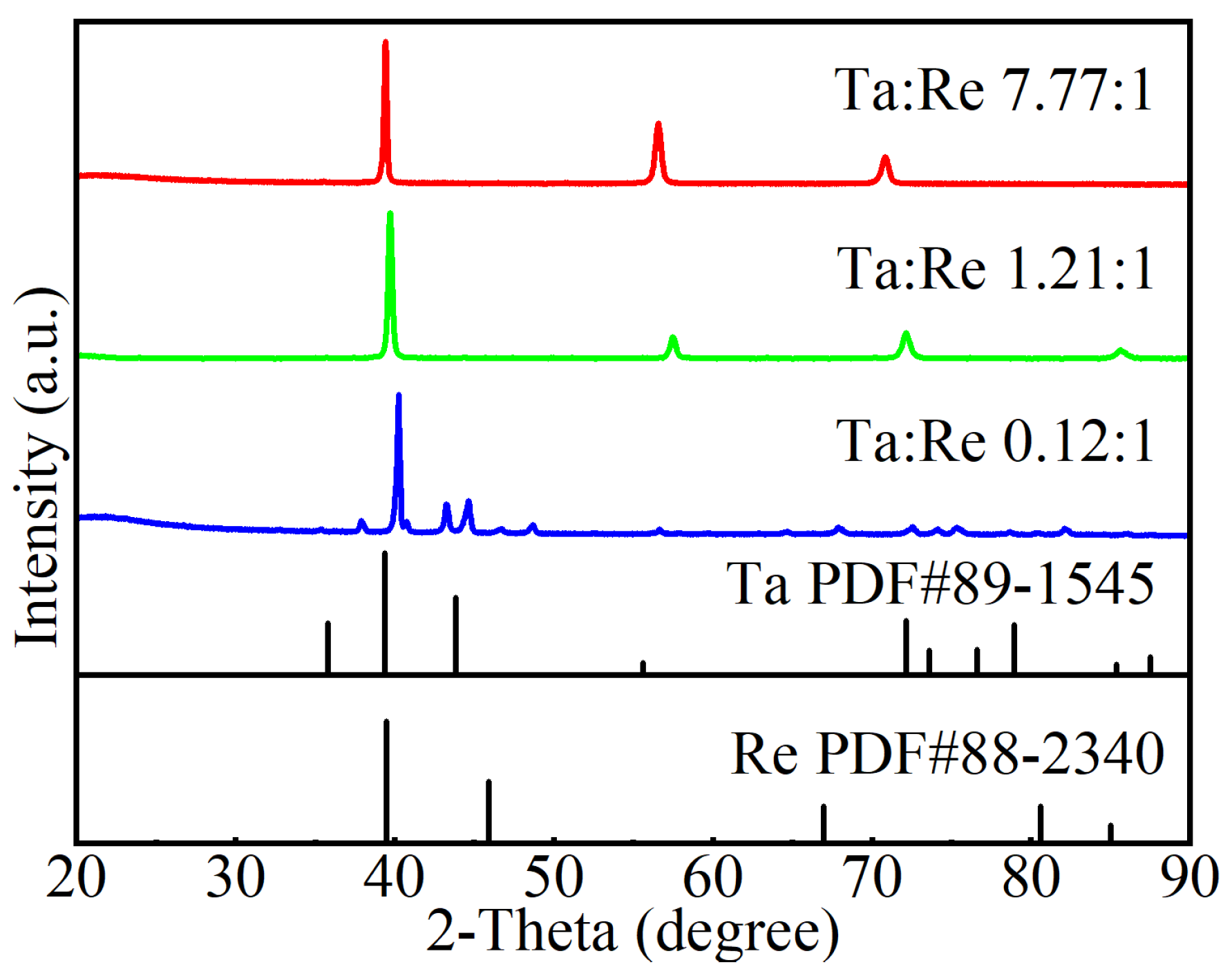
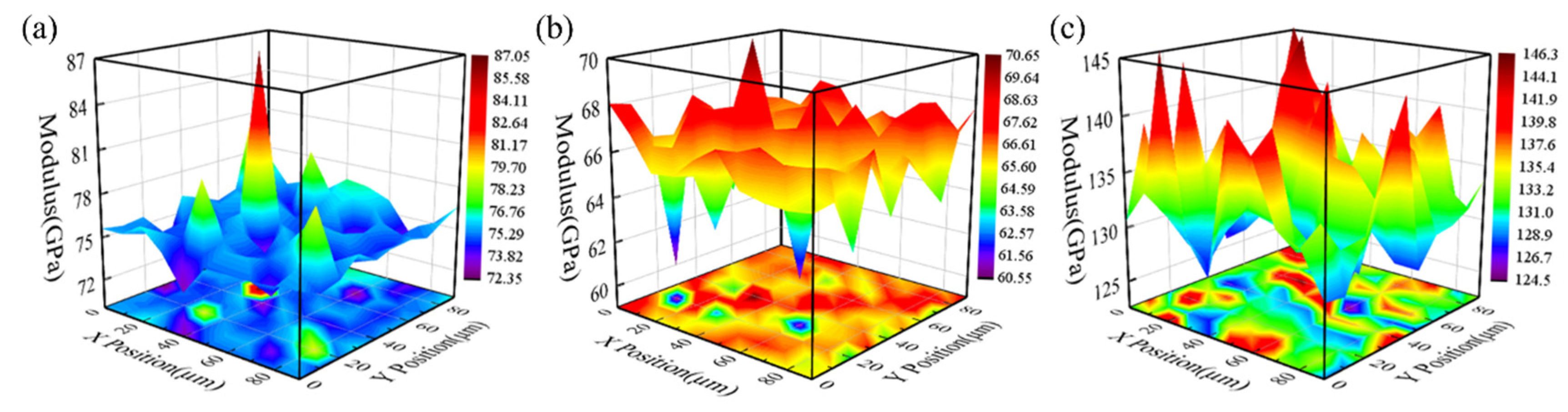
X-ray diffraction (XRD) analysis was performed using a Shimadzu XRD 6000 diffractometer (Shimadzu Corporation, Kyoto, Japan) at room temperature. CuKα radiation, generated by a Cu X-ray tube operating at 15 mA and 30 kV, was used. The samples were scanned over a Bragg angle 2θ range from 20° to 90° at a rate of 2° per minute. Characterization was conducted with a JEOL SU3900 Scanning Electron Microscope (SEM) (JEOL, Kyoto, Japan) equipped with an Energy Dispersive Spectrometer (EDS), using an acceleration voltage of 20 kV. Nanoindentation tests were performed using a UMIS machine from Fischer-Cripps Laboratories, which is equipped with a Berkovich indenter and has a maximum load capacity of 400 mN.
5. Conclusions
In this study, we successfully developed the EAM potential function for Ta-Re alloys using the force-matching method. The bulk structure lattice constants and surface formation energies per unit area of Ta-Re alloys, as obtained from the potential function, are 12.196 Å, E100 = 0.16 × 10−2 eV, E110 = 0.10 × 10−2 eV, and E111 = 0.08 × 10−2 eV. The corresponding error values are 0.015 Å, 0.04 × 10−2 eV, 0.02 × 10−2 eV, and 0.01 × 10−2 eV, respectively, when compared with first-principles calculations. We also determined the atom-averaged binding energies for two cluster structures, Ta-rich (Ta39Re20) and Re-rich (Ta20Re39), with errors ranging from 1.64% to 1.98%. Ta-Re alloys with three compositions (Ta48Re06, Ta30Re24, Ta06Re48) were prepared, and their bulk moduli were measured. The experimentally measured bulk modulus values exhibited a trend of initially decreasing and then increasing, which was consistent with the trend observed in the calculated values. These results validate the accuracy and reliability of the EAM potential functions in characterizing various properties of Ta-Re alloy crystals, surfaces, defects, and clusters. The findings of this study provide valuable theoretical insights into the modification of properties in Ta/Re layered composites.

 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}