
Chemical Bonding and Dynamic Structural Fluxionality of a Boron-Based B8Al3+ Cluster


Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
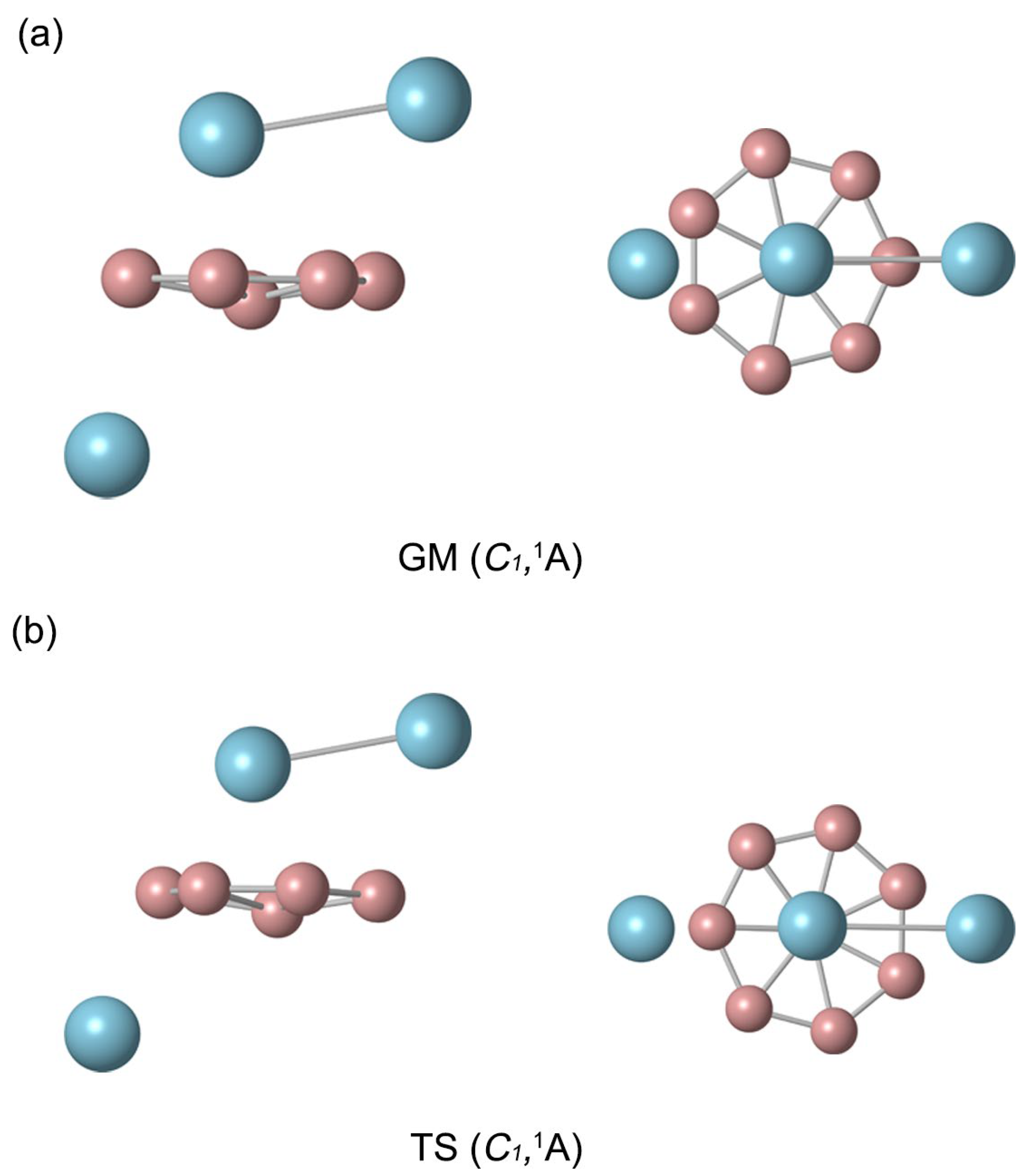
2.1. Global Minimum of B8Al3+ Cluster
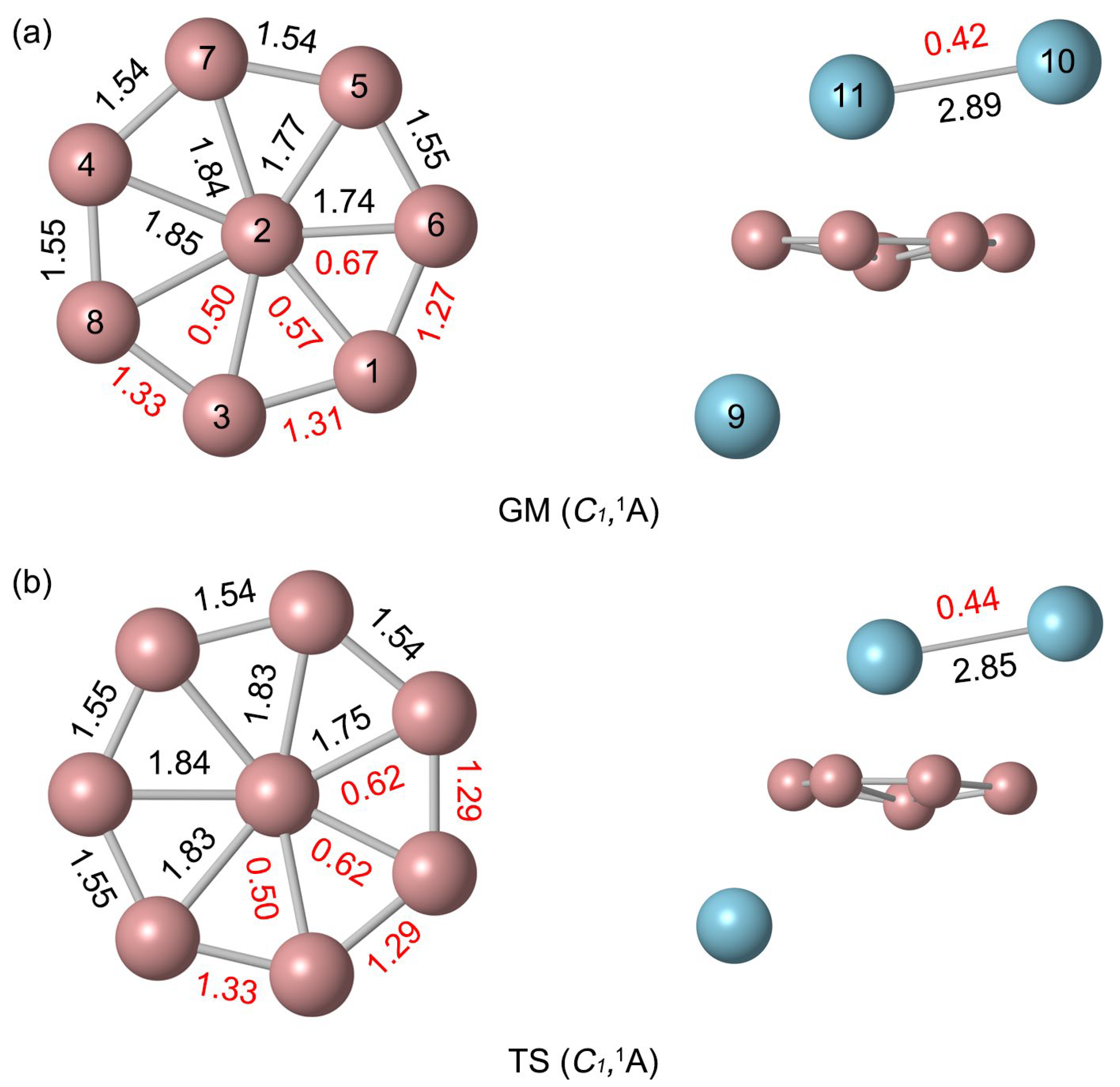
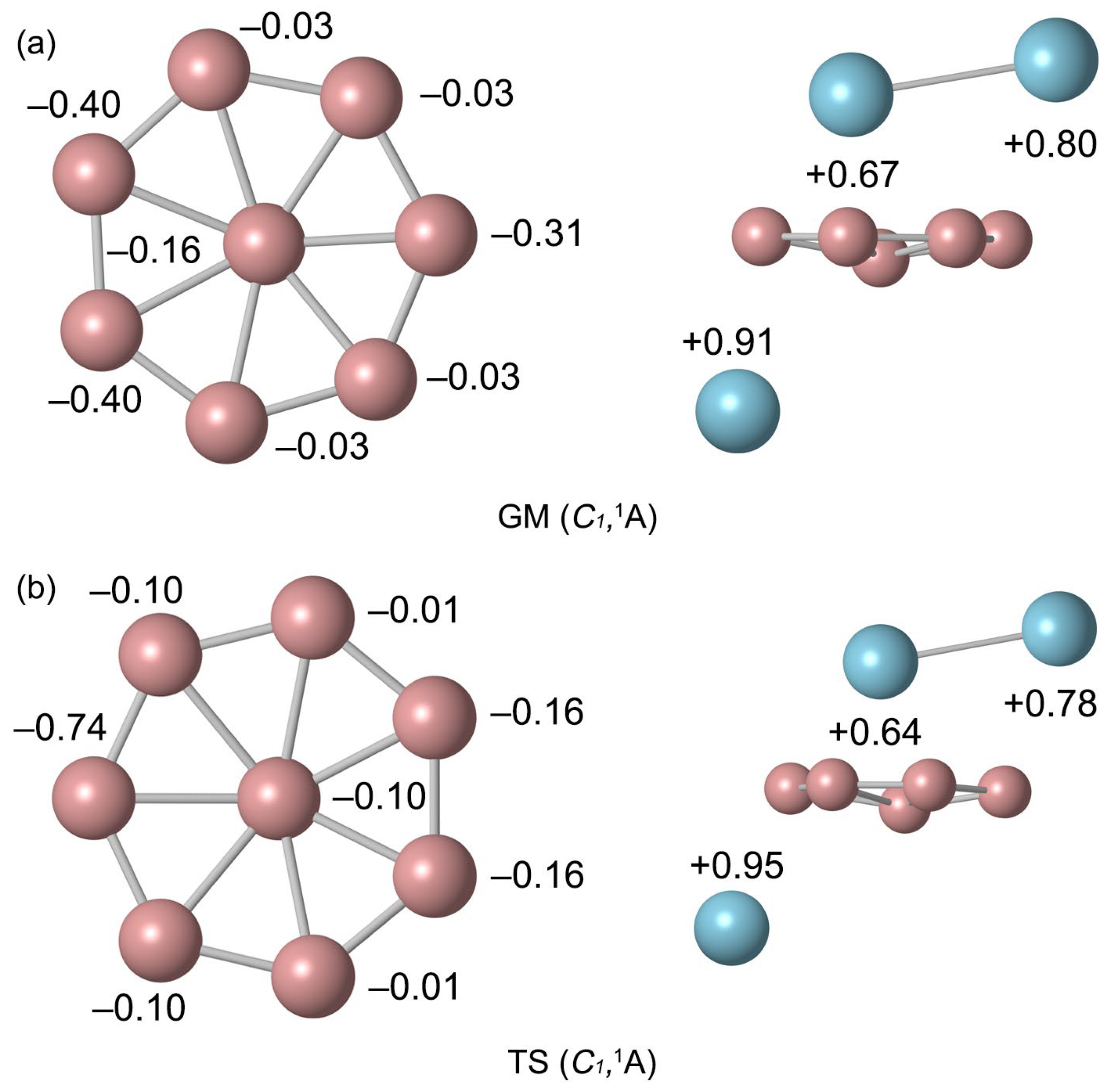
2.2. Bond Distances, Wiberg Bond Indices, and Natural Atomic Charges
3. Discussion
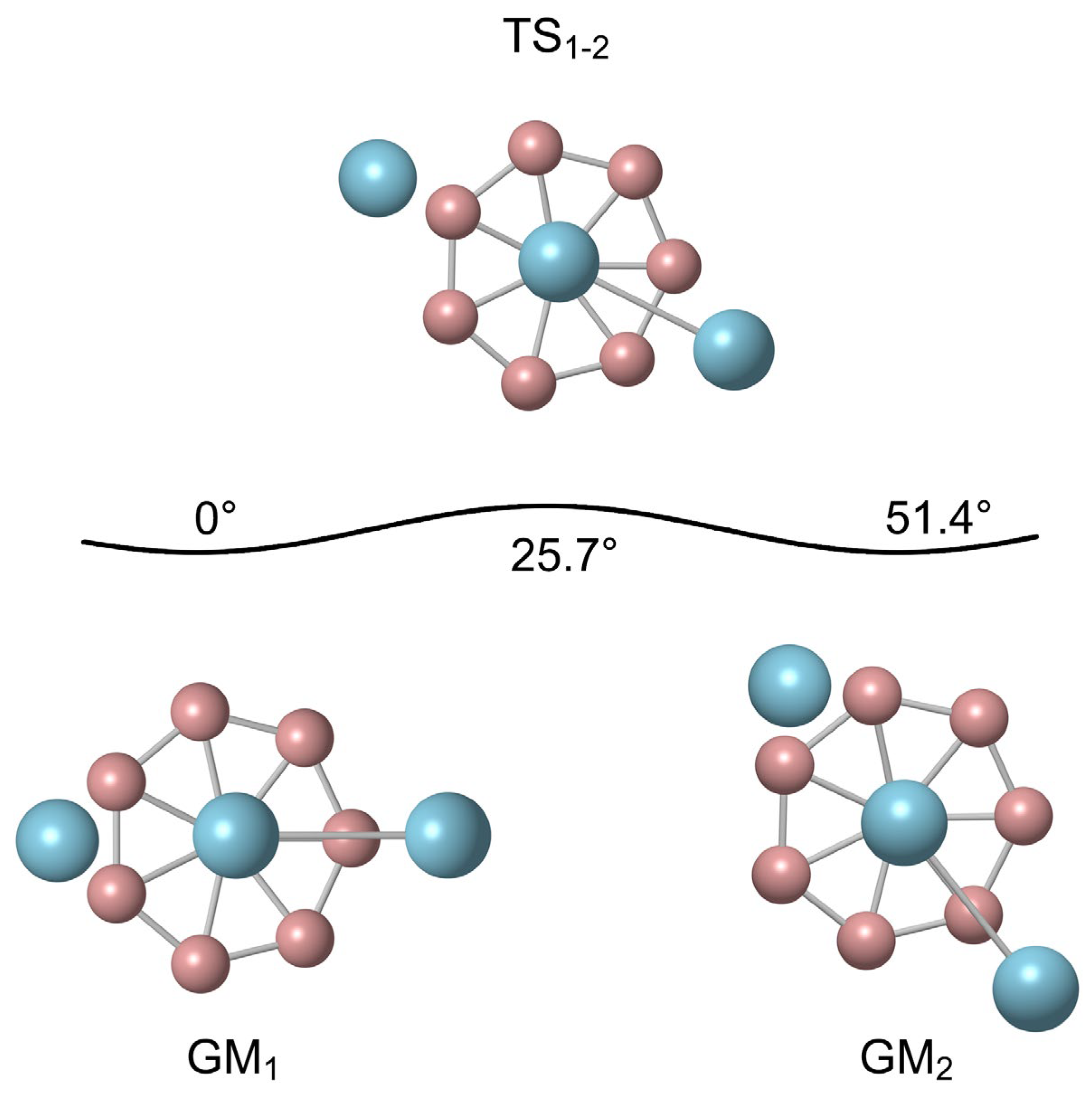
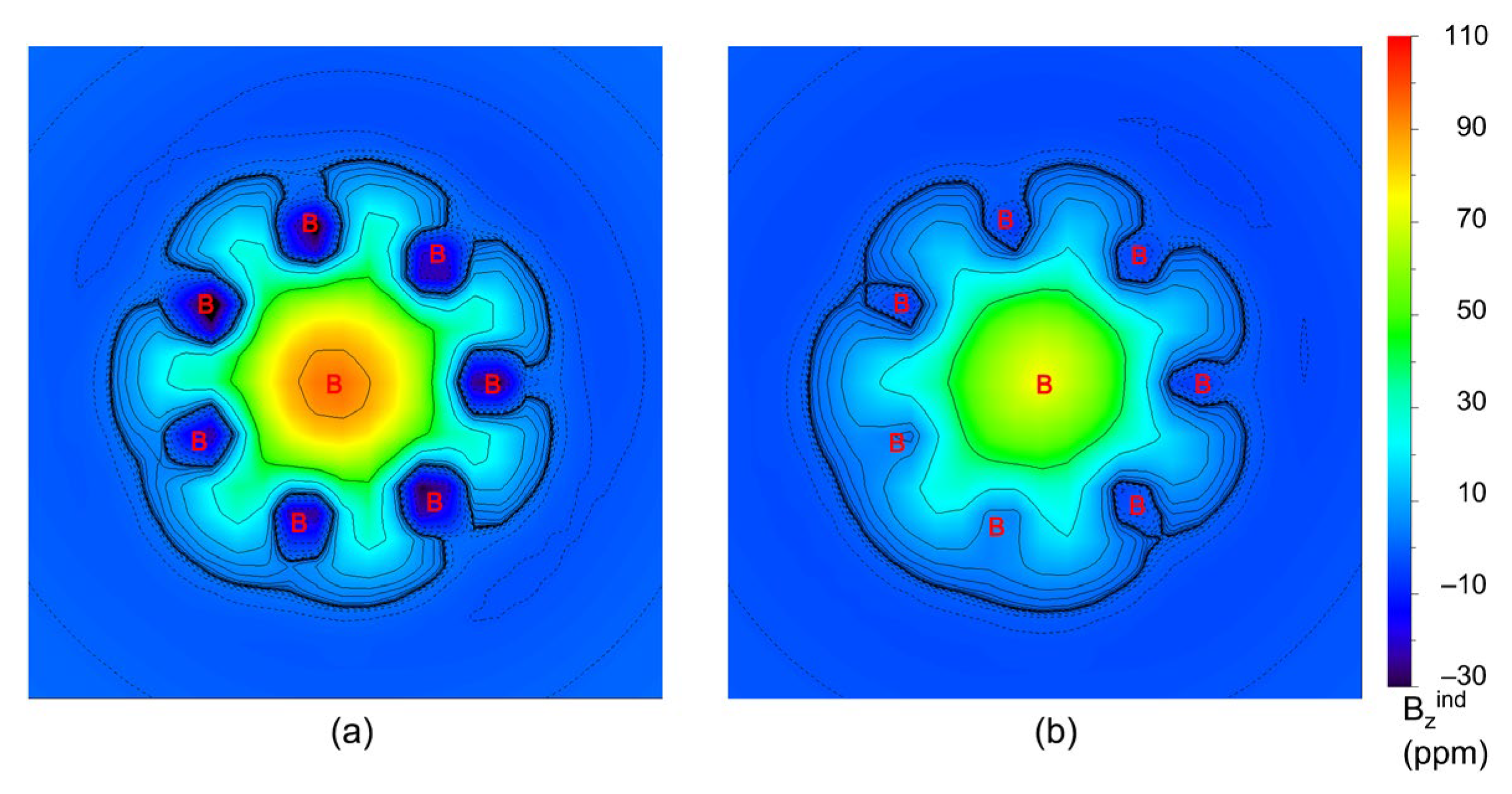
3.1. Dynamic Structural Fluxionality
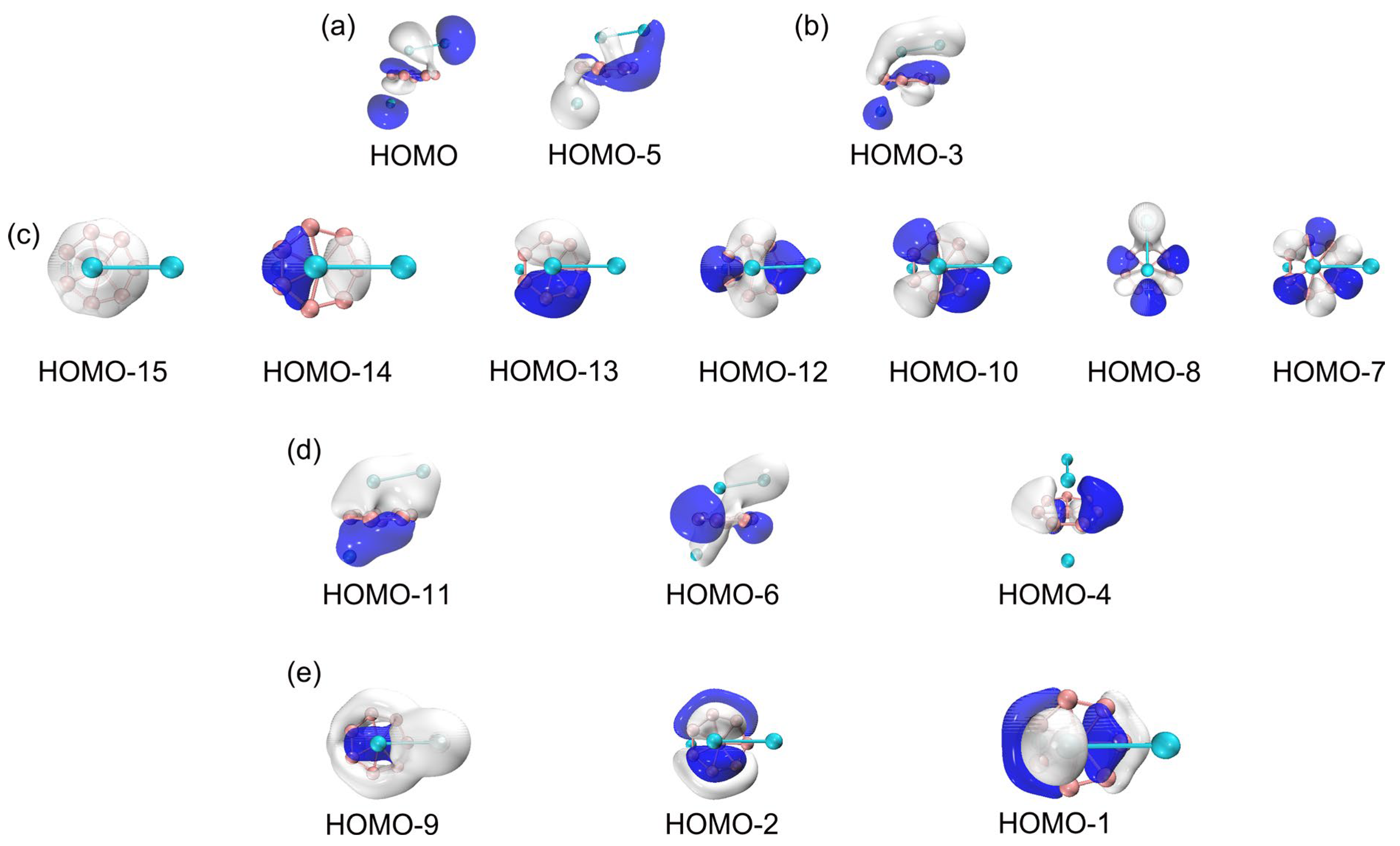
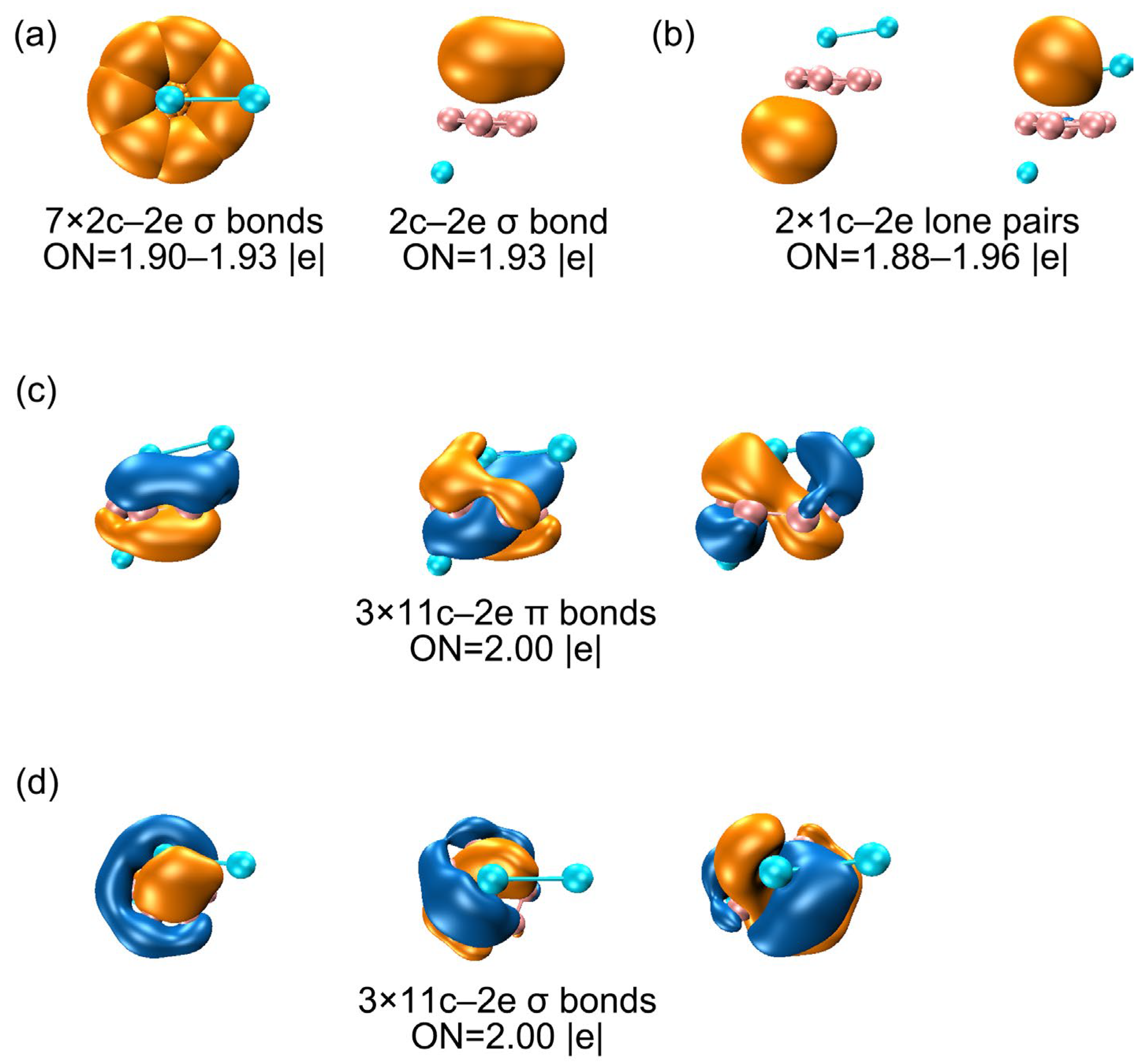
3.2. Chemical Bonding
4. Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ruatta, S.A.; Hamley, L.; Anderson, S.L. Dynamics of boron cluster ion reactions with deuterium: Adduct formation and decay. J. Chem. Phys. 1989, 91, 226–239. [Google Scholar] [CrossRef]
- Boustani, I.; Quandt, A.; Hernández, E.; Rubio, A. New boron based nanostructured materials. J. Chem. Phys. 1999, 110, 3176–3185. [Google Scholar] [CrossRef]
- Fowler, J.E.; Ugalde, J.M. The curiously stable B13+ cluster and its neutral and anionic counterparts: The advantages of planarity. J. Phys. Chem. A 2000, 104, 397–403. [Google Scholar] [CrossRef]
- Aihara, J.I. B13+ is highly aromatic. J. Phys. Chem. A 2001, 105, 5486–5489. [Google Scholar] [CrossRef]
- Aihara, J.I.; Kanno, H.; Ishida, T. Aromaticity of planar boron clusters confirmed. J. Am. Chem. Soc. 2005, 127, 13324–13330. [Google Scholar] [CrossRef] [PubMed]
- Zhai, H.-J.; Kiran, B.; Li, J.; Wang, L.-S. Hydrocarbon analogues of boron clusters-planarity, aromaticity and antiaromaticity. Nat. Mater. 2003, 2, 827–833. [Google Scholar] [CrossRef] [PubMed]
- Zhai, H.-J.; Alexandrova, A.N.; Birch, K.A.; Boldyrev, A.I.; Wang, L.-S. Hepta- and octacoordinate boron in molecular wheels of eight- and nine- atom boron clusters: Observation and confirmation. Angew. Chem. Int. Ed. 2003, 42, 6004–6008. [Google Scholar] [CrossRef] [PubMed]
- Oger, E.; Crawford, N.R.M.; Kelting, R.; Weis, P.; Kappes, M.M.; Ahlrichs, R. Boron cluster cations: Transition from planar to cylindrical structures. Angew. Chem. Int. Ed. 2007, 46, 8503–8506. [Google Scholar] [CrossRef] [PubMed]
- Alexandrova, A.N.; Boldyrev, A.I.; Zhai, H.-J.; Wang, L.-S. All-boron aromatic clusters as potential new inorganic ligands and building blocks in chemistry (review). Coord. Chem. Rev. 2006, 250, 2811–2866. [Google Scholar] [CrossRef]
- Li, W.-L.; Chen, Q.; Tian, W.-J.; Bai, H.; Zhao, Y.-F.; Hu, H.-S.; Li, J.; Zhai, H.-J.; Li, S.-D.; Wang, L.-S. The B35 cluster with a double-hexagonal vacancy: A new and more flexible structural motif for borophene. J. Am. Chem. Soc. 2014, 136, 12257–12260. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-J.; Zhao, Y.-F.; Li, W.-L.; Jian, T.; Chen, Q.; You, X.-R.; Ou, T.; Zhao, X.-Y.; Zhai, H.-J.; Li, S.-D.; et al. Observation and characterization of the smallest borospherene, B28− and B28. J. Chem. Phys. 2016, 144, 064307. [Google Scholar] [CrossRef] [PubMed]
- Sergeeva, A.P.; Zubarev, D.Y.; Zhai, H.-J.; Boldyrev, A.I.; Wang, L.-S. A photoelectron spectroscopic and theoretical study of B16− and B162−: An all-boron naphthalene. J. Am. Chem. Soc. 2008, 130, 7244–7246. [Google Scholar] [CrossRef] [PubMed]
- Zhai, H.-J.; Zhao, Y.-F.; Li, W.-L.; Chen, Q.; Bai, H.; Hu, H.-S.; Piazza, Z.A.; Tian, W.-J.; Lu, H.-G.; Wu, Y.-B.; et al. Observation of an all-boron fullerene. Nat. Chem. 2014, 6, 727–731. [Google Scholar] [CrossRef]
- Mannix, A.J.; Zhou, X.-F.; Kiraly, B.; Wood, J.D.; Alducin, D.; Myers, B.D.; Liu, X.-L.; Fisher, B.L.; Santiago, U.; Guest, J.R.; et al. Synthesis of borophenes: Anisotropic, two-dimensional boron polymorphs. Science 2015, 350, 1513–1516. [Google Scholar] [CrossRef]
- Feng, B.; Zhang, J.; Zhong, Q.; Li, W.; Li, S.; Li, H.; Cheng, P.; Meng, S.; Chen, L.; Wu, K. Experimental realization of two-dimensional boron sheets. Nat. Chem. 2016, 8, 563–568. [Google Scholar] [CrossRef] [PubMed]
- Li, W.-L.; Jian, T.; Chen, X.; Li, H.-R.; Chen, T.-T.; Luo, X.-M.; Li, S.-D.; Li, J.; Wang, L.-S. Observation of a metal-centered B2-Ta@B18− tubular molecular rotor and a perfect Ta@B20− boron drum with the record coordination number of twenty. Chem. Commun. 2017, 53, 1587–1590. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Moreno, D.; Osorio, E.; Castro, A.C.; Pan, S.; Chattaraj, P.K.; Heine, T.; Merino, G. Structure and bonding of IrB12−: Converting a rigid boron B12 platelet to a Wankel motor. RSC Adv. 2016, 6, 27177–27182. [Google Scholar] [CrossRef]
- Popov, I.A.; Li, W.-L.; Piazza, Z.A.; Boldyrev, A.I.; Wang, L.-S. Complexes between planar boron clusters and transition metals: A photoelectron spectroscopy and ab initio study of CoB12− and RhB12−. J. Phys. Chem. A 2014, 118, 8098–8105. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.; Li, H.-R.; Zhao, X.-Y.; Lu, X.-Q.; Mu, Y.-W.; Lu, H.-G.; Li, S.-D. Fluxional bonds in planar B19−, tubular Ta@B20−, and cage-like B39−. J. Comput. Chem. 2018, 40, 966–970. [Google Scholar] [CrossRef] [PubMed]
- Yue, R.-X.; Gao, S.-J.; Han, P.-F.; Zhai, H.-J. Chemical bonding and dynamic structural fluxionality of a boron-based Al2B8 binary cluster: The robustness of a doubly 6π/6σ aromatic [B8]2− molecular. RSC Adv. 2023, 13, 1964–1973. [Google Scholar] [CrossRef]
- Chen, W.-J.; Chen, T.-T.; Chen, Q.; Lu, H.-G.; Zhao, X.-Y.; Ma, Y.-Y.; Yan, Q.-Q.; Yuan, R.-N.; Li, S.-D.; Wang, L.S. Boron-lead multiple bonds in the PbB2O– and PbB3O2– clusters. Commun. Chem. 2022, 25, 00643. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-J.; Zhao, X.-Y.; Chen, Q.; Zhai, H.-J.; Li, S.-D. B11−: A moving subnanoscale tank tread. Nanoscale 2015, 7, 16054–16060. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Guajardo, G.; Sergeeva, A.P.; Boldyrev, A.I.; Heine, T.; Ugalde, J.M.; Merino, G. Unravelling phenomenon of internal rotation in B13+ through chemical bonding analysis. Chem. Commun. 2011, 47, 6242–6244. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Halla, J.O.C.; Islas, R.; Heine, T.; Merino, G. B19−: An aromatic wankel motor. Angew. Chem. Int. Ed. 2010, 49, 5668–5671. [Google Scholar] [CrossRef]
- Huang, W.; Sergeeva, A.P.; Zhai, H.-J.; Averkiev, B.B.L.; Wang, L.-S.; Boldyrev, A.I. A concentric planar doubly π-aromatic B19− cluster. Nat. Chem. 2010, 2, 202206. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.-C.; Feng, L.-Y.; Wang, Y.-J.; Jalife, S.; Vásquez-Espinal, A.; Cabellos, J.L.; Pan, S.; Merino, G.; Zhai, H.-J. Coaxial triple-layered versus helical Be6B11− clusters: Dual structural fluxionality and multifold aromaticity. Angew. Chem. Int. Ed. 2017, 56, 10174–10177. [Google Scholar] [CrossRef] [PubMed]
- Han, P.-F.; Wang, Y.-J.; Feng, L.-Y.; Gao, S.-J.; Sun, Q.; Zhai, H.-J. Chemical bonding and dynamic structural fluxionality of a boron-based Na5B7 sandwich cluster. Molecules 2023, 28, 3276. [Google Scholar] [CrossRef] [PubMed]
- Han, P.-F.; Sun, Q.; Zhai, H.-J. Boron-based inverse sandwich V2B7− cluster: Double π/σ aromaticity, metal-metal bonding, and chemical analogy to planar hypercoordinate molecular wheels. Molecules 2023, 28, 4721. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-J.; Feng, L.-Y.; Yan, M.; Zhai, H.-J. Be3B11− cluster: A dynamically fluxional berylo-borospherene. Phys. Chem. Chem. Phys. 2023, 25, 2846–2852. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.; Yan, G.-R.; Liu, Y.-Q.; Cui, Z.-H. Two-layer molecular rotors: A zinc dimer rotating over planar hypercoordinate motifs. J. Comput. Chem. 2023, 30, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-J.; Feng, L.-Y.; Xu, L.; Hou, X.-R.; Li, N.; Miao, C.-Q.; Zhai, H.-J. Boron-based ternary Rb6Be2B6 cluster featuring unique sandwich geometry and a naked hexagonal boron ring. Phys. Chem. Chem. Phys. 2020, 22, 20043. [Google Scholar] [CrossRef] [PubMed]
- Bera, P.P.; Sattelmeyer, K.W.; Saunders, M.; Schaefer, H.F., III; Schleyer, P.v.R. Mindless chemistry. J. Phys. Chem. 2006, 110, 4287–4290. [Google Scholar] [CrossRef] [PubMed]
- Saunders, M. Stochastic search for isomers on a quantum mechanical surface. J. Comput. Chem. 2004, 25, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Yañez, O.; Vásquez-Espinal, A.; Pino-Rios, R.; Ferraro, F.; Pan, -S.; Osorio, E.; Merino, G.; Tiznado, W. Exploiting electronic strategies to stabilize a planar tetracoordinate carbon in cyclic aromatic hydrocarbons. Chem. Commun. 2017, 53, 12112–12115. [Google Scholar] [CrossRef] [PubMed]
- Peterson, K.A.; Figgen, D.; Goll, E.; Stoll, H.; Dolg, M. Systematically convergent basis sets with relativistic pseudopotentials. II. small-core pseudopotentials and correlation consistent basis sets for the post-d group 16–18 elements. J. Chem. Phys. 2003, 119, 11113–11123. [Google Scholar] [CrossRef]
- Purvis, G.D., III; Bartlett, R.J. A full coupled-cluster singles and doubles model: The inclusion of disconnected triples. J. Chem. Phys. 1982, 76, 1910–1918. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Scuseria, G.E.; Schaefer, H.F., III. Is coupled cluster singles and doubles (CCSD) more computationally intensive than quadratic configuration interaction (QCISD)? J. Chem. Phys. 1989, 90, 3700–3703. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Zubarev, D.Y.; Boldyrev, A.I. Developing paradigms of chemical bonding: Adaptive natural density partitioning. Phys. Chem. Chem. Phys. 2008, 10, 5207–5217. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Wannere, C.S.; Corminboeuf, C.; Puchta, R.; Schleyer, P.v.R. Nucleus-independent chemical shifts (NICS) as an aromaticity criterion. Chem. Rev. 2005, 105, 3842–3888. [Google Scholar] [CrossRef] [PubMed]
- Klod, S.; Kleinpeter, E. Ab initio calculation of the anisotropy effect of multiple bonds and the ring current effect of arenes-application in conformational and configurational analysis. J. Chem. Soc. Perkin Trans. 2001, 2, 1893–1898. [Google Scholar]
- Lu, T.; Chen, F.-W. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- VandeVondele, J.; Krack, M.; Mohamed, F.; Parrinello, M.; Chassaing, T.; Hutter, J. Quickstep: Fast and accurate density functional calculations using a mixed gaussian and plane waves approach. Comput. Phys. Commun. 2005, 167, 103–128. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. GAUSSIAN 09, Revision D.01, Gaussian, Inc.: Wallingford, CT, USA, 2009.
- Dennington, R.; Keith, T.; Millam, J. GaussView, Version 5; Semichem, Inc.: Shawnee Mission, KS, USA, 2009.
- Legault, C.Y. CYLview, 1.0b, Universitè de Sherbrooke. 2009. Available online: https://www.cylview.org. (accessed on 1 September 2024).
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, S.-J.; Yu, T.-L. Chemical Bonding and Dynamic Structural Fluxionality of a Boron-Based B8Al3+ Cluster. Molecules 2024, 29, 5961. https://doi.org/10.3390/molecules29245961
Gao S-J, Yu T-L. Chemical Bonding and Dynamic Structural Fluxionality of a Boron-Based B8Al3+ Cluster. Molecules. 2024; 29(24):5961. https://doi.org/10.3390/molecules29245961
Chicago/Turabian StyleGao, Shu-Juan, and Tan-Lai Yu. 2024. "Chemical Bonding and Dynamic Structural Fluxionality of a Boron-Based B8Al3+ Cluster" Molecules 29, no. 24: 5961. https://doi.org/10.3390/molecules29245961
APA StyleGao, S.-J., & Yu, T.-L. (2024). Chemical Bonding and Dynamic Structural Fluxionality of a Boron-Based B8Al3+ Cluster. Molecules, 29(24), 5961. https://doi.org/10.3390/molecules29245961