

Determination of Tire Wear Particle-Type Polymers by Combination of Quantitative Nuclear Magnetic Resonance Spectroscopy and Soxhlet Extraction


Abstract
1. Introduction
2. Results and Discussion
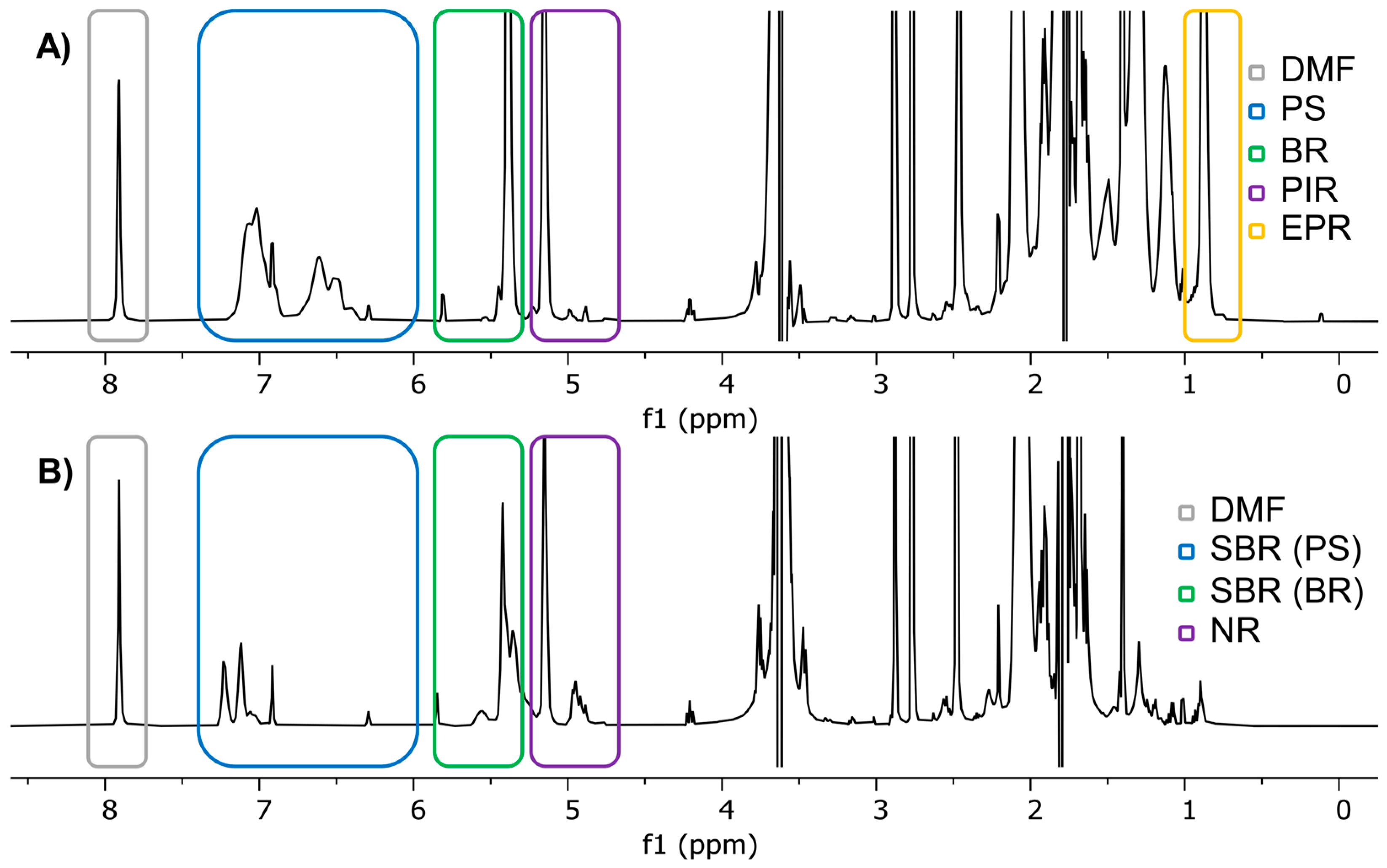
2.1. qNMR Validation
2.2. TWP Extraction
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Brahney, J.; Mahowald, N.; Prank, M.; Cornwell, G.; Klimont, Z.; Matsui, H.; Prather, K.A. Constraining the atmospheric limb of the plastic cycle. Proc. Natl. Acad. Sci. USA 2021, 118, e2020719118. [Google Scholar] [CrossRef] [PubMed]
- Baensch-Baltruschat, B.; Kocher, B.; Stock, F.; Reifferscheid, G. Tyre and road wear particles (TRWP)—A review of generation, properties, emissions, human health risk, ecotoxicity, and fate in the environment. Sci. Total Environ. 2020, 733, 137823. [Google Scholar] [CrossRef] [PubMed]
- Wagner, S.; Klöckner, P.; Reemtsma, T. Aging of tire and road wear particles in terrestrial and freshwater environments—A review on processes, testing, analysis and impact. Chemosphere 2022, 288, 132467. [Google Scholar] [CrossRef] [PubMed]
- Sommer, F.; Dietze, V.; Baum, A.; Sauer, J.; Gilge, S.; Maschowski, C.; Gieré, R. Tire Abrasion as a Major Source of Microplastics in the Environment. Aerosol Air Qual. Res. 2018, 18, 2014–2028. [Google Scholar] [CrossRef]
- Goßmann, I.; Halbach, M.; Scholz-Böttcher, B.M. Car and truck tire wear particles in complex environmental samples—A quantitative comparison with “traditional” microplastic polymer mass loads. Sci. Total Environ. 2021, 773, 145667. [Google Scholar] [CrossRef] [PubMed]
- Klöckner, P.; Seiwert, B.; Eisentraut, P.; Braun, U.; Reemtsma, T.; Wagner, S. Characterization of tire and road wear particles from road runoff indicates highly dynamic particle properties. Water Res. 2020, 185, 116262. [Google Scholar] [CrossRef] [PubMed]
- Ziajahromi, S.; Lu, H.-C.; Drapper, D.; Hornbuckle, A.; Leusch, F.D.L. Microplastics and Tire Wear Particles in Urban Stormwater: Abundance, Characteristics, and Potential Mitigation Strategies. Environ. Sci. Technol. 2023, 57, 12829–12837. [Google Scholar] [CrossRef] [PubMed]
- Mennekes, D.; Nowack, B. Tire wear particle emissions: Measurement data where are you? Sci. Total Environ. 2022, 830, 154655. [Google Scholar] [CrossRef] [PubMed]
- Primpke, S.; Christiansen, S.H.; Cowger, W.; De Frond, H.; Deshpande, A.; Fischer, M.; Holland, E.B.; Meyns, M.; O’Donnell, B.A.; Ossmann, B.E.; et al. Critical Assessment of Analytical Methods for the Harmonized and Cost-Efficient Analysis of Microplastics. Appl. Spectrosc. 2020, 74, 1012–1047. [Google Scholar] [CrossRef]
- Kovochich, M.; Liong, M.; Parker, J.A.; Oh, S.C.; Lee, J.P.; Xi, L.; Kreider, M.L.; Unice, K.M. Chemical mapping of tire and road wear particles for single particle analysis. Sci. Total Environ. 2021, 757, 144085. [Google Scholar] [CrossRef] [PubMed]
- Rødland, E.S.; Gustafsson, M.; Jaramillo-Vogel, D.; Järlskog, I.; Müller, K.; Rauert, C.; Rausch, J.; Wagner, S. Analytical challenges and possibilities for the quantification of tire-road wear particles. TrAC 2023, 165, 117121. [Google Scholar] [CrossRef]
- Peez, N.; Janiska, M.C.; Imhof, W. The First Application of Quantitative 1H-NMR-Spectroscopy as a Simple and Fast Method of Identification and Quantification of Microplastic Particles (PE, PET and PS). Anal. Bioanal. Chem. 2019, 411, 823–833. [Google Scholar] [CrossRef] [PubMed]
- Peez, N.; Becker, J.; Ehlers, S.M.; Fritz, M.; Fischer, C.B.; Koop, J.H.E.; Winkelmann, C.; Imhof, W. Quantitative Analysis of PET Microplastics in Environmental Model Samples Using Quantitative NMR Spectroscopy: Validation of an Optimized and Consistent Clean-up Method. Anal. Bioanal. Chem. 2019, 411, 7409–7418. [Google Scholar] [CrossRef]
- Peez, N.; Imhof, W. Quantitative 1H-NMR Spectroscopy as an Efficient Method for Identification and Quantification of PVC, ABS and PA Microparticles. Analyst 2020, 145, 5363–5371. [Google Scholar] [CrossRef] [PubMed]
- Peez, N.; Rinesch, T.; Kolz, J.; Imhof, W. Applicable and cost-efficient microplastic analysis by quantitative 1H-NMR spectroscopy using benchtop NMR and NoD methods. Magn. Res. Chem. 2022, 60, 172–183. [Google Scholar] [CrossRef]
- Günther, M.; Imhof, W. Simultaneous quantification of microplastic particles by NoD 1H-qNMR from samples comprising different polymer types. Analyst 2023, 148, 1151–1161. [Google Scholar] [CrossRef] [PubMed]
- Günther, M.; Imhof, W. Highly selective solid liquid extraction of microplastic mixtures as a pre-preparation tool for quantitative nuclear magnetic resonance spectroscopy studies. Analyst 2024, 149, 5800–5811. [Google Scholar] [CrossRef]
- Nelson, T.F.; Remke, S.C.; Kohler, H.-P.E.; McNeill, K.; Sander, M. Quantification of Synthetic Polyesters from Biodegradable Mulch Films in Soils. Environ. Sci. Technol. 2020, 54, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Dukek, P.; Schleheck, D.; Kovermann, M. High-Resolution NMR Spectroscopic Approaches to Quantify PET Microplastics Pollution in Environmental Freshwater Samples. Chemosphere 2024, 367, 143657. [Google Scholar] [CrossRef] [PubMed]
- Bellasi, A.; Binda, G.; Pozzi, A.; Boldrocchi, G.; Bettinetti, R. The extraction of microplastics from sediments: An overview of existing methods and the proposal of a new and green alternative. Chemosphere 2021, 278, 130357. [Google Scholar] [CrossRef]
- He, D.; Zhang, X.; Hu, J. Methods for separating microplastics from complex solid matrices: Comparative analysis. J. Hazard. Mater. 2021, 409, 124640. [Google Scholar] [CrossRef] [PubMed]
- Cashman, M.A.; Ho, K.T.; Boving, T.B.; Russo, S.; Robinson, S.; Burgess, R.M. Comparison of microplastic isolation and extraction procedures from marine sediments. Mar. Pollut. Bull. 2020, 159, 111507. [Google Scholar] [CrossRef]
- Fuller, S.; Gautam, A. A Procedure for Measuring Microplastics using Pressurized Fluid Extraction. Environ. Sci. Technol. 2016, 50, 5774–5780. [Google Scholar] [CrossRef]
- Dierkes, G.; Lauschke, T.; Becher, S.; Schumacher, H.; Földi, C.; Ternes, T. Quantification of microplastics in environmental samples via pressurized liquid extraction and pyrolysis-gas chromatography. Anal. Bioanal. Chem. 2019, 411, 6959–6968. [Google Scholar] [CrossRef] [PubMed]
- Macko, T.; Pasch, H.; Wang, Y. Liquid Chromatographic Separation of Olefin Oligomers and its Relation to Separation of Polyolefins—An Overview. Macromol. Symp. 2009, 282, 93–100. [Google Scholar] [CrossRef]
- Steinmetz, Z.; Kintzi, A.; Muñoz, K.; Schaumann, G.E. A simple method for the selective quantification of polyethylene, polypropylene, and polystyrene plastic debris in soil by pyrolysis-gas chromatography/mass spectrometry. J. Anal. Appl. Pyrolysis 2020, 147, 104803. [Google Scholar] [CrossRef]
- Comins, D.L.; Joseph, S.P. Encyclopedia of Reagents for Organic Synthesis; John Wiley & Sons Ltd.: Chichester, UK, 2001. [Google Scholar]
- Hero, D.; Kali, G. New, Aqueous Radical (Co)Polymerization of Olefins at Low Temperature and Pressure. Processes 2020, 8, 688. [Google Scholar] [CrossRef]
- Rolere, S.; Liengprayoon, S.; Vaysse, L.; Sainte-Beuve, J.; Bonfils, F. Investigating natural rubber composition with Fourier Transform Infrared (FT-IR) spectroscopy: A rapid and non-destructive method to determine both protein and lipid contents simultaneously. Polym. Test 2015, 43, 83–93. [Google Scholar] [CrossRef]
- Castelvetro, V.; Corti, A.; Bianchi, S.; Giacomelli, G.; Manariti, A.; Vinciguerra, V. Microplastics in fish meal: Contamination level analyzed by polymer type, including polyester (PET), polyolefins, and polystyrene. Environ. Pollut. 2021, 273, 115792. [Google Scholar] [CrossRef] [PubMed]
- Chiantore, O.; di Cortemiglia, M.P.L.; Guaita, M.; Rendina, G. Thermal degradation of polybutadiene, 1. Reactions at temperatures lower than 250 °C. Makromol. Chem. 1989, 190, 3143–3152. [Google Scholar]
- Rhieu, S.Y.; Urbas, A.A.; Lippa, K.A.; Reipa, V. Quantitative measurements of glutathione in yeast cell lysate using 1H NMR. Anal. Bioanal. Chem. 2013, 405, 4963–4968. [Google Scholar] [CrossRef] [PubMed]
- Barthlott, L.; Scharinger, A.; Golombek, P.; Kuballa, T.; Lachenmeier, D.W. A Quantitative 1H NMR Method for Screening Cannabinoids in CBD Oils. Toxics 2021, 9, 136. [Google Scholar] [CrossRef] [PubMed]
- Rauert, C.; Rødland, E.S.; Okoffo, D.; Reid, M.J.; Meland, S.; Thomas, K.V. Challenges with Quantifying Tire Road Wear Particles: Recognizing the Need for Further Refinement of the ISO Technical Specification. Environ. Sci. Technol. Lett. 2021, 8, 231–236. [Google Scholar] [CrossRef]
- MestReNova, Version 14.1.1-24571; Mestrelab Research S.L.: San Diego, CA, USA, 2019.

{kind=link}
| MP Type | Range c [mg/mL] | Linearity R2 | RMSD | LOD [µg/mL] | LOQ [µg/mL] |
|---|---|---|---|---|---|
| PS | 0.30–1.51 | 0.99994 | 0.00002 | 12.84 | 42.80 |
| BR | 0.50–2.49 | 0.99995 | 0.00003 | 1.87 | 6.23 |
| PIR | 0.50–2.50 | 0.99983 | 0.00002 | 5.37 | 17.89 |
| EPR | 0.20–0.98 | 0.99952 | 0.00016 | 1.09 | 3.63 |
| MP Type | Massgrav. [mg] | Masscalc. [mg] | Accuracy [%] | Precision [%] |
|---|---|---|---|---|
| PS | 1.34 | 1.32 | 98.6 | 99.8 |
| 1.02 | 1.00 | 98.0 | 99.7 | |
| 0.46 | 0.48 | 104.5 | 99.9 | |
| BR | 2.42 | 2.35 | 97.3 | 99.8 |
| 1.46 | 1.41 | 96.5 | 99.8 | |
| 0.85 | 0.82 | 96.7 | 99.9 | |
| PIR | 2.20 | 2.15 | 97.6 | 99.3 |
| 1.68 | 1.64 | 97.3 | 99.0 | |
| 0.65 | 0.62 | 95.9 | 98.9 | |
| EPR | 0.85 | 0.83 | 97.3 | 99.9 |
| 0.57 | 0.56 | 97.6 | 99.1 | |
| 0.27 | 0.24 | 89.0 | 99.4 |
| MP Type | Range c [mg/mL] | Linearity R2 | RMSD | LOD [µg/mL] | LOQ [µg/mL] |
|---|---|---|---|---|---|
| SBR (PS) | 0.12–0.59 | 0.99956 | 0.00004 | 23.09 | 76.97 |
| SBR (BR) | 0.38–1.91 | 0.99979 | 0.00010 | 8.89 | 29.63 |
| NR | 0.50–2.50 | 0.99830 | 0.00047 | 5.11 | 17.04 |
| MP Type | Massgrav. [mg] | Masscalc. [mg] | Accuracy [%] | Precision [%] |
|---|---|---|---|---|
| SBR (PS) | 0.38 | 0.38 | 98.7 | 99.2 |
| 0.59 | 0.58 | 98.4 | 99.7 | |
| 0.57 | 0.56 | 97.8 | 99.9 | |
| SBR (BR) | 1.23 | 1.19 | 96.4 | 98.5 |
| 1.91 | 1.86 | 97.3 | 99.3 | |
| 1.84 | 1.78 | 96.5 | 99.7 | |
| NR | 2.34 | 2.29 | 97.7 | 99.4 |
| 1.74 | 1.73 | 99.2 | 99.7 | |
| 0.81 | 0.80 | 99.2 | 99.5 |
| MP Type | Massgrav. [mg] | Masscalc.,orig. [mg] | Accuracyorig. [%] | Masscalc.,alt. [mg] | Accuracyalt. [%] |
|---|---|---|---|---|---|
| NR | 2.34 | 2.29 | 97.7 | 2.10 | 89.7 |
| 1.74 | 1.73 | 99.2 | 1.60 | 91.8 | |
| 0.81 | 0.80 | 99.2 | 0.74 | 91.2 | |
| PIR | 2.20 | 2.15 | 97.6 | 2.33 | 110.3 |
| 1.68 | 1.64 | 97.3 | 1.75 | 109.6 | |
| 0.65 | 0.62 | 95.9 | 0.68 | 109.8 |
| MP Type | Massgrav. [mg] | Masscalc. [mg] | Recovery Rate [%] | Precision [%] |
|---|---|---|---|---|
| SBR (PS) | 0.57 | 0.52 | 91.3 | 98.4 |
| SBR (BR) | 1.86 | 1.63 | 87.8 | 99.8 |
| BR | 2.46 | 2.25 | 91.5 | 99.2 |
| NR | 2.47 | 1.91 | 77.2 | 98.7 |
| PIR | 2.47 | 1.93 | 78.3 | 99.5 |
| EPR | 0.96 | 1.15 | 120.1 | 94.1 |
| EPR* | 0.96 | 0.87 | 90.9 | 92.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Günther, M.; Kirimlioglu Sayilik, G.; Imhof, W. Determination of Tire Wear Particle-Type Polymers by Combination of Quantitative Nuclear Magnetic Resonance Spectroscopy and Soxhlet Extraction. Molecules 2024, 29, 5899. https://doi.org/10.3390/molecules29245899
Günther M, Kirimlioglu Sayilik G, Imhof W. Determination of Tire Wear Particle-Type Polymers by Combination of Quantitative Nuclear Magnetic Resonance Spectroscopy and Soxhlet Extraction. Molecules. 2024; 29(24):5899. https://doi.org/10.3390/molecules29245899
Chicago/Turabian StyleGünther, Marcel, Gizem Kirimlioglu Sayilik, and Wolfgang Imhof. 2024. "Determination of Tire Wear Particle-Type Polymers by Combination of Quantitative Nuclear Magnetic Resonance Spectroscopy and Soxhlet Extraction" Molecules 29, no. 24: 5899. https://doi.org/10.3390/molecules29245899
APA StyleGünther, M., Kirimlioglu Sayilik, G., & Imhof, W. (2024). Determination of Tire Wear Particle-Type Polymers by Combination of Quantitative Nuclear Magnetic Resonance Spectroscopy and Soxhlet Extraction. Molecules, 29(24), 5899. https://doi.org/10.3390/molecules29245899