

Initial Examinations of the Diastereoselectivity and Chemoselectivity of Intramolecular Silyl Nitronate [3+2] Cycloadditions with Alkenyl/Alkynyl Nitroethers

Abstract

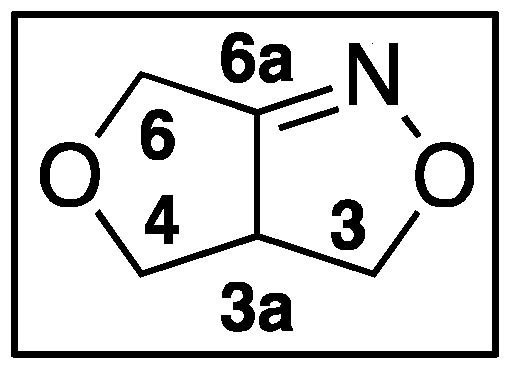
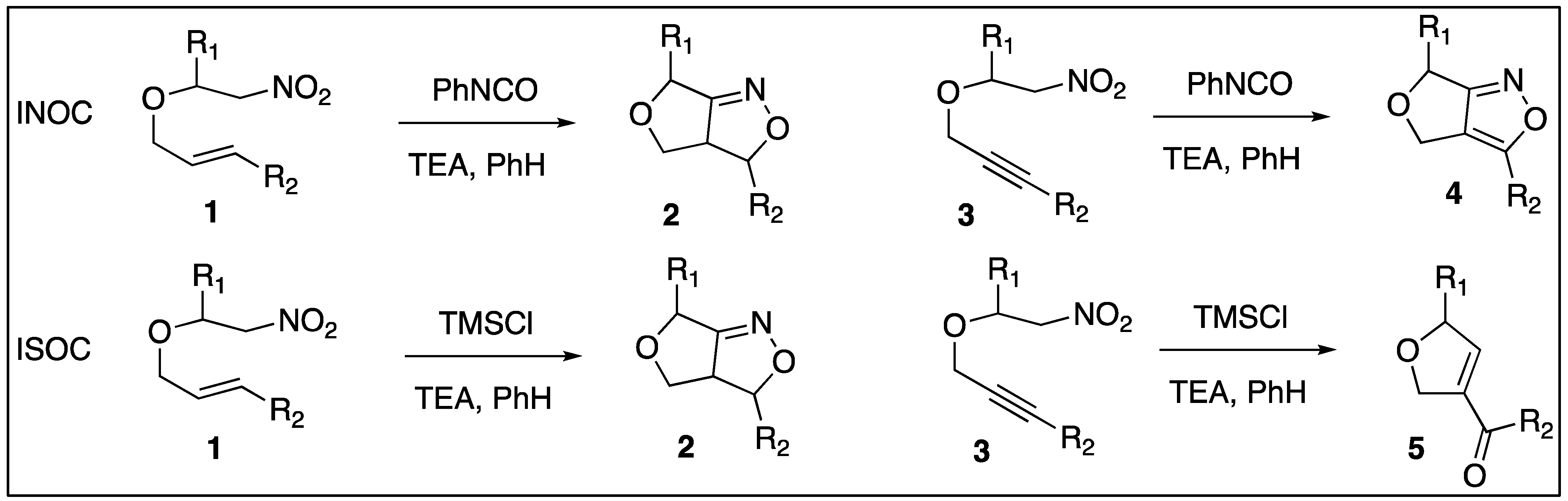
1. Introduction
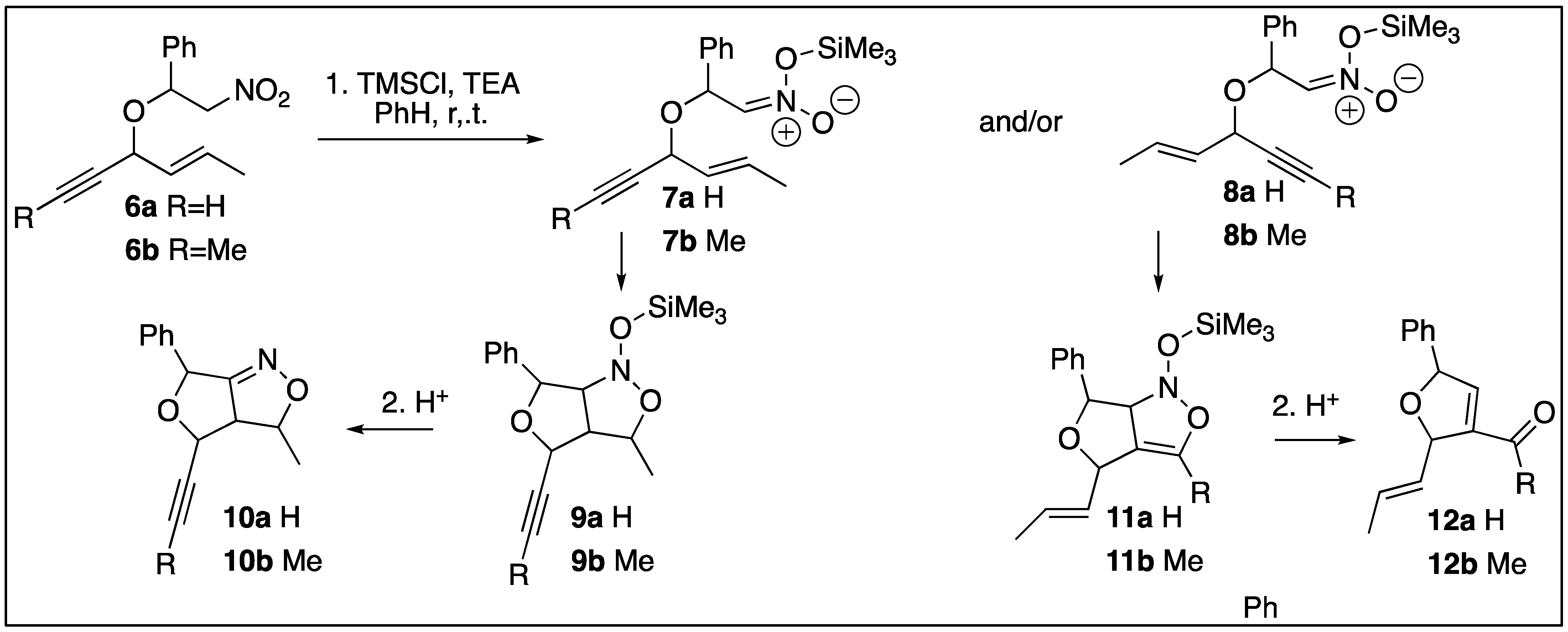
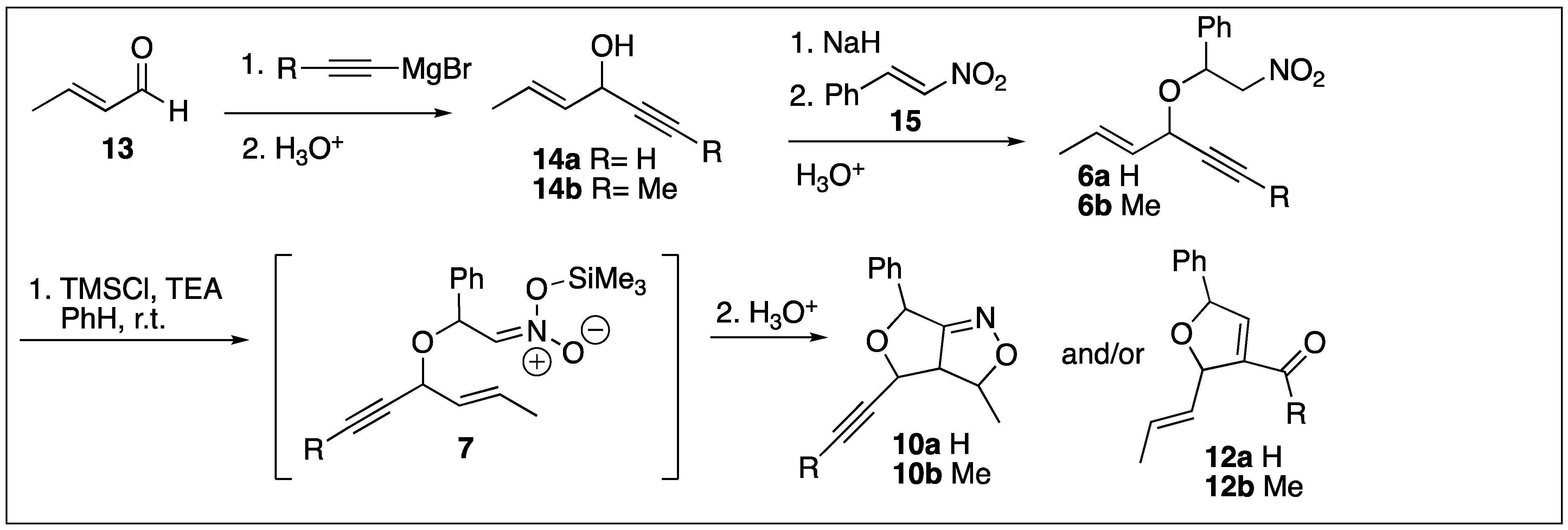
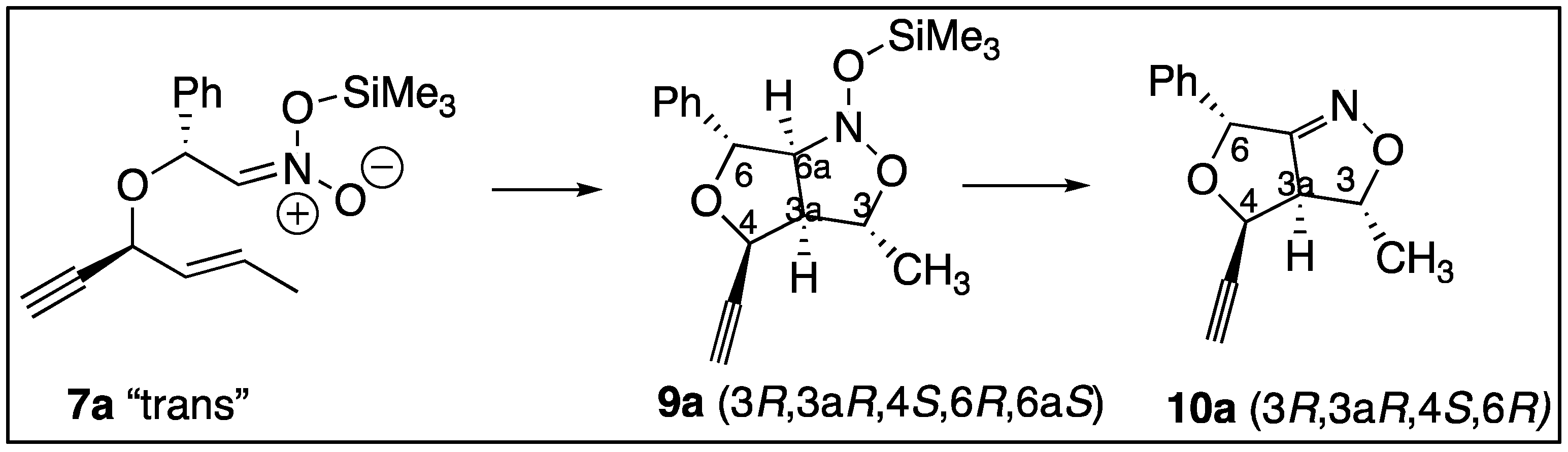
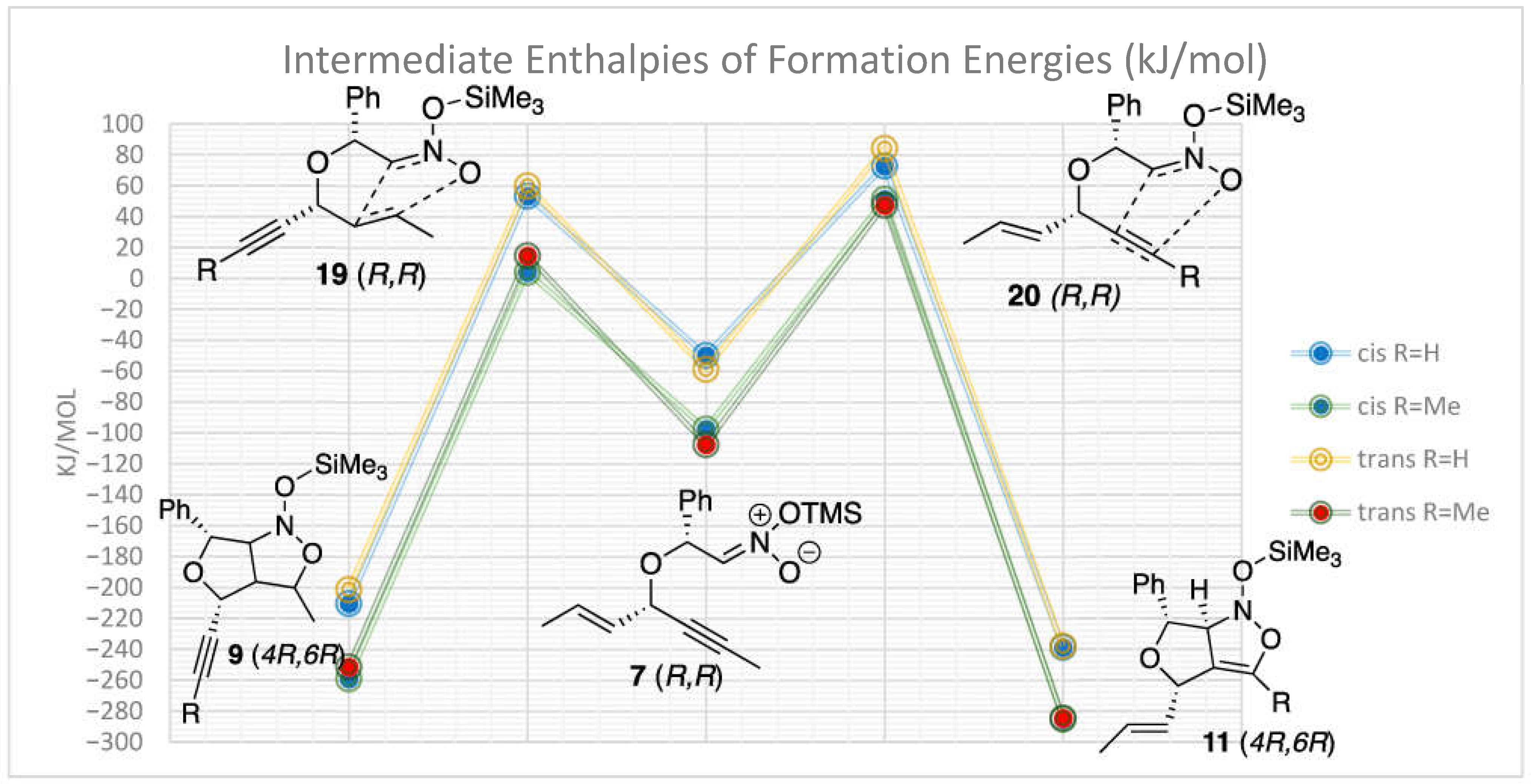
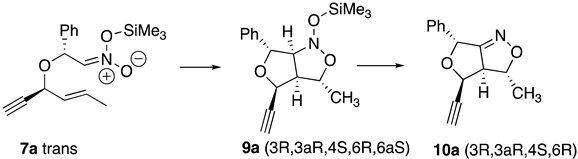
2. Results
3. Discussion
4. Materials and Methods
4.1. Physical and Spectral Data
- Low-resolution mass spectra were examined on a Hewlett Packard G1800A GCD system (Agilent Technologies, Santa Clara, CA, USA).
- Proton and carbon NMR spectra were obtained either on a JEOL (400 MHz) spectrometer (JEOL Ltd., Peabody, MA, USA) or a Varian (300 MHz) system (Varian, Inc., Palo Alto, CA, USA). Listed proton NMR data are given in the following order: ppm (multiplicity, coupling constants, integrated number of protons, and assignment). Listed carbon NMR data are given by chemical shift and assignment. All the spectra were run in deuterated chloroform with TMS (Trimethylsilyl) unless stated otherwise.
- Infrared spectra were recorded on a Nicolet Avatar 361 FT-IR (Thermo Nicolet Corporation, Waltham, MA, USA) with Gateway 2000 data system and were either neat NaCl plates (liquids) or KBr pellets (solids).
4.2. Chromatography
- Flash column chromatography refers to the resolution technique of W. Clark Still [24]. A glass column is filled with a slurry of dry 40–62 μm silica gel and solvent. The same solvent is used as an eluent to push a sample through the column, with pressure from a nitrogen inlet to speed the elution to a rate of 2 in./min.
- TLC refers to thin-layer chromatography, which was performed on Sigma Chemical Co. (Livonia, MI, USA) plates made of 250 μm silica gel on polyester with a 254 nm fluorescent indicator added. Visualization was performed via iodine chambers or UV lamp.
- Capillary gas chromatography (GC) was performed on an HP G1800A GCMS (Agilent Technologies, Santa Clara, CA, USA), equipped with an HP-5 Crosslinked 5% PH ME Siloxane column (Agilent Technologies, Santa Clara, CA, USA), 30 m × 0.25 mm × 0.25 μm.
4.3. Solvents
4.4. Reactions
- Concentration under reduced pressure refers to solvent removal on a Büchi RE 111 rotary evaporator (BüCHI Labortechnik AG, Flawil, Switzerland) connected to a water aspirator and an ethylene glycol cooling system.
- All reactions were run under a nitrogen atmosphere unless otherwise stated.
- Unless otherwise stated, all other solvents and reagents were reagent grade and used without further purification.
4.5. General Procedures
4.5.1. General Procedure A: Alkenynols
4.5.2. General Procedure B: Nitroethers (6)
4.5.3. General Procedure C: Intramolecular Silyl Nitronate Cycloaddition (ISNC)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Young, D.G.J.; Goemez-Bengoa, E.; Hoveyda, A.H. Diastereoselective Intramolecular Cycloaddition of Vinylsilanes and Silyl Nitronates. Effective Control of Remote Acyclic Asymmetry. J. Org. Chem. 1999, 64, 692–693. [Google Scholar] [CrossRef] [PubMed]
- Marrugo, H.; Dogbeavou, R.; Breau, L. Diastereoselective Synthesis of 2-Isoxazolines via Silaketal Tethered 1,3-Dipolar Cycloadditions. Tetrahedron Lett. 1999, 40, 8979–8983. [Google Scholar] [CrossRef]
- Hassner, A.; Friedman, O.; Dehaen, W. Cycloadditions, 55.—Substituent Effects in Tandem Intramolecular Silyl Nitronate Olefin Cycloadditions (ISOC) Leading to Functionalized Tetrahydrofurans. Liebigs Ann. 1997, 1997, 587–594. [Google Scholar] [CrossRef]
- Galley, G.; Jones, P.G.; Pätzel, M. Enantiomerically Pure Isoxazolines by Stereoselective 1,3-Dipolar Cycloaddition of Silyl Nitronates. Tetrahedron Asymmetry 1996, 7, 2073–2082. [Google Scholar] [CrossRef]
- Kim, B.H.; Lee, J.Y.; Kim, K.; Whang, D. Asymmetric Induction in Silyl Nitronate Cycloadditions to Oppolzer’s Chiral Sultam Derivatives. Tetrahedron Asymmetry 1991, 2, 27–30. [Google Scholar] [CrossRef]
- Torsell, K.; Zeuthen, O. Reactions of T-Butyl Nitrones and Trimethylsilyl Nitronates. Synthesis and Reactions of Isoxazolidines and 2-Isoxazolines. Acta Chem. Scand. B 1978, 32, 118–124. [Google Scholar] [CrossRef]
- Gottlieb, L.; Hassner, A. Cycloadditions. 53. Stereoselective Synthesis of Functionalized Pyrrolidines via Intramolecular 1,3-Dipolar Silyl Nitronate Cycloaddition. J. Org. Chem. 1995, 60, 3759–3763. [Google Scholar] [CrossRef]
- Dehaen, W.; Hassner, A. Stereoselectivity in Intramolecular 1,3-Dipolar Cycloadditions. Nitrile Oxides versus Silyl Nitronates. Tetrahedron Lett. 1990, 31, 743–746. [Google Scholar] [CrossRef]
- Kumar, A.; Fernandes, J.; Kumar, P. Synthesis and Biological Evaluation of Some Novel Isoxazoline Derivatives of Carbostyril. World J. Pharm. Pharmacuetical Sci. 2014, 3, 1267–1277. [Google Scholar]
- Kaur, K.; Kumar, V.; Sharma, A.K.; Gupta, G.K. Isoxazoline Containing Natural Products as Anticancer Agents: A Review. Eur. J. Med. Chem. 2014, 77, 121–133. [Google Scholar] [CrossRef]
- Zhou, X.; Hohman, A.E.; Hsu, W.H. Current Review of Isoxazoline Ectoparasiticides Used in Veterinary Medicine. J. Vet. Pharmacol. Ther. 2022, 45, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Indorkar, D.; Chourasia, O.P.; Limaye, S.N. Synthesis, Characterization, Antimicrobial, Antifungal Activity of Some s-Triazine Derivatives of Isoxazoline, Pyrazoline and PC Model Computational Studies. Res. J. Pharm. Sci. 2012, 1, 10–16. [Google Scholar]
- Duffy, J.L.; Kurth, M.J. A Novel Intramolecular Silyl Nitronate Cycloaddition Route to Dihydrofuraldehydes and Dihydropyranaldehydes. J. Org. Chem. 1994, 59, 3783–3785. [Google Scholar] [CrossRef]
- Grandbois, M.L.; Betsch, K.J.; Buchanan, W.D.; Duffy-Matzner, J.L. Synthesis of Novel 2H,5H-Dihydrofuran-3-Yl Ketones via ISNC Reactions. Tetrahedron Lett. 2009, 50, 6446–6449. [Google Scholar] [CrossRef]
- Namboothiri, I.N.N.; Hassner, A.; Gottlieb, H.E. A Highly Stereoselective One-Pot Tandem Consecutive 1,4-Addition−Intramolecular 1,3-Dipolar Cycloaddition Strategy for the Construction of Functionalized Five- and Six-Membered Carbocycles. J. Org. Chem. 1997, 62, 485–492. [Google Scholar] [CrossRef]
- Cheng, Y.; Wang, J.; Hu, Z.; Zhong, S.; Huang, N.; Zhao, Y.; Tao, Y.; Liang, Y. Preparation of Norfloxacin-Grafted Chitosan Antimicrobial Sponge and Its Application in Wound Repair. Int. J. Biol. Macromol. 2022, 210, 243–251. [Google Scholar] [CrossRef]
- Duffy, J.L.; Kurth, J.A.; Kurth, M.J. Lithium, Potassium and Sodium Alkoxides: Donors in the Michael Addition Reaction of α-Nitroolefins. Tetrahedron Lett. 1993, 34, 1259–1260. [Google Scholar] [CrossRef]
- Kim, H.R.; Kim, H.J.; Duffy, J.L.; Olmstead, M.M.; Ruhlandt-Senge, K.; Kurth, M.J. Double Diastereoselectivity in the Intramolecular Nitrile Oxide-Olefin Cycloaddition (INOC) Reaction. Tetrahedron Lett. 1991, 32, 4259–4262. [Google Scholar] [CrossRef]
- Duffy, J.L. An Investigation into the Diastereoselectivity of the Intramolecular Nitrile Oxide Olefin Cycloaddition vs. the Intramolecular Silyl Nitronate Olefin Cycloaddition. Ph.D. Thesis, University of California, Davis, CA, USA, 1993. [Google Scholar]
- Kim, H.R.; Kim, K.M.; Kim, J.N.; Ryu, E.K. Regioselectivity and Stereoselectivity in the Intramolecular Nitrile Oxide-Olefin Cycloaddition (INOC) Reaction and the Intramolecular Silyl Nitronate-Olefin Cycloaddition (ISOC) Reaction. Synth. Commun. 1994, 24, 1107–1116. [Google Scholar] [CrossRef]
- Chiacchio, M.A.; Legnani, L. Density Functional Theory Calculations: A Useful Tool to Investigate Mechanisms of 1,3-Dipolar Cycloadditions Reactions. Int. J. Mol. Sci. 2024, 25, 1298. [Google Scholar] [CrossRef]
- Stewart, J.J.P. Optimization of Parameters for Semiempirical Methods V: Modification of NDDO Approximations and Application to 70 Elements. J. Mol. Model. 2007, 13, 1173–1213. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.J.P. Optimization of Parameters for Semiempirical Methods VI: More Modifications to the NDDO Approximations and Re-Optimization of Parameters. J. Mol. Model. 2013, 19, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Gairaud, C.B.; Lappin, G.R. The Synthesis of ι-Nitrostyrenes. J. Org. Chem. 1953, 18, 1–3. [Google Scholar] [CrossRef]
- Kącka-Zych, A. Push-pull nitronates in the [3+2] cycloaddition with nitroethylene: Molecular Electron Density Theory Study. J. Mol. Graph. Model. 2020, 97, 107549. [Google Scholar] [CrossRef]
- Kącka-Zych, A.; Jasiński, R. Understanding the molecular mechanism of the stereoselective conversion of N-trialkylsilyloxy nitronates into bicyclic isoxazoline derivatives. New J. Chem. 2021, 45, 9491–9500. [Google Scholar] [CrossRef]


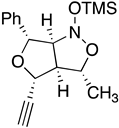
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 10a Major | 10a Minor | 10b Major | 10b Minor |
|---|---|---|---|
 | 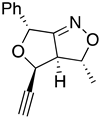 |  |  |
| (±)-(3R,3aR,4R,6R) | (±)-(3R,3aR,4S,6R) | (±)-(3R,3aR,4R,6R) | (±)-(3R,3aR,4S,6R) |
| I 3R 3aR 4R 6R 6aS | II 3S 3aS 4R 6R 6aR | III 3S 3aS 4R 6R 6aS | IV 3R 3aR 4R 6R 6aR |
| Intermediates | |||
| ∆Hf˙= −210.26 kJ/mol | −189.11 kJ/mol | −132.63 kJ/mol | −117.22 kJ/mol |
 | 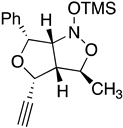 | 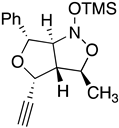 | 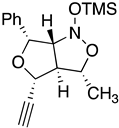 |
| Transition states | |||
| ‡ alkene up/imino up | ‡ alkene down/imino down | ‡ alkene down/imino up | ‡ alkene up/imino down |
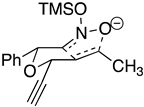 | 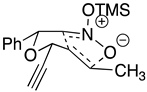 | 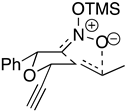 |  |
| Rxn Scheme | Transition State |
|---|---|
 | 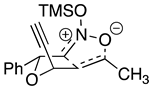 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stevens, K.; Li, S.K.; Kaufman, E.; Schull, A.; Hassebroek, K.; Stevens, J.; Grandbois, M.; Viste, A.; Duffy-Matzner, J. Initial Examinations of the Diastereoselectivity and Chemoselectivity of Intramolecular Silyl Nitronate [3+2] Cycloadditions with Alkenyl/Alkynyl Nitroethers. Molecules 2024, 29, 5816. https://doi.org/10.3390/molecules29245816
Stevens K, Li SK, Kaufman E, Schull A, Hassebroek K, Stevens J, Grandbois M, Viste A, Duffy-Matzner J. Initial Examinations of the Diastereoselectivity and Chemoselectivity of Intramolecular Silyl Nitronate [3+2] Cycloadditions with Alkenyl/Alkynyl Nitroethers. Molecules. 2024; 29(24):5816. https://doi.org/10.3390/molecules29245816
Chicago/Turabian StyleStevens, Katelyn, Shik Ki Li, Emily Kaufman, Annika Schull, Katie Hassebroek, Joseph Stevens, Matthew Grandbois, Arlen Viste, and Jetty Duffy-Matzner. 2024. "Initial Examinations of the Diastereoselectivity and Chemoselectivity of Intramolecular Silyl Nitronate [3+2] Cycloadditions with Alkenyl/Alkynyl Nitroethers" Molecules 29, no. 24: 5816. https://doi.org/10.3390/molecules29245816
APA StyleStevens, K., Li, S. K., Kaufman, E., Schull, A., Hassebroek, K., Stevens, J., Grandbois, M., Viste, A., & Duffy-Matzner, J. (2024). Initial Examinations of the Diastereoselectivity and Chemoselectivity of Intramolecular Silyl Nitronate [3+2] Cycloadditions with Alkenyl/Alkynyl Nitroethers. Molecules, 29(24), 5816. https://doi.org/10.3390/molecules29245816