
Symmetry Breaking of Electronic Structure upon the π→π* Excitation in Anthranilic Acid Homodimer


Abstract

1. Introduction
2. Computational Details
3. Results and Discussion
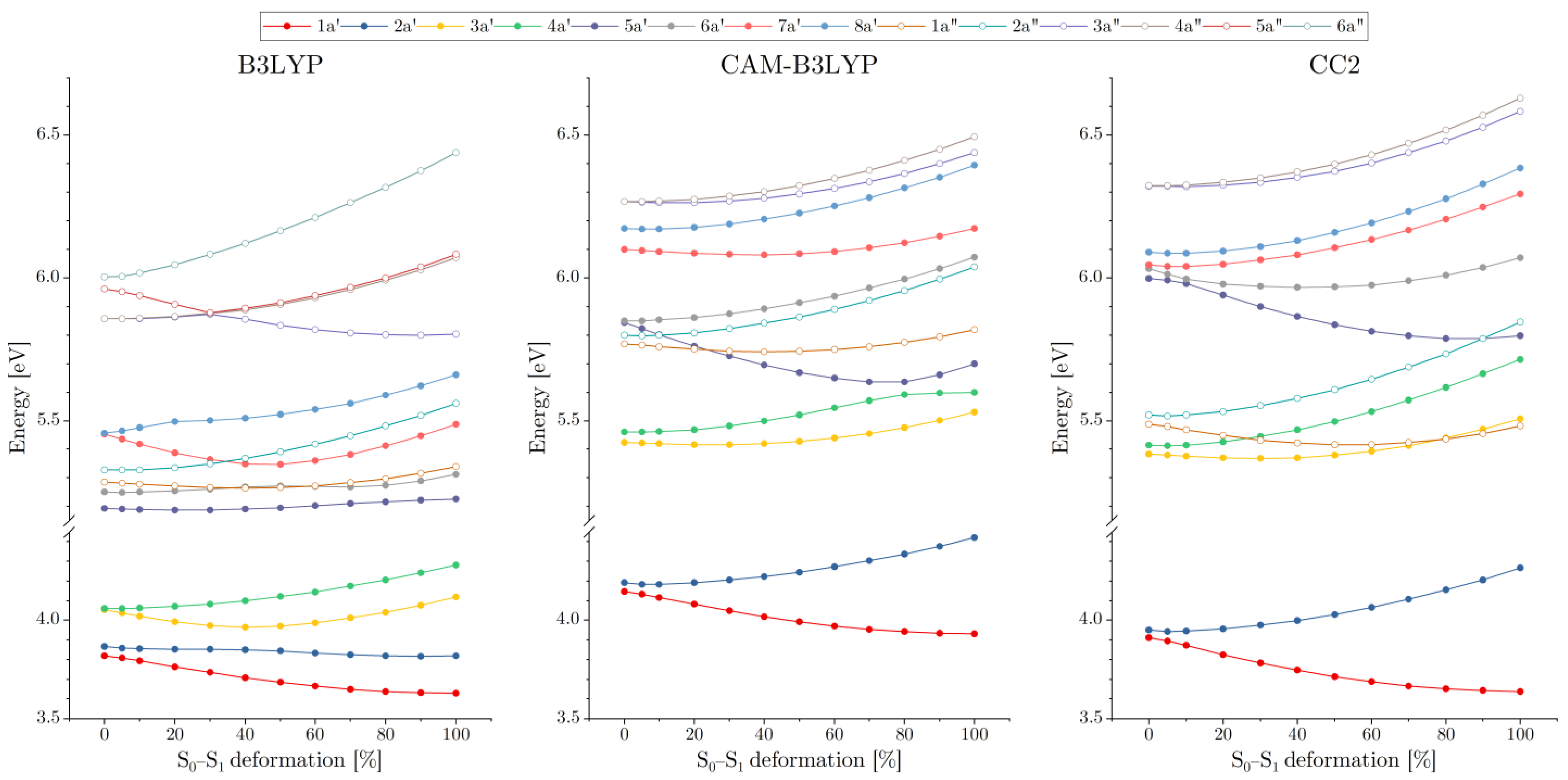
3.1. Excited Electronic States of AA2
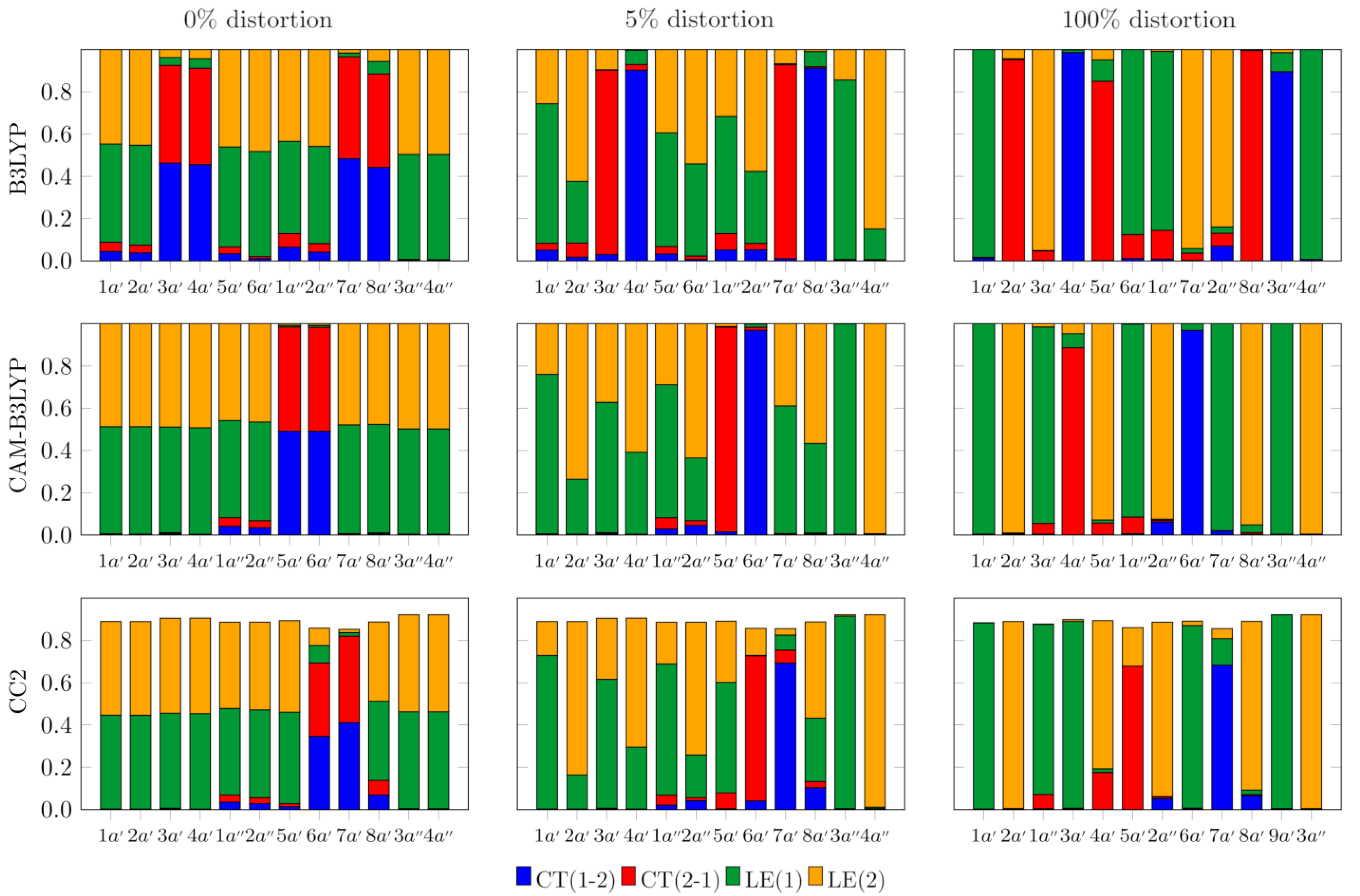
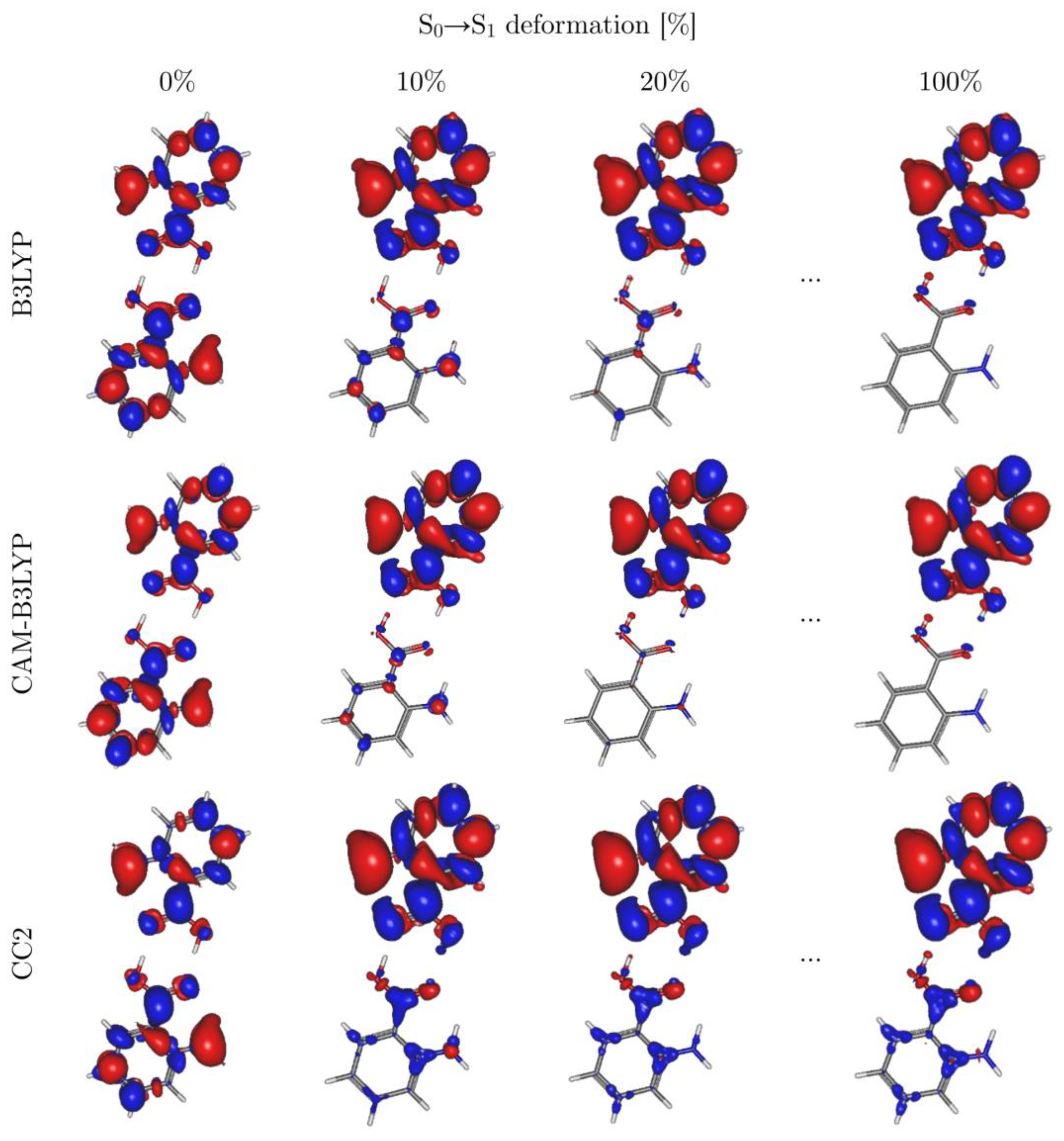
3.2. Symmetry Breaking of the Electronic Structure in the S1 (π→π*) Excites State of AA2: Crucial Role of Small Geometry Distortions
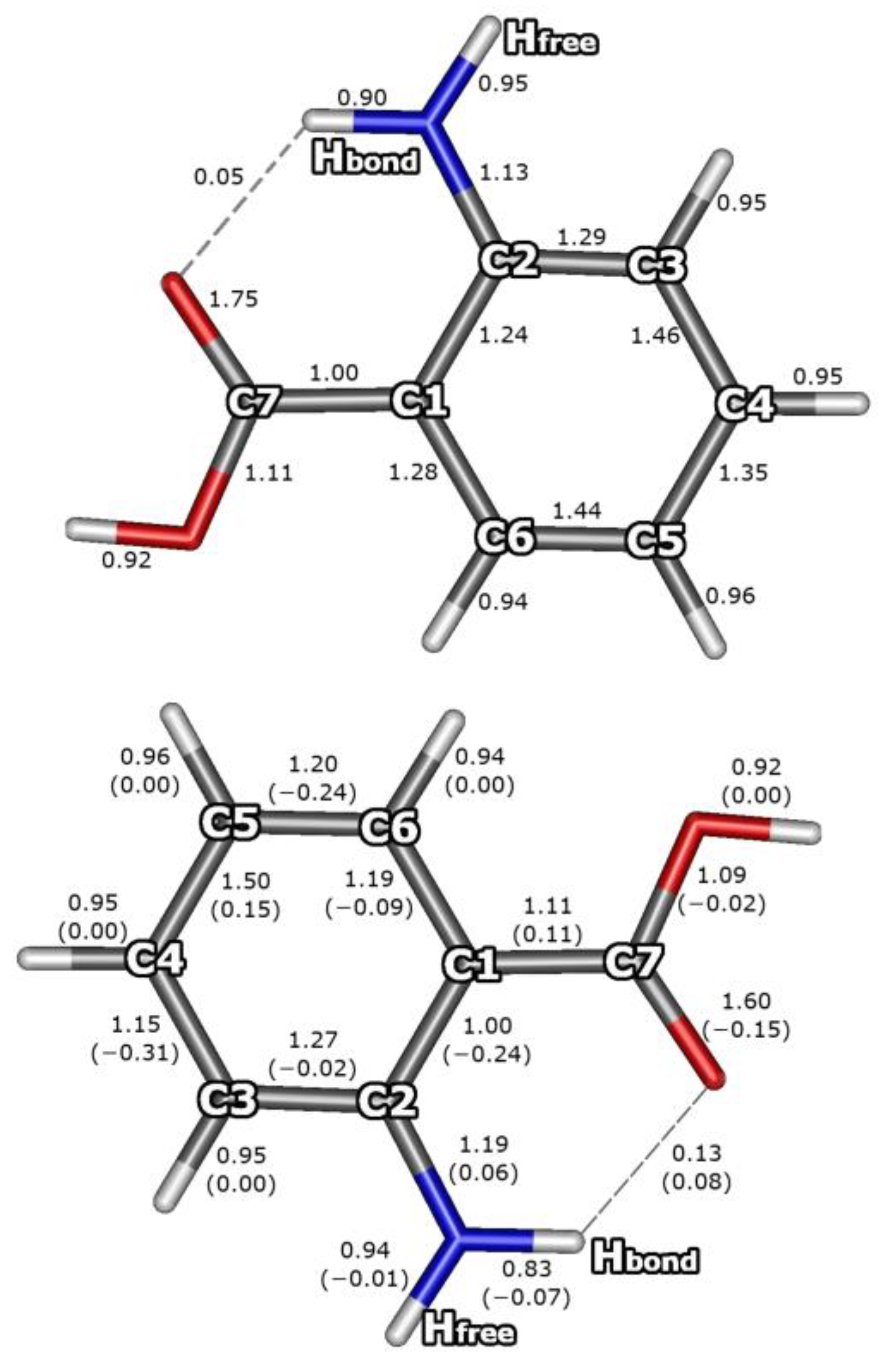
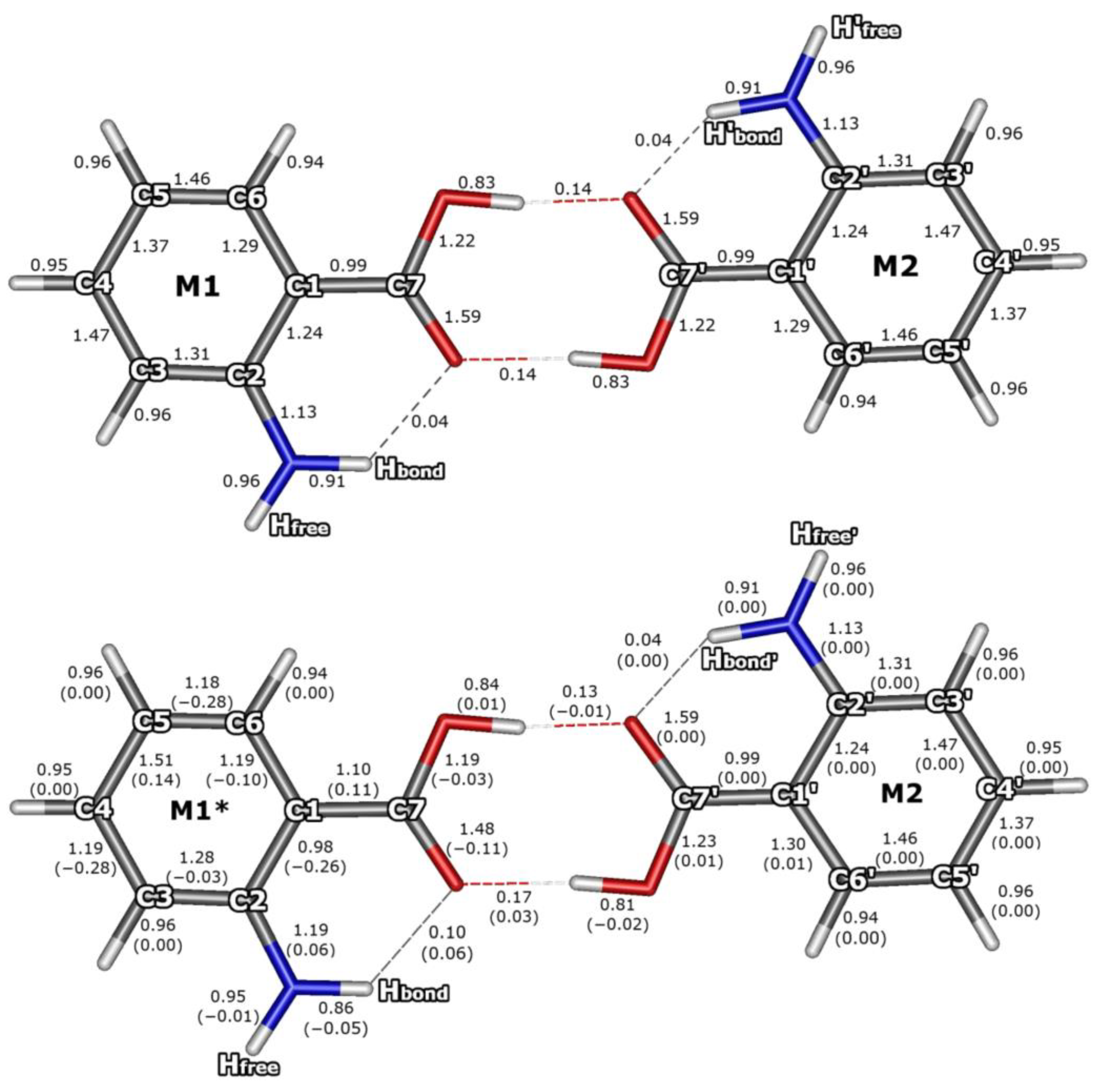
3.3. Bond Orders for the S0 and S1 Electronic States of AA
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Baum, J.C.; McClure, D.S. The ultraviolet transitions of benzoic acid. 1. Interpretation of the singlet absorption spectrum. J. Am. Chem. Soc. 1979, 101, 2335–2339. [Google Scholar] [CrossRef]
- Baum, J.C.; McClure, D.S. The ultraviolet transitions of benzoic acid. 2. Hydrogen bonding in the ground and excited states. J. Am. Chem. Soc. 1979, 101, 2340–2343. [Google Scholar] [CrossRef]
- Baum, J.C. The ultraviolet transitions of benzoic acid. 3. Effects of hydrogen bonding on the emission properties. J. Am. Chem. Soc. 1980, 102, 716–719. [Google Scholar] [CrossRef]
- Baum, J.C.; McClure, D.S. The ultraviolet transitions of benzoic acid. 4. High-resolution spectral studies of hydrogen bonding in the excited states of the benzoic acid dimer. J. Am. Chem. Soc. 1980, 102, 720–727. [Google Scholar] [CrossRef]
- Tomioka, Y.; Abe, H.; Mikami, N.; Ito, M. Electronic spectra of benzoic acid in a supersonic free jet. J. Phys. Chem. 1984, 88, 2263–2270. [Google Scholar] [CrossRef]
- Poeltl, D.E.; McVey, J.K. Laser induced fluorescence excitation spectrum of jet-cooled benzoic acid dimers. J. Chem. Phys. 1983, 78, 4349–4355. [Google Scholar] [CrossRef]
- Florio, G.M.; Sibert, E.L.; Zwier, T.S. Fluorescence-dip IR spectra of jet-cooled benzoic acid dimer in its ground and first excited singlet states. Faraday Discuss. 2001, 118, 315–330. [Google Scholar] [CrossRef]
- Nandi, C.K.; Chakraborty, T. Hydrogen bond-induced vibronic mode mixing in benzoic acid dimer: A laser-induced fluorescence study. J. Chem. Phys. 2004, 120, 8521–8527. [Google Scholar] [CrossRef]
- Nandi, C.K.; Hazra, M.K.; Chakraborty, T. Vibrational coupling in carboxylic acid dimers. J. Chem. Phys. 2005, 123, 124310. [Google Scholar] [CrossRef]
- Bakker, J.M.; Aleese, L.M.; von Helden, G.; Meijer, G. The infrared absorption spectrum of the gas phase neutral benzoic acid monomer and dimer. J. Chem. Phys. 2003, 119, 11180–11185. [Google Scholar] [CrossRef]
- Ebata, T.; Fujii, A.; Mikami, N. Vibrational spectroscopy of small-sized hydrogen-bonded clusters and their ions. Int. Rev. Phys. Chem. 1998, 17, 331–361. [Google Scholar] [CrossRef]
- Poeltl, D.E.; McVey, J.K. Excited-state dynamics of hydrogen-bonded dimers of benzoic acid. J. Chem. Phys. 1984, 80, 1801–1811. [Google Scholar] [CrossRef]
- Oppenlander, A.; Rambaud, C.; Trommsdorff, H.P.; Vial, J.C. Translational tunneling of protons in benzoic-acid crystals. Phys. Rev. Lett. 1989, 63, 1432–1435. [Google Scholar] [CrossRef]
- Rambaud, C.; Trommsdorff, H.P. Cooperative proton transfer and tunneling in dye doped benzoic acid crystals. Chem. Phys. Lett. 1999, 306, 124–132. [Google Scholar] [CrossRef]
- Remmers, K.; Meerts, W.L.; Ozier, I. Proton tunneling in the benzoic acid dimer studied by high resolution ultraviolet spectroscopy. J. Chem. Phys. 2000, 112, 10890–10894. [Google Scholar] [CrossRef]
- Ottiger, P.; Leutwyler, S.; Köppel, H. Vibrational quenching of excitonic splittings in H-bonded molecular dimers: The electronic Davydov splittings cannot match experiment. J. Chem. Phys. 2012, 136, 174308. [Google Scholar] [CrossRef]
- Ottiger, P.; Köppel, H.; Leutwyler, S. Excitonic splittings in molecular dimers: Why static ab initio calculations cannot match them. Chem. Sci. 2015, 6, 6059–6068. [Google Scholar] [CrossRef]
- Ottiger, P.; Leutwyler, S. Excitonic splitting and coherent electronic energy transfer in the gas-phase benzoic acid dimer. J. Chem. Phys. 2012, 137, 204303–204313. [Google Scholar] [CrossRef]
- Lahmani, F.; Zehnacker-Rentien, A. Spectroscopic study of jet-cooled heterodimers of salicylic acid with acetic and trifluoroacetic acids. Chem. Phys. Lett. 1997, 271, 6–14. [Google Scholar] [CrossRef]
- Yahagi, T.; Fujii, A.; Ebata, T.; Mikami, N. Infrared Spectroscopy of the OH Stretching Vibrations of Jet-Cooled Salicylic Acid and Its Dimer in S0 and S1. J. Phys. Chem. A 2001, 105, 10673–10680. [Google Scholar] [CrossRef]
- Maheshwary, S.; Lourderaj, U.; Sathyamurthy, N. Ab Initio Quantum Chemical Investigation of the Ground and Excited States of Salicylic Acid Dimer. J. Phys. Chem. A 2006, 110, 12662–12669. [Google Scholar] [CrossRef] [PubMed]
- Southern, C.A.; Levy, D.H.; Stearns, J.A.; Florio, G.M.; Longarte, A.; Zwier, T.S. Spectroscopic Consequences of Localized Electronic Excitation in Anthranilic Acid Dimer. J. Phys. Chem. A 2004, 108, 4599–4609. [Google Scholar] [CrossRef]
- Kolek, P.; Andrzejak, M.; Uchacz, T.; Goclon, J.; Pogocki, D.; Kisała, J.; Bankiewicz, B.; Szlachcic, P.; Tulej, M. LIF spectrum for the localized S0 → S1(ππ*) excitation in the H-bonded anthranilic acid dimer: Symmetry breaking or coupling of vibrations. Spectrochim. Acta A Mol. Biomole. Spectrosc. 2024, 319, 124491. [Google Scholar] [CrossRef]
- Bisht, P.B.; Petek, H.; Yoshihara, K.; Nagashima, U. Excited state enol-keto tautomerization in salicylic acid: A supersonic free jet study. J. Chem. Phys. 1995, 103, 5290–5307. [Google Scholar] [CrossRef]
- Lahmani, F.; Zehnacker-Rentien, A. Effect of Substitution on the Photoinduced Intramolecular Proton Transfer in Salicylic Acid. J. Phys. Chem. A 1997, 101, 6141–6147. [Google Scholar] [CrossRef]
- Sobolewski, A.L.; Domcke, W. Ab initio study of excited-state intramolecular proton dislocation in salicylic acid. Chem. Phys. 1998, 232, 257–265. [Google Scholar] [CrossRef]
- Sobolewski, A.L.; Domcke, W. Abinitio potential-energy functions for excited state intramolecular proton transfer: A comparative study of o-hydroxybenzaldehyde, salicylic acid and 7-hydroxy-1-indanone. Phys. Chem. Chem. Phys. 1999, 1, 3065–3072. [Google Scholar] [CrossRef]
- Southern, C.A.; Levy, D.H.; Florio, G.M.; Longarte, A.; Zwier, T.S. Electronic and Infrared Spectroscopy of Anthranilic Acid in a Supersonic Jet. J. Phys. Chem. A 2003, 107, 4032–4040. [Google Scholar] [CrossRef]
- Stearns, J.A.; Das, A.; Zwier, T.S. Hydrogen atom dislocation in the excited state of anthranilic acid: Probing the carbonyl stretch fundamental and the effects of water complexation. Phys. Chem. Chem. Phys. 2004, 6, 2605–2610. [Google Scholar] [CrossRef]
- Vauthey, E. Photoinduced symmetry-breaking charge separation. ChemPhysChem 2012, 13, 2001–2011. [Google Scholar] [CrossRef]
- Hong, Y.; Schlosser, F.; Kim, W.; Würthner, F.; Kim, D. Ultrafast symmetry-breaking charge separation in a perylene bisimide dimer enabled by vibronic coupling and breakdown of adiabaticity. J. Am. Chem. Soc. 2022, 144, 15539–15548. [Google Scholar] [CrossRef]
- Lin, C.; Kim, T.; Schultz, J.D.; Young, R.M.; Wasielewski, M.R. Accelerating symmetry-breaking charge separation in a perylenediimide trimer through a vibronically coherent dimer intermediate. Nat. Chem. 2022, 14, 786–793. [Google Scholar] [CrossRef]
- Menzel, J.P.; de Groot, H.J.M.; Buda, F. Photoinduced electron transfer in donor-acceptor complexes: Isotope effect and dynamic symmetry breaking. J. Phys. Chem. Lett. 2019, 10, 6504–6511. [Google Scholar] [CrossRef]
- Kolesov, B.A. Unusual behavior of benzoic acid at low temperature: Raman spectroscopic study. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2015, 142, 320–323. [Google Scholar] [CrossRef]
- Demkin, A.G.; Kolesov, B.A. Tautomeric hydrogen bond in dimers of ibuprofen. J. Phys. Chem. A 2019, 123, 5537–5541. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; He, Y.; Kong, W. Two-color two-photon REMPI and ZEKE spectroscopy of supersonically cooled o-aminobenzoic acid. Chem. Phys. Lett. 2004, 398, 351–356. [Google Scholar] [CrossRef]
- Kolek, P.; Lesniewski, S.; Pirowska, K.; Najbar, J. LIF excitation spectra for S0 → S1 transition of anthranilic acid: Detailed studies. J. Mol. Spectrosc. 2008, 249, 100–112. [Google Scholar] [CrossRef]
- Lesniewski, S.; Kolek, P.; Pirowska, K.; Sobolewski, A.L.; Najbar, J. Franck-Condon analysis of laser-induced fluorescence excitation spectrum of anthranilic acid: Evaluation of geometry change upon S0 → S1 excitation. J. Chem. Phys. 2009, 130, 054307. [Google Scholar] [CrossRef]
- Kolek, P.; Andrzejak, M.; Uchacz, T.; Szlachcic, P. Consistent Franck-Condon modeling of geometry changes for the S0 → S1(ππ*) excitation in anthranilic acid: LIF spectroscopy aided by CC2 or TDDFT vibrations. J. Quant. Spectrosc. Radiat. Transf. 2020, 242, 106747. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297. [Google Scholar] [CrossRef]
- Christiansen, O.; Koch, H.; Jørgensen, P. The second-order approximate coupled cluster singles and doubles model CC2. Chem. Phys. Lett. 1995, 243, 409–418. [Google Scholar] [CrossRef]
- Hättig, C. Geometry optimizations with the coupled-cluster model CC2 using the resolution-of-the-identity approximation. J. Chem. Phys. 2003, 118, 7751–7761. [Google Scholar] [CrossRef]
- Köhn, A.; Hättig, C. Analytic gradients for excited states in the coupled-cluster model CC2 employing the resolution-of-the-identity approximation. J. Chem. Phys. 2003, 119, 5021–5036. [Google Scholar] [CrossRef]
- Friese, D.H.; Hättig, C.; Koßmann, J. Analytic Molecular Hessian Calculations for CC2 and MP2 Combined with the Resolution of Identity Approximation. J. Chem. Theory Comput. 2013, 9, 1469–1480. [Google Scholar] [CrossRef]
- Hättig, C.; Weigend, F. CC2 excitation energy calculations on large molecules using the resolution of the identity approximation. J. Chem. Phys. 2000, 113, 5154–5161. [Google Scholar] [CrossRef]
- Koch, H.; Jørgensen, P. Coupled cluster response functions. J. Chem. Phys. 1990, 93, 3333–3344. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B Condens. Matter Mater. Phys. 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Miehlich, B.; Savin, A.; Stoll, H.; Preuss, H. Results obtained with the correlation energy density functionals of Becke and Lee, Yang, and Parr. Chem. Phys. Lett. 1989, 157, 200–206. [Google Scholar] [CrossRef]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Angeli, C.; Cimiraglia, R.; Evangelisti, S.; Leininger, T.; Malrieu, J.P. Introduction of n-electron valence states for multireference perturbation theory. J. Chem. Phys. 2001, 114, 10252–10264. [Google Scholar] [CrossRef]
- Angeli, C.; Cimiraglia, R.; Malrieu, J.P. n-electron valence state perturbation theory: A spinless formulation and an efficient implementation of the strongly contracted and of the partially contracted variants. J. Chem. Phys. 2002, 117, 9138–9153. [Google Scholar] [CrossRef]
- Mayer, I. Bond orders and valences from ab initio wave functions. Int. J. Quantum Chem. 1986, 29, 477–483. [Google Scholar] [CrossRef]
- Mayer, I. Bond order and valence indices: A personal account. J. Comput. Chem. 2007, 28, 204–221. [Google Scholar] [CrossRef]
- Ahlrichs, R.; Bär, M.; Baron, H.P.; Bauernschmitt, R.; Böcker, S.; Ehrig, M.; Eichkorn, K.; Elliott, S.; Furche, F.; Haase, F.; et al. TurboMole—Program System for Ab Initio Electronic Structure Calculations, Version 5.2; Universität Karlsruhe: Karlsruhe, Germany, 2000. Available online: https://www.turbomole.org (accessed on 21 November 2024).
- Neese, F. The ORCA program system. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Plasser, F. TheoDORE: A toolbox for a detailed and automated analysis of electronic excited state computations. J. Chem. Phys. 2020, 152, 084108. [Google Scholar] [CrossRef]
- Sarkar, R.; Loos, P.F.; Boggio-Pasqua, M.; Jacquemin, D. Assessing the Performances of CASPT2 and NEVPT2 for Vertical Excitation Energies. J. Chem. Theory Comput. 2022, 18, 2418–2436. [Google Scholar] [CrossRef]
- Polo, V.; Kraka, E.; Cremer, D. Electron correlation and the self-interaction error of density functional theory. Mol. Phys. 2002, 100, 1771–1790. [Google Scholar] [CrossRef]
- He, Y.; Gräfenstein, J.; Kraka, E.; Cremer, D. What correlation effects are covered by density functional theory? Mol. Phys. 2000, 98, 1639–1658. [Google Scholar] [CrossRef]
- Mori-Sánchez, P.; Cohen, A.J.; Yang, W. Many-electron self-interaction error in approximate density functionals. J. Chem. Phys. 2006, 125, 201102. [Google Scholar] [CrossRef] [PubMed]
- Andrzejak, M.; Szczepanik, D.W.; Orzeł, Ł. The lowest triplet states of bridged cis-2,2′-bithiophenes—Theory vs. experiment. Phys. Chem. Chem. Phys. 2015, 17, 5328–5337. [Google Scholar] [CrossRef]
- Schreiber, M.; Silva-Junior, M.R.; Sauer, S.P.A.; Thiel, W. Benchmarks for electronically excited states: CASPT2, CC2, CCSD, and CC3. J. Chem. Phys. 2008, 128, 134110. [Google Scholar] [CrossRef]
- Tozer, D.J. Relationship between long-range charge-transfer excitation energy error and integer discontinuity in Kohn-Sham theory. J. Chem. Phys. 2003, 119, 12697–12699. [Google Scholar] [CrossRef]
- Dreuw, A.; Head-Gordon, M. Single-Reference ab Initio Methods for the Calculation of Excited States of Large Molecules. Chem. Rev. 2005, 105, 4009–4037. [Google Scholar] [CrossRef] [PubMed]
- Dreuw, A.; Head-Gordon, M. Failure of Time-Dependent Density Functional Theory for Long-Range Charge-Transfer Excited States: The Zinc bacteriochlorin-Bacteriochlorin and Bacteriochlorophyll-Spheroidene Complexes. J. Am. Chem. Soc. 2004, 126, 4007–4016. [Google Scholar] [CrossRef]
- Grimme, S.; Parac, M. Substantial errors from time-dependent density functional theory for the calculation of excited states of large π systems. ChemPhysChem 2003, 4, 292–295. [Google Scholar] [CrossRef]
- Prlj, A.; Curchod, B.F.E.; Fabrizio, A.; Floryan, L.; Corminboeuf, C. Qualitatively Incorrect Features in the TDDFT Spectrum of Thiophene-Based Compounds. J. Phys. Chem. Lett. 2015, 6, 13–21. [Google Scholar] [CrossRef]
- Andrzejak, M.; Kolek, P. Theoretical modeling of deuteration-induced shifts of the 0-0 bands in absorption spectra of selected aromatic amines: The role of the double-well potential. J. Phys. Chem. A 2013, 117, 12770–12782. [Google Scholar] [CrossRef]
- Kolek, P.; Lesniewski, S.; Andrzejak, M.; Gora, M.; Cias, P.; Wegrzynowicz, A.; Najbar, J. LIF excitation spectra for S0 → S1 transition of deuterated anthranilic acid (COOD, ND2) in supersonic-jet expansion. J. Mol. Spectrosc. 2010, 264, 129–136. [Google Scholar] [CrossRef]
- Kolek, P.; Andrzejak, M.; Najbar, J.; Ostrowska-Kopec, M.; Piotrowska, I. Isotopic effects in the S1 excited state of anthranilic acid deuterated in various positions in substituent groups: Supersonic-jet LIF spectroscopy and CC2 ab initio study. Chem. Phys. 2015, 450, 46–58. [Google Scholar] [CrossRef]
- Plasser, F.; Wormit, M.; Dreuw, A. New tools for the systematic analysis and visualization of electronic excitations. I. Formalism. J. Chem. Phys. 2014, 141, 024106. [Google Scholar] [CrossRef] [PubMed]
- Plasser, F.; Bäppler, S.A.; Wormit, M.; Dreuw, A. New tools for the systematic analysis and visualization of electronic excitations. II. Applications. J. Chem. Phys. 2014, 141, 024107. [Google Scholar] [CrossRef] [PubMed]
- Plasser, F. Entanglement entropy of electronic excitations. J. Chem. Phys. 2016, 144, 194107. [Google Scholar] [CrossRef]
- Plasser, F.; Lischka, H. Analysis of excitonic and charge transfer interactions from quantum chemical calculations. J. Chem. Theory Comput. 2012, 8, 2777–2789. [Google Scholar] [CrossRef]
- Scholes, G.D.; Ghiggino, K.P. Electronic interactions and interchromophore excitation transfer. J. Phys. Chem. 1994, 98, 4580–4590. [Google Scholar] [CrossRef]
- Lim, E.C.; East, A.L.L. Naphthalene dimer: Electronic states, excimers, and triplet decay. J. Chem. Phys. 2000, 113, 8981–8994. [Google Scholar]
- Grimme, S.; Neese, F. Double-hybrid density functional theory for excited electronic states of molecules. J. Chem. Phys. 2007, 127, 154116. [Google Scholar] [CrossRef]
- Wang, Y.; Li, Y.; Chen, J.; Zhang, I.Y.; Xu, X. Doubly Hybrid Functionals Close to Chemical Accuracy for Both Finite and Extended Systems: Implementation and Test of XYG3 and XYGJ-OS. JACS Au 2021, 1, 543–549. [Google Scholar] [CrossRef]
- Santra, G.; Calinsky, R.; Martin, J.M.L. Benefits of Range-Separated Hybrid and Double-Hybrid Functionals for a Large and Diverse Data Set of Reaction Energies and Barrier Heights. J. Phys. Chem. A 2022, 126, 5492–5505. [Google Scholar] [CrossRef] [PubMed]
- Casanova-Páez, M.; Dardis, M.B.; Goerigk, L. ωB2PLYP and ωB2GPPLYP: The First Two Double-Hybrid Density Functionals with Long-Range Correction Optimized for Excitation Energies. J. Chem. Theory Comput. 2019, 15, 4735–4744. [Google Scholar] [CrossRef] [PubMed]
- Pauling, L.; Brockway, L.O.; Beach, H.Y. The dependence of interatomic distance on single bond-double bond resonance. J. Am. Chem. Soc. 1935, 57, 2705. [Google Scholar] [CrossRef]
- Pauling, L. Atomic radii and interatomic distances in metals. J. Am. Chem. Soc. 1947, 69, 542. [Google Scholar] [CrossRef]
- Pauling, L. The Nature of the Chemical Bond; Cornell University Press: New York, NY, USA, 1960. [Google Scholar]
- Bridgeman, A.J.; Cavigliasso, G.; Ireland, L.R.; Rothery, J. The Mayer bond order as a tool in inorganic chemistry. J. Chem. Soc. Dalton Trans. 2001, 14, 2095–2108. [Google Scholar] [CrossRef]
- Cooper, D.L.; Ponec, R.; Karadakov, P.B. Investigating István Mayer’s “improved” definitions of bond orders and free valence for correlated singlet-state wave functions. Int. J. Quantum Chem. 2022, 122, e26612. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Monomer | Dimer | |||
|---|---|---|---|---|
| B3LYP | 3.890 | 3.670 (3.561) | 3.820 | 3.629 (3.512) |
| CAM-B3LYP | 4.198 | 3.966 (3.877) | 4.146 | 3.932 (3.837) |
| CC2 | 3.972 | 3.684 (3.552) | 3.912 | 3.638 (3.493) |
| CC2 a | 4.041 | 3.695 (3.536) | 3.986 | 3.649 (3.489) |
| CASSCF/NEVPT2 b | 3.999 | 3.787 (3.655) | 4.000 | 3.759 (3.614) |
| Experimental | 3.545 [28,36,37] | 3.502 [22,23] | ||
| B3LYP | CAM-B3LYP | CC2 | ||||
|---|---|---|---|---|---|---|
| S0 | S1 | S0 | S1 | S0 | S1 | |
| 1a′ | 0.235 | 0.115 | 0.296 | 0.150 | 0.278 | 0.138 |
| 2a′ | 0.000 * | 0.007 * | 0.000 | 0.137 | 0.000 | 0.133 |
| 3a′ | 0.000 * | 0.107 | 0.085 | 0.071 | 0.121 | 0.074 |
| 4a′ | 0.011 | 0.001 * | 0.000 | 0.000 * | 0.000 | 0.044 |
| 5a′ | 0.153 | 0.054 | 0.000 * | 0.032 | 1.395 | 0.012 * |
| 6a′ | 0.000 | 0.080 | 0.001 * | 0.003 * | 0.000 * | 0.792 |
| 7a′ | 0.000 | 0.046 | 1.281 | 0.837 | 0.011 * | 0.094 * |
| 8a′ | 0.035 | 0.002 | 0.000 | 0.432 | 0.000 | 0.478 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Andrzejak, M.; Zams, J.; Goclon, J.; Kolek, P. Symmetry Breaking of Electronic Structure upon the π→π* Excitation in Anthranilic Acid Homodimer. Molecules 2024, 29, 5562. https://doi.org/10.3390/molecules29235562
Andrzejak M, Zams J, Goclon J, Kolek P. Symmetry Breaking of Electronic Structure upon the π→π* Excitation in Anthranilic Acid Homodimer. Molecules. 2024; 29(23):5562. https://doi.org/10.3390/molecules29235562
Chicago/Turabian StyleAndrzejak, Marcin, Joanna Zams, Jakub Goclon, and Przemysław Kolek. 2024. "Symmetry Breaking of Electronic Structure upon the π→π* Excitation in Anthranilic Acid Homodimer" Molecules 29, no. 23: 5562. https://doi.org/10.3390/molecules29235562
APA StyleAndrzejak, M., Zams, J., Goclon, J., & Kolek, P. (2024). Symmetry Breaking of Electronic Structure upon the π→π* Excitation in Anthranilic Acid Homodimer. Molecules, 29(23), 5562. https://doi.org/10.3390/molecules29235562