
Hybrid Metal Catalysts as Valuable Tools in Organic Synthesis: An Overview of the Recent Advances in Asymmetric C─C Bond Formation Reactions



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Different Hybrid Catalysts in C─C Bond Formation Reactions
2.1. Hybrid Catalysis Applied to Carbene Insertion Reactions
2.2. Michael Addition Reactions Catalyzed by Hybrid Catalysts
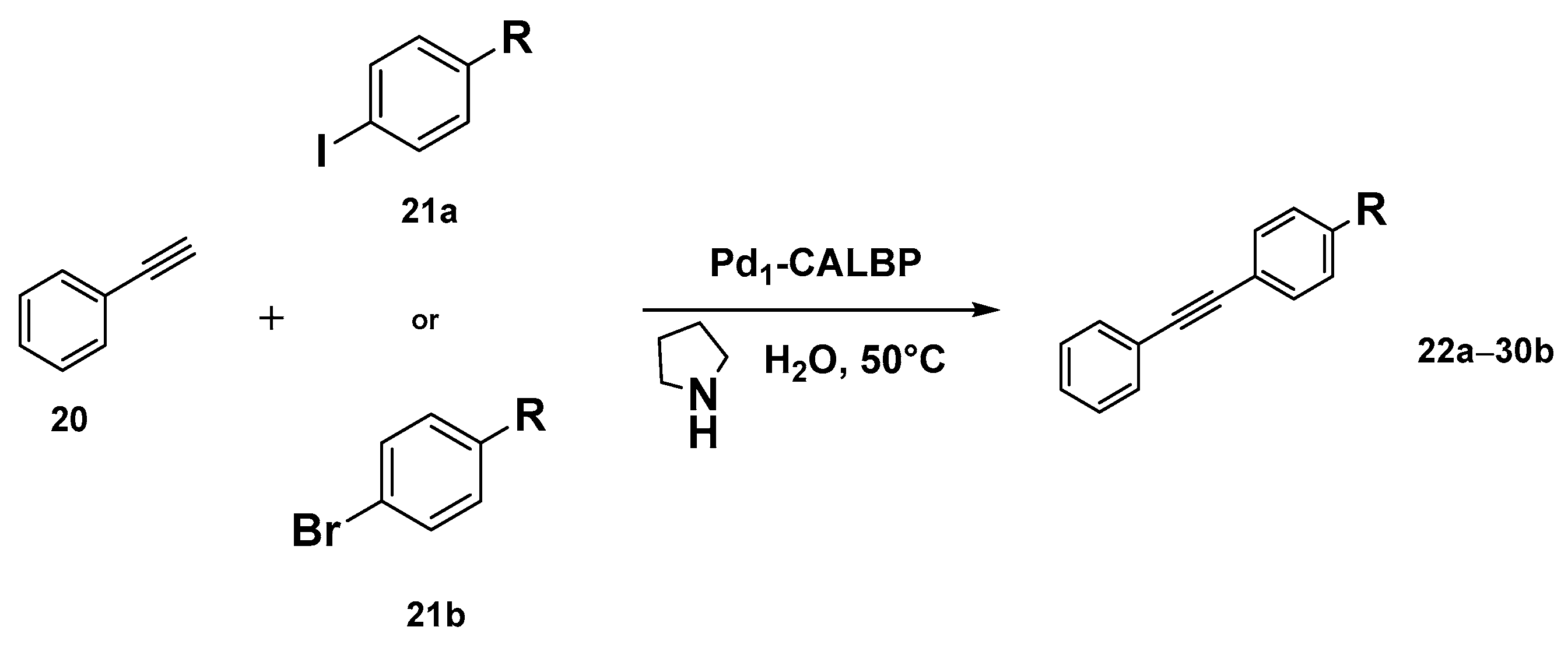
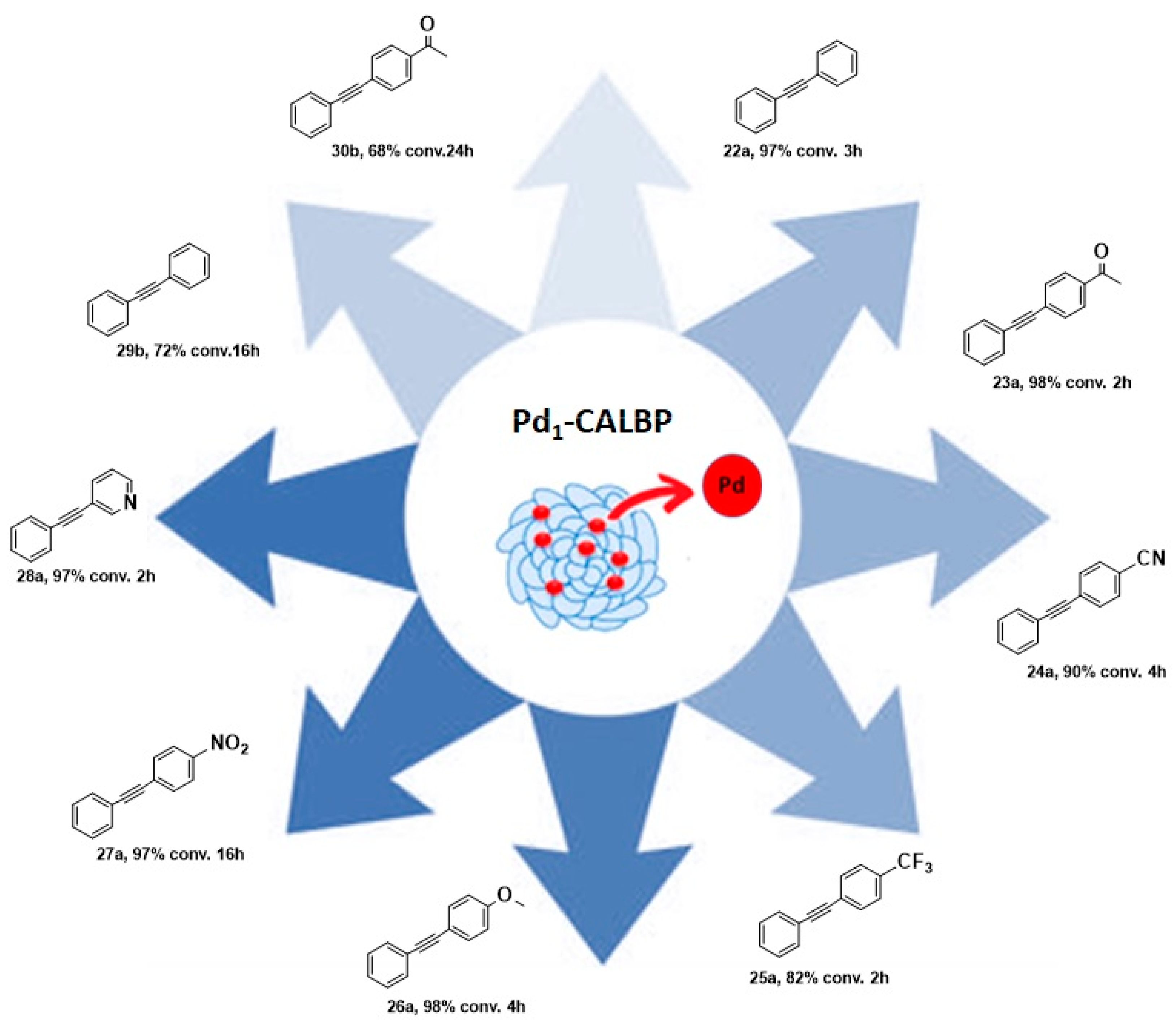
2.3. New Hybrid Catalytic Approaches in Friedel–Crafts Alkylation and Sonogashira Cross-Coupling
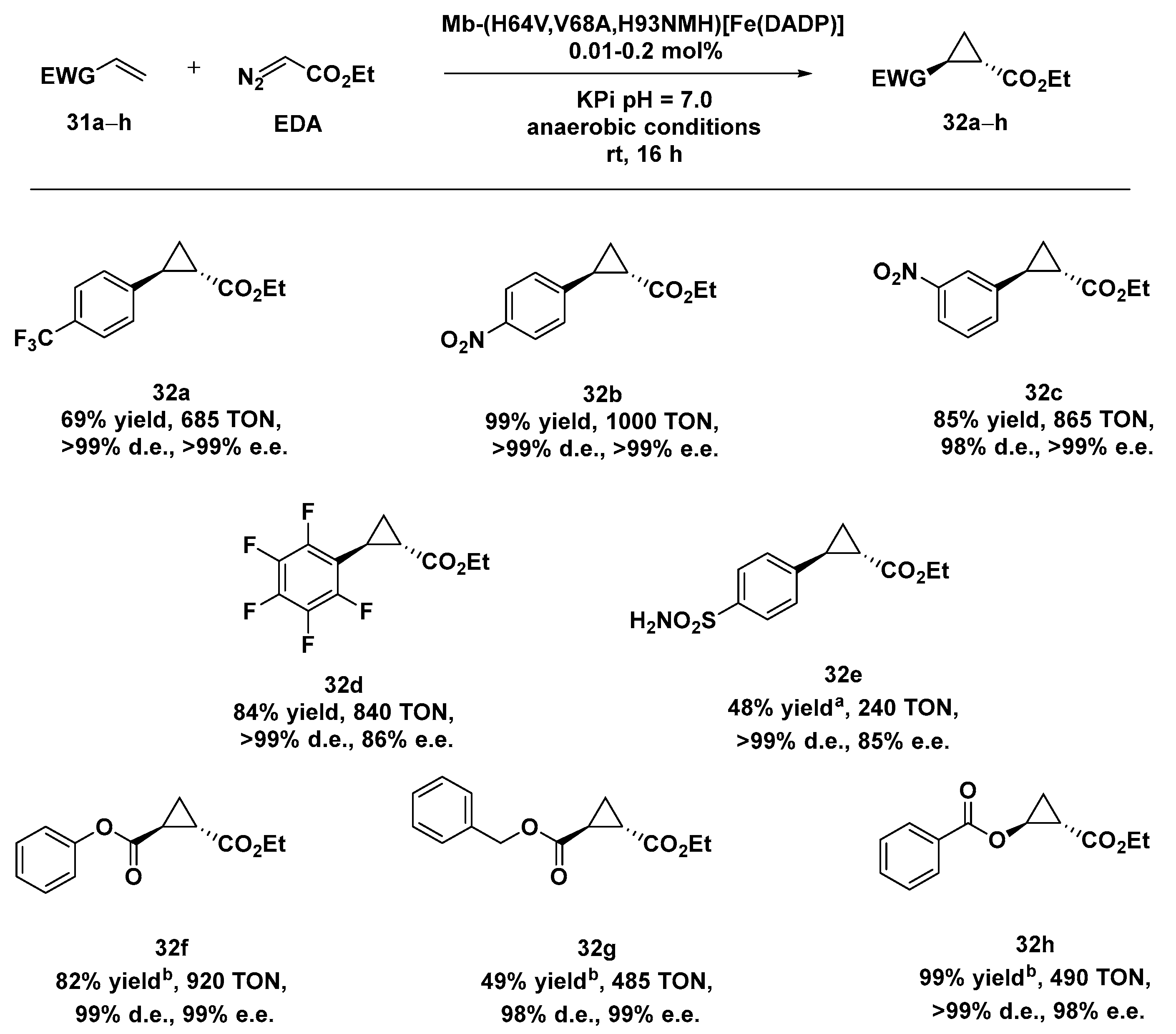
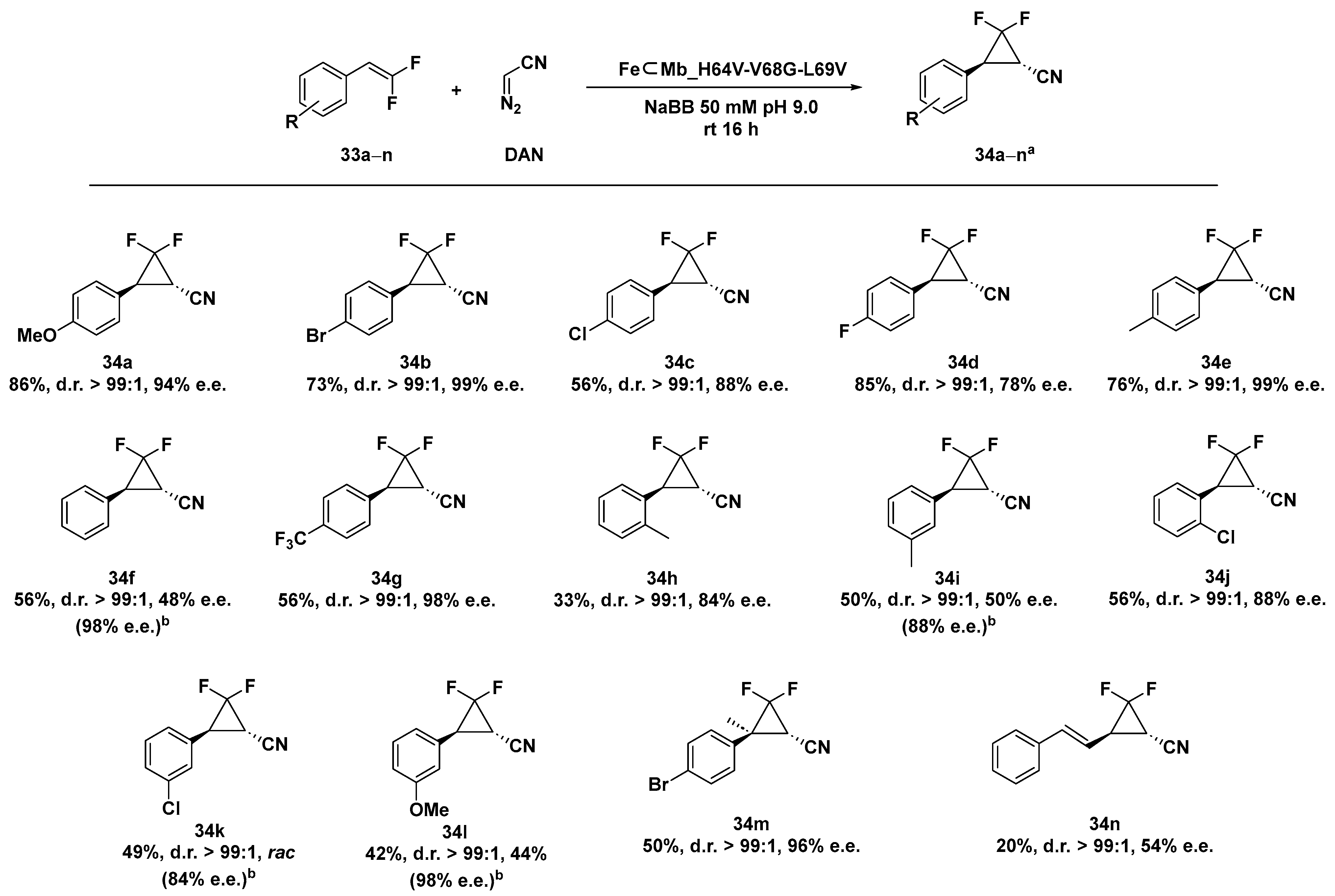
2.4. ArMs and MPs in Cyclopropanation Reactions
3. Conclusions and Future Directions
Funding
Conflicts of Interest
References
- Isahak, W.N.R.W.; Al-Amiery, A. Catalysts driving efficiency and innovation in thermal reactions: A comprehensive review. Green. Technol. Sustain. 2024, 2, 100078. [Google Scholar] [CrossRef]
- James, C.C.; de Bruin, B.; Reek, J.N.H. Transition Metal Catalysis in Living Cells: Progress, Challenges, and Novel Supramolecular Solutions. Angew. Chem. Int. Ed. 2023, 62, e202306645. [Google Scholar] [CrossRef] [PubMed]
- Docherty, J.H.; Lister, T.M.; McArthur, G.; Findlay, M.T.; Domingo-Legarda, P.; Kenyon, J.; Choudhary, S.; Larrosa, I. Transition-Metal-Catalyzed C–H Bond Activation for the Formation of C–C Bonds in Complex Molecules. Chem. Rev. 2023, 123, 7692–7760. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Yu, H.; Stolarzewicz, I.A.; Tang, W. Enantioselective Transformations in the Synthesis of Therapeutic Agents. Chem. Rev. 2023, 123, 9397–9446. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Jiang, S.; Jin, Z.; Li, T. Application of Asymmetric Catalysis in Chiral Pesticide Active Molecule Synthesis. J. Agric. Food Chem. 2024, 72, 17153–17165. [Google Scholar] [CrossRef] [PubMed]
- Lakhani, P.; Modi, C.K. Shaping enantiochemistry: Recent advances in enantioselective reactions via heterogeneous chiral catalysis. Mol. Catal. 2023, 548, 113429. [Google Scholar] [CrossRef]
- Steinlandt, P.S.; Zhang, L.; Meggers, E. Metal stereogenicity in asymmetric transition metal catalysis. Chem. Rev. 2023, 123, 4764–4794. [Google Scholar] [CrossRef]
- Marianov, A.N.; Jiang, Y.; Baiker, A.; Huang, J. Homogeneous and heterogeneous strategies of enantioselective hydrogenation: Critical evaluation and future prospects. Chem. Catal. 2023, 3, 100631. [Google Scholar] [CrossRef]
- Nuzhdin, A.L.; Bukhtiyarova, M.V.; Bukhtiyarova, G.A. Organic synthesis in flow mode by selective liquid-phase hydrogenation over heterogeneous non-noble metal catalysts. Org. Biomol. Chem. 2024, 22, 7936–7950. [Google Scholar] [CrossRef]
- Gupta, A.; Gupta, R.; Arora, G.; Yadav, P.; Sharma, R.K. Heterogeneous Catalysis under Continuous Flow Conditions. Curr. Org. Chem. 2023, 27, 1090–1110. [Google Scholar] [CrossRef]
- González-Granda, S.; Albarrán-Velo, J.; Lavandera, I.; Gotor-Fernández, V. Expanding the Synthetic Toolbox through Metal–Enzyme Cascade Reactions. Chem. Rev. 2023, 123, 5297–5346. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Pradhan, T.K.; Samanta, R. Recent Progress on Transition Metal Catalyzed Macrocyclizations Based on C−H Bond Activation at Heterocyclic Scaffolds. Chem.-Asian J. 2024, 19, e202400397. [Google Scholar] [CrossRef] [PubMed]
- Song, F.; Wang, B.; Shi, Z.-J. Transition-Metal-Catalyzed C–C Bond Formation from C–C Activation. Acc. Chem. Res. 2023, 56, 2867–2886. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X. Cyclization Strategies in Carbonyl–Olefin Metathesis: An Up-to-Date Review. Molecules 2024, 29, 4861. [Google Scholar] [CrossRef]
- Jose, J.; Diana, E.J.; Mathew, T.V. Chapter 9—Olefin metathesis homogeneous catalysts. In Homogeneous Hydrogenation and Metathesis Reactions; Rahimpour, M.R., Makarem, M.A., Roostaie, T., Meshksar, M., Eds.; Elsevier: Amsterdam, The Netherlands, 2025; Volume 5, pp. 191–210. [Google Scholar]
- Hoveyda, A.H.; Qin, C.; Sui, X.Z.; Liu, Q.; Li, X.; Nikbakht, A. Taking Olefin Metathesis to the Limit: Stereocontrolled Synthesis of Trisubstituted Alkenes. Acc. Chem. Res. 2023, 56, 2426–2446. [Google Scholar] [CrossRef]
- Fu, D.; Wang, Z.; Liu, Q.; Prettyman, S.J.; Solan, G.A.; Sun, W.-H. Organometallic Mn(I) Complexes in Asymmetric Catalytic (Transfer) Hydrogenation and Related Transformations. ChemCatChem 2024, 16, e202301567. [Google Scholar] [CrossRef]
- Ansari, M.F.; Anshika; Sortais, J.-B.; Elangovan, S. Transition-Metal-Catalysed Transfer Hydrogenation Reactions with Glycerol and Carbohydrates as Hydrogen Donors. Eur. J. Org. Chem. 2024, 27, e202301278. [Google Scholar] [CrossRef]
- Lückemeier, L.; Pierau, M.; Glorius, F. Asymmetric arene hydrogenation: Towards sustainability and application. Chem. Soc. Rev. 2023, 52, 4996–5012. [Google Scholar] [CrossRef]
- Facchetti, G.; Cesarotti, E.; Pellizzoni, M.; Zerla, D.; Rimoldi, I. “In situ” Activation of Racemic RuII Complexes: Separation of trans and cis Species and Their Application in Asymmetric Reduction. Eur. J. Inorg. Chem. 2012, 2012, 4365–4370. [Google Scholar] [CrossRef]
- Facchetti, G.; Fusè, M.; Pecoraro, T.; Nava, D.; Rimoldi, I. New sp3 diphosphine-based rhodium catalysts for the asymmetric conjugate addition of aryl boronic acids to 3-azaarylpropenones. New J. Chem. 2021, 45, 18769–18775. [Google Scholar] [CrossRef]
- Islam, M.T.; Bitu, N.A.; Chaki, B.M.; Hossain, M.J.; Asraf, M.A.; Hossen, M.F.; Kudrat-E-Zahan, M.; Latif, M.A. Water-soluble Schiff base ligands and metal complexes: An overview considering green solvent. RSC Adv. 2024, 14, 25256–25272. [Google Scholar] [CrossRef] [PubMed]
- Baricelli, P.J.; Pereira, J.C.; Rosales, M. Aqueous-biphasic catalysis: A technological alternative for the use of organometallic complexes in hydrogenation and hydroformylation reactions with possible industrial application. Catal. Today 2025, 443, 114969. [Google Scholar] [CrossRef]
- Yan, Q.; Wu, X.; Jiang, H.; Wang, H.; Xu, F.; Li, H.; Zhang, H.; Yang, S. Transition metals-catalyzed amination of biomass feedstocks for sustainable construction of N-heterocycles. Coord. Chem. Rev. 2024, 502, 215622. [Google Scholar] [CrossRef]
- Habib, U.; Ahmad, F.; Awais, M.; Naz, N.; Aslam, M.; Urooj, M.; Moqeem, A.; Tahseen, H.; Waqar, A.; Sajid, M. Sustainable Catalysis: Navigating Challenges and Embracing Opportunities for a Greener Future. J. Chem. Environ. 2023, 2, 14–53. [Google Scholar] [CrossRef]
- Wilson, M.E.; Whitesides, G.M. Conversion of a protein to a homogeneous asymmetric hydrogenation catalyst by site-specific modification with a diphosphinerhodium (I) moiety. J. Am. Chem. Soc. 1978, 100, 306–307. [Google Scholar] [CrossRef]
- Morita, I.; Ward, T.R. Recent advances in the design and optimization of artificial metalloenzymes. Curr. Opin. Chem. Biol. 2024, 81, 102508. [Google Scholar] [CrossRef]
- LIU, X.-y.; HUANG, C.-q.; JIN, X.-r.; LUO, Y.-z. Research Progress of Artificial Metalloenzymes. China Biotechnol. 2023, 43, 72–84. [Google Scholar]
- Jeong, W.J.; Lee, J.; Eom, H.; Song, W.J. A Specific Guide for Metalloenzyme Designers: Introduction and Evolution of Metal-Coordination Spheres Embedded in Protein Environments. Acc. Chem. Res. 2023, 56, 2416–2425. [Google Scholar] [CrossRef]
- Wittwer, M.; Markel, U.; Schiffels, J.; Okuda, J.; Sauer, D.F.; Schwaneberg, U. Engineering and emerging applications of artificial metalloenzymes with whole cells. Nat. Catal. 2021, 4, 814–827. [Google Scholar] [CrossRef]
- Lee, J.; Yang, M.; Song, W.J. The expanded landscape of metalloproteins by genetic incorporation of noncanonical amino acids. Bull. Korean Chem. Soc. 2023, 44, 23–34. [Google Scholar] [CrossRef]
- Yu, Y.; Hu, C.; Xia, L.; Wang, J. Artificial Metalloenzyme Design with Unnatural Amino Acids and Non-Native Cofactors. ACS Catal. 2018, 8, 1851–1863. [Google Scholar] [CrossRef]
- Miller, A.H.; Blagova, E.V.; Large, B.; Booth, R.L.; Wilson, K.S.; Duhme-Klair, A.-K. Catch-and-Release: The Assembly, Immobilization, and Recycling of Redox-Reversible Artificial Metalloenzymes. ACS Catal. 2024, 14, 3218–3227. [Google Scholar] [CrossRef] [PubMed]
- Facchetti, G.; Bucci, R.; Fusè, M.; Erba, E.; Gandolfi, R.; Pellegrino, S.; Rimoldi, I. Alternative Strategy to Obtain Artificial Imine Reductase by Exploiting Vancomycin/D-Ala-D-Ala Interactions with an Iridium Metal Complex. Inorg. Chem. 2021, 60, 2976–2982. [Google Scholar] [CrossRef] [PubMed]
- Duran-Meza, E.; Araya-Secchi, R.; Romero-Hasler, P.; Soto-Bustamante, E.A.; Castro-Fernandez, V.; Castillo-Caceres, C.; Monasterio, O.; Diaz-Espinoza, R. Metal Ions Can Modulate the Self-Assembly and Activity of Catalytic Peptide Amyloids. Langmuir 2024, 40, 6094–6106. [Google Scholar] [CrossRef] [PubMed]
- Leone, L.; De Fenza, M.; Esposito, A.; Maglio, O.; Nastri, F.; Lombardi, A. Peptides and metal ions: A successful marriage for developing artificial metalloproteins. J. Pept. Sci. 2024, 30, e3606. [Google Scholar] [CrossRef]
- Kang, X.; Wang, L.; Liu, B.; Zhou, S.; Li, Y.; Yang, S.-L.; Yao, R.; Qiao, L.; Wang, X.; Gong, W.; et al. Mechanically rigid metallopeptide nanostructures achieved by highly efficient folding. Nat. Synth. 2024, 3, 835–845. [Google Scholar] [CrossRef]
- Bonczidai-Kelemen, D.; Tóth, K.; Fábián, I.; Lihi, N. The role of the terminal cysteine moiety in a metallopeptide mimicking the active site of the NiSOD enzyme. Dalton Trans. 2024, 53, 1648–1656. [Google Scholar] [CrossRef]
- Han, J.; Yan, X.; Yoon, J. Self-assembled Peptide-based Biocatalyst. In Peptide Self-Assembly and Engineering; Wiley-VCH GmbH: Weinheim, Germany, 2024; Volume 2, pp. 421–448. [Google Scholar]
- Yao, M.; Dong, S.; Xu, X. Asymmetric Carbene Transformations for the Construction of All-Carbon Quaternary Centers. Chem.—A Eur. J. 2024, 30, e202304299. [Google Scholar] [CrossRef]
- Balhara, R.; Chatterjee, R.; Jindal, G. Mechanism and stereoselectivity in metal and enzyme catalyzed carbene insertion into X–H and C (sp 2)–H bonds. Chem. Soc. Rev. 2024. [Google Scholar] [CrossRef]
- Pan, Y.; Wang, Y.; Karmakar, S.; Sivaguru, P.; Liu, Z. Catalytic alkane C–H functionalization by carbene insertion of unactivated C (sp3)–H bonds. Org. Chem. Front. 2024, 11, 3777–3799. [Google Scholar] [CrossRef]
- Díaz-Requejo, M.M.; Pérez, P.J. The TpxM Core in Csp3–H Bond Functionalization Reactions: Comparing Carbene, Nitrene, and Oxo Insertion Processes (Tpx = Scorpionate Ligand; M = Cu, Ag). Eur. J. Inorg. Chem. 2020, 2020, 879–885. [Google Scholar] [CrossRef]
- Rumo, C.; Stein, A.; Klehr, J.; Tachibana, R.; Prescimone, A.; Häussinger, D.; Ward, T.R. An Artificial Metalloenzyme Based on a Copper Heteroscorpionate Enables sp3 C–H Functionalization via Intramolecular Carbene Insertion. J. Am. Chem. Soc. 2022, 144, 11676–11684. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, L.M.C.; Consol, P.; Chen, Y. Drug Discovery in the Field of β-Lactams: An Academic Perspective. Antibiotics 2024, 13, 59. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Molina, J.M.; Belderrain, T.R.; Pérez, P.J. Trispyrazolylborate coinage metals complexes: Structural features and catalytic transformations. Coord. Chem. Rev. 2019, 390, 171–189. [Google Scholar] [CrossRef]
- Facchetti, G.; Rimoldi, I. 8-Amino-5,6,7,8-tetrahydroquinoline in iridium(iii) biotinylated Cp* complex as artificial imine reductase. New J. Chem. 2018, 42, 18773–18776. [Google Scholar] [CrossRef]
- Pellizzoni, M.; Facchetti, G.; Gandolfi, R.; Fusè, M.; Contini, A.; Rimoldi, I. Evaluation of Chemical Diversity of Biotinylated Chiral 1,3-Diamines as a Catalytic Moiety in Artificial Imine Reductase. ChemCatChem 2016, 8, 1665–1670. [Google Scholar] [CrossRef]
- Liang, A.D.; Serrano-Plana, J.; Peterson, R.L.; Ward, T.R. Artificial Metalloenzymes Based on the Biotin-Streptavidin Technology: Enzymatic Cascades and Directed Evolution. Acc. Chem. Res. 2019, 52, 585–595. [Google Scholar] [CrossRef]
- Mukherjee, P.; Sairaman, A.; Deka, H.J.; Jain, S.; Mishra, S.K.; Roy, S.; Bhaumik, P.; Maiti, D. Enantiodivergent synthesis of isoindolones catalysed by a Rh(III)-based artificial metalloenzyme. Nat. Synth. 2024, 3, 835–845. [Google Scholar] [CrossRef]
- Zhao, J.; Bachmann, D.G.; Lenz, M.; Gillingham, D.G.; Ward, T.R. An artificial metalloenzyme for carbene transfer based on a biotinylated dirhodium anchored within streptavidin. Catal. Sci. Technol. 2018, 8, 2294–2298. [Google Scholar] [CrossRef]
- Robles, V.M.; Dürrenberger, M.; Heinisch, T.; Lledós, A.; Schirmer, T.; Ward, T.R.; Maréchal, J.-D. Structural, Kinetic, and Docking Studies of Artificial Imine Reductases Based on Biotin–Streptavidin Technology: An Induced Lock-and-Key Hypothesis. J. Am. Chem. Soc. 2014, 136, 15676–15683. [Google Scholar] [CrossRef]
- Natoli, S.N.; Hartwig, J.F. Noble−Metal Substitution in Hemoproteins: An Emerging Strategy for Abiological Catalysis. Acc. Chem. Res. 2019, 52, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Key, H.M.; Dydio, P.; Clark, D.S.; Hartwig, J.F. Abiological catalysis by artificial haem proteins containing noble metals in place of iron. Nature 2016, 534, 534–537. [Google Scholar] [CrossRef] [PubMed]
- Dydio, P.; Key, H.M.; Nazarenko, A.; Rha, J.Y.-E.; Seyedkazemi, V.; Clark, D.S.; Hartwig, J.F. An artificial metalloenzyme with the kinetics of native enzymes. Science 2016, 354, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Lemon, C.M. Diversifying the functions of heme proteins with non-porphyrin cofactors. J. Inorg. Biochem. 2023, 246, 112282. [Google Scholar] [CrossRef]
- Pott, M.; Tinzl, M.; Hayashi, T.; Ota, Y.; Dunkelmann, D.; Mittl, P.R.E.; Hilvert, D. Noncanonical Heme Ligands Steer Carbene Transfer Reactivity in an Artificial Metalloenzyme. Angew. Chem. Int. Ed. 2021, 60, 15063–15068. [Google Scholar] [CrossRef]
- Gu, Y.; Natoli, S.N.; Liu, Z.; Clark, D.S.; Hartwig, J.F. Site-Selective Functionalization of (sp3)C−H Bonds Catalyzed by Artificial Metalloenzymes Containing an Iridium-Porphyrin Cofactor. Angew. Chem. Int. Ed. 2019, 58, 13954–13960. [Google Scholar] [CrossRef]
- Bloomer, B.J.; Natoli, S.N.; Garcia-Borràs, M.; Pereira, J.H.; Hu, D.B.; Adams, P.D.; Houk, K.N.; Clark, D.S.; Hartwig, J.F. Mechanistic and structural characterization of an iridium-containing cytochrome reveals kinetically relevant cofactor dynamics. Nat. Catal. 2023, 6, 39–51. [Google Scholar] [CrossRef]
- Mukherjee, A.; Roy, S. Non-Electrostatic Basis for an Artificial Metalloenzyme Catalysis. bioRxiv 2024. [Google Scholar] [CrossRef]
- Huang, X.; Besset, T.; Jubault, P.; Couve-Bonnaire, S. Fluorinated Substrates as Michael Acceptors Towards Fine Chemicals. Adv. Synth. Catal. 2023, 365, 2467–2486. [Google Scholar] [CrossRef]
- Deehan, T.; Hellier, P.; Ladommatos, N. The influence of Michael acceptors on the structural reactivity of renewable fuels. Sustain. Energy Fuels 2024, 8, 4168–4182. [Google Scholar] [CrossRef]
- Samanta, B.; Shankar Panda, B.; Mohapatra, S.; Nayak, S. Current Developments in Michael Addition Reaction using Heterocycles as Convenient Michael Donors. Asian J. Org. Chem. 2024, 13, e202400193. [Google Scholar] [CrossRef]
- Morita, Y.; Kubo, H.; Matsumoto, R.; Fujieda, N. A thiopyridine-bound mirror-image copper center in an artificial non-heme metalloenzyme. J. Inorg. Biochem. 2024, 260, 112694. [Google Scholar] [CrossRef] [PubMed]
- Fujieda, N.; Ichihashi, H.; Yuasa, M.; Nishikawa, Y.; Kurisu, G.; Itoh, S. Cupin Variants as a Macromolecular Ligand Library for Stereoselective Michael Addition of Nitroalkanes. Angew. Chem. 2020, 132, 7791–7794. [Google Scholar] [CrossRef]
- Fujieda, N.; Nakano, T.; Taniguchi, Y.; Ichihashi, H.; Sugimoto, H.; Morimoto, Y.; Nishikawa, Y.; Kurisu, G.; Itoh, S. A Well-Defined Osmium–Cupin Complex: Hyperstable Artificial Osmium Peroxygenase. J. Am. Chem. Soc. 2017, 139, 5149–5155. [Google Scholar] [CrossRef] [PubMed]
- Tabares, L.C.; Daniel, D.T.; Vázquez-Ibar, J.L.; Kouklovsky, C.; Alezra, V.; Un, S. Using the Noncanonical Metallo-Amino Acid [Cu(II)(2,2′-Bipyridin-5-yl)]-alanine to Study the Structures of Proteins. J. Phys. Chem. Lett. 2023, 14, 3368–3375. [Google Scholar] [CrossRef]
- Lamartina, C.W.; Chartier, C.A.; Hirano, J.M.; Shah, N.H.; Rovis, T. Crafting Unnatural Peptide Macrocycles via Rh(III)-Catalyzed Carboamidation. J. Am. Chem. Soc. 2024, 146, 20868–20877. [Google Scholar] [CrossRef]
- Learte-Aymamí, S.; Martínez-Castro, L.; González-González, C.; Condeminas, M.; Martin-Malpartida, P.; Tomás-Gamasa, M.; Baúlde, S.; Couceiro, J.R.; Maréchal, J.-D.; Macias, M.J.; et al. De Novo Engineering of Pd-Metalloproteins and Their Use as Intracellular Catalysts. JACS Au 2024, 4, 2630–2639. [Google Scholar] [CrossRef]
- Singh, K.; Kaur, A.; Goyal, B.; Goyal, D. Harnessing the Therapeutic Potential of Peptides for Synergistic Treatment of Alzheimer’s Disease by Targeting Aβ Aggregation, Metal-Mediated Aβ Aggregation, Cholinesterase, Tau Degradation, and Oxidative Stress. ACS Chem. Neurosci. 2024, 15, 2545–2564. [Google Scholar] [CrossRef]
- Wang, C.; Shao, S.; Li, N.; Zhang, Z.; Zhang, H.; Liu, B. Advances in Alzheimer’s Disease-Associated Aβ Therapy Based on Peptide. Int. J. Mol. Sci. 2023, 24, 13110. [Google Scholar] [CrossRef]
- Fujieda, N.; Tonomura, A.; Mochizuki, T.; Itoh, S. Asymmetric Michael addition catalysed by copper–amyloid complexes. RSC Adv. 2024, 14, 206–210. [Google Scholar] [CrossRef]
- Kjaergaard, C.H.; Qayyum, M.F.; Wong, S.D.; Xu, F.; Hemsworth, G.R.; Walton, D.J.; Young, N.A.; Davies, G.J.; Walton, P.H.; Johansen, K.S.; et al. Spectroscopic and computational insight into the activation of O2 by the mononuclear Cu center in polysaccharide monooxygenases. Proc. Natl. Acad. Sci. USA 2014, 111, 8797–8802. [Google Scholar] [CrossRef] [PubMed]
- Himes, R.A.; Barnese, K.; Karlin, K.D. One is Lonely and Three is a Crowd: Two Coppers Are for Methane Oxidation. Angew. Chem. Int. Ed. 2010, 49, 6714–6716. [Google Scholar] [CrossRef]
- Iacovino, L.G.; Pinzi, L.; Facchetti, G.; Bortolini, B.; Christodoulou, M.S.; Binda, C.; Rastelli, G.; Rimoldi, I.; Passarella, D.; Di Paolo, M.L.; et al. Promising Non-cytotoxic Monosubstituted Chalcones to Target Monoamine Oxidase-B. ACS Med. Chem. Lett. 2021, 12, 1151–1158. [Google Scholar] [CrossRef] [PubMed]
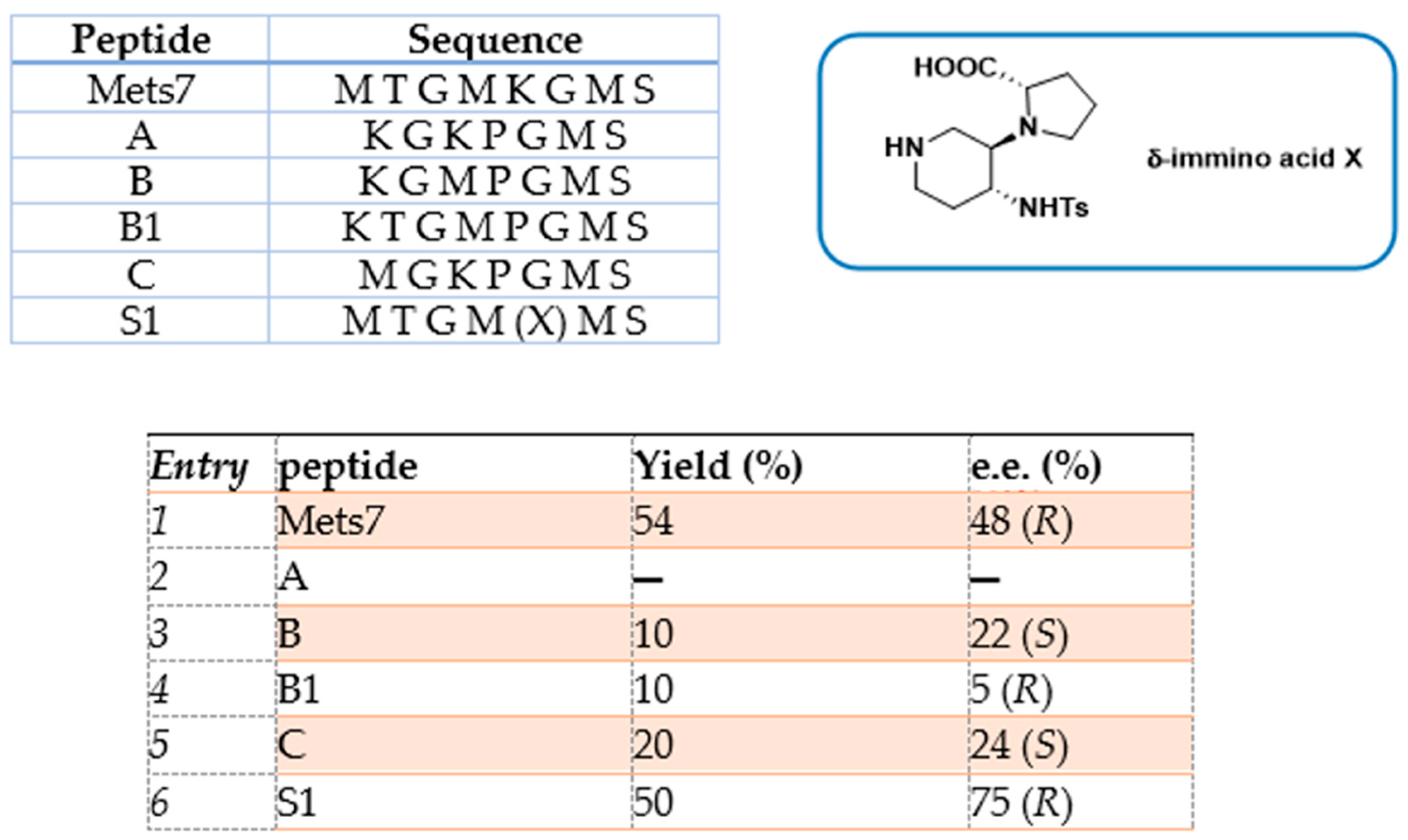
- Pellegrino, S.; Facchetti, G.; Contini, A.; Gelmi, M.L.; Erba, E.; Gandolfi, R.; Rimoldi, I. Ctr-1 Mets7 motif inspiring new peptide ligands for Cu(i)-catalyzed asymmetric Henry reactions under green conditions. RSC Adv. 2016, 6, 71529–71533. [Google Scholar] [CrossRef]
- Coffetti, G.; Moraschi, M.; Facchetti, G.; Rimoldi, I. The Challenging Treatment of Cisplatin-Resistant Tumors: State of the Art and Future Perspectives. Molecules 2023, 28, 3407. [Google Scholar] [CrossRef] [PubMed]
- Arnesano, F.; Natile, G. Interference between copper transport systems and platinum drugs. Semin. Cancer Biol. 2021, 76, 173–188. [Google Scholar] [CrossRef]
- Pellegrino, S.; Contini, A.; Gelmi, M.L.; Lo Presti, L.; Soave, R.; Erba, E. Asymmetric Modular Synthesis of a Semirigid Dipeptide Mimetic by Cascade Cycloaddition/Ring Rearrangement and Borohydride Reduction. J. Org. Chem. 2014, 79, 3094–3102. [Google Scholar] [CrossRef]
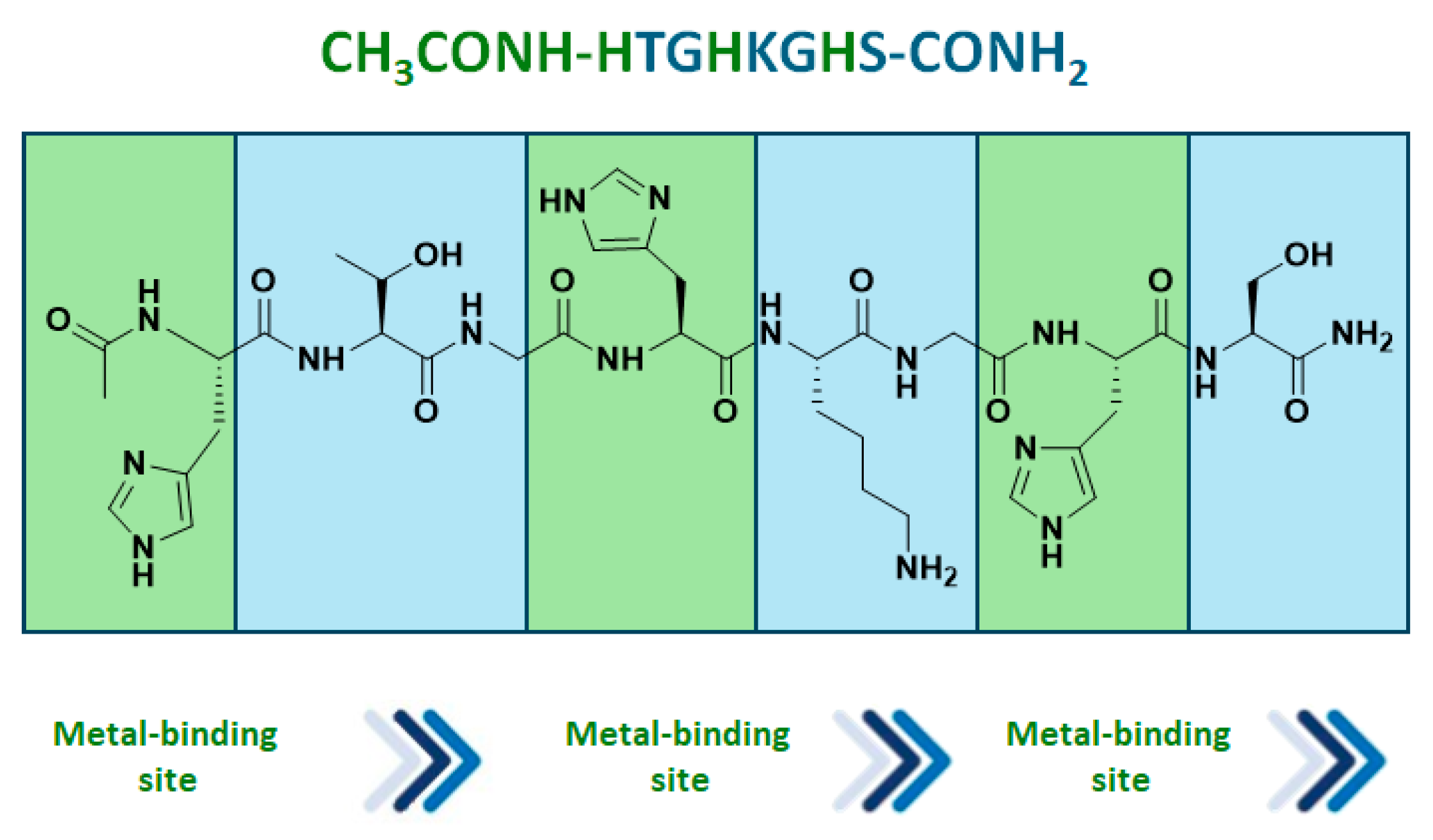
- Rimoldi, I.; Bucci, R.; Feni, L.; Santagostini, L.; Facchetti, G.; Pellegrino, S. Exploring the copper binding ability of Mets7 hCtr-1 protein domain and His7 derivative: An insight in Michael addition catalysis. J. Pept. Sci. 2021, 27, e3289. [Google Scholar] [CrossRef]
- Mavlankar, N.A.; Maulik, A.; Pal, A. Metal co-factors to enhance catalytic activity of short prion-derived peptide sequences. Pept. Catal. Incl. Catal. Amyloids 2024, 697, 473. [Google Scholar]
- Tao, K.; Wu, H.; Adler-Abramovich, L.; Zhang, J.; Fan, X.; Wang, Y.; Zhang, Y.; Tofail, S.A.M.; Mei, D.; Li, J.; et al. Aromatic short peptide architectonics: Assembly and engineering. Prog. Mater. Sci. 2024, 142, 101240. [Google Scholar] [CrossRef]
- Facchetti, G.; Vitoria, J.G.; Moraschi, M.; Bucci, R.; Abel, A.C.; Pieraccini, S.; Pellegrino, S.; Rimoldi, I. Exploitation of Dimeric Cyclic Cysteine as Helix Inducer in Ultra-Short Peptides for Cu(II)-Catalyzed Asymmetric Michael Addition on Chalcones. Eur. J. Org. Chem. 2023, 26, e202300240. [Google Scholar] [CrossRef]
- Oliva, F.; Bucci, R.; Tamborini, L.; Pieraccini, S.; Pinto, A.; Pellegrino, S. Bicyclic pyrrolidine-isoxazoline γ amino acid: A constrained scaffold for stabilizing α-turn conformation in isolated peptides. Front. Chem. 2019, 7, 133. [Google Scholar] [CrossRef] [PubMed]
- Heravi, M.M.; Zadsirjan, V.; Saedi, P.; Momeni, T. Applications of Friedel–Crafts reactions in total synthesis of natural products. RSC Adv. 2018, 8, 40061–40163. [Google Scholar] [CrossRef]
- Ohata, J. Friedel–Crafts reactions for biomolecular chemistry. Org. Biomol. Chem. 2024, 22, 3544–3558. [Google Scholar] [CrossRef]
- Haapalainen, A.M.; van Aalten, D.M.; Meriläinen, G.; Jalonen, J.E.; Pirilä, P.; Wierenga, R.K.; Hiltunen, J.K.; Glumoff, T. Crystal structure of the liganded SCP-2-like domain of human peroxisomal multifunctional enzyme type 2 at 1.75 Å resolution. J. Mol. Biol. 2001, 313, 1127–1138. [Google Scholar] [CrossRef]
- Van Stappen, C.; Deng, Y.; Liu, Y.; Heidari, H.; Wang, J.-X.; Zhou, Y.; Ledray, A.P.; Lu, Y. Designing Artificial Metalloenzymes by Tuning of the Environment beyond the Primary Coordination Sphere. Chem. Rev. 2022, 122, 11974–12045. [Google Scholar] [CrossRef] [PubMed]
- Jarvis, A.G.; Obrecht, L.; Deuss, P.J.; Laan, W.; Gibson, E.K.; Wells, P.P.; Kamer, P.C.J. Enzyme Activity by Design: An Artificial Rhodium Hydroformylase for Linear Aldehydes. Angew. Chem. Int. Ed. 2017, 56, 13596–13600. [Google Scholar] [CrossRef]
- Doble, M.V.; Jarvis, A.G.; Ward, A.C.C.; Colburn, J.D.; Götze, J.P.; Bühl, M.; Kamer, P.C.J. Artificial Metalloenzymes as Catalysts for Oxidative Lignin Degradation. ACS Sustain. Chem. Eng. 2018, 6, 15100–15107. [Google Scholar] [CrossRef]
- Kuckhoff, T.; Brewster, R.C.; Ferguson, C.T.J.; Jarvis, A.G. Reactivity Tuning of Metal-Free Artificial Photoenzymes through Binding Site Specific Bioconjugation. Eur. J. Org. Chem. 2023, 26, e202201412. [Google Scholar] [CrossRef]
- Pandya, C.; Sivaramakrishna, A. Organoruthenium-bipyridyl complexes—A platform for diverse chemistry and applications. Coord. Chem. Rev. 2024, 504, 215655. [Google Scholar] [CrossRef]
- Hu, Z.; Liang, J.; Su, T.; Zhang, D.; Li, H.; Gao, X.; Yao, W.; Song, X. Minimizing the Anticodon-Recognized Loop of Methanococcus jannaschii Tyrosyl-tRNA Synthetase to Improve the Efficiency of Incorporating Noncanonical Amino Acids. Biomolecules 2023, 13, 610. [Google Scholar] [CrossRef] [PubMed]
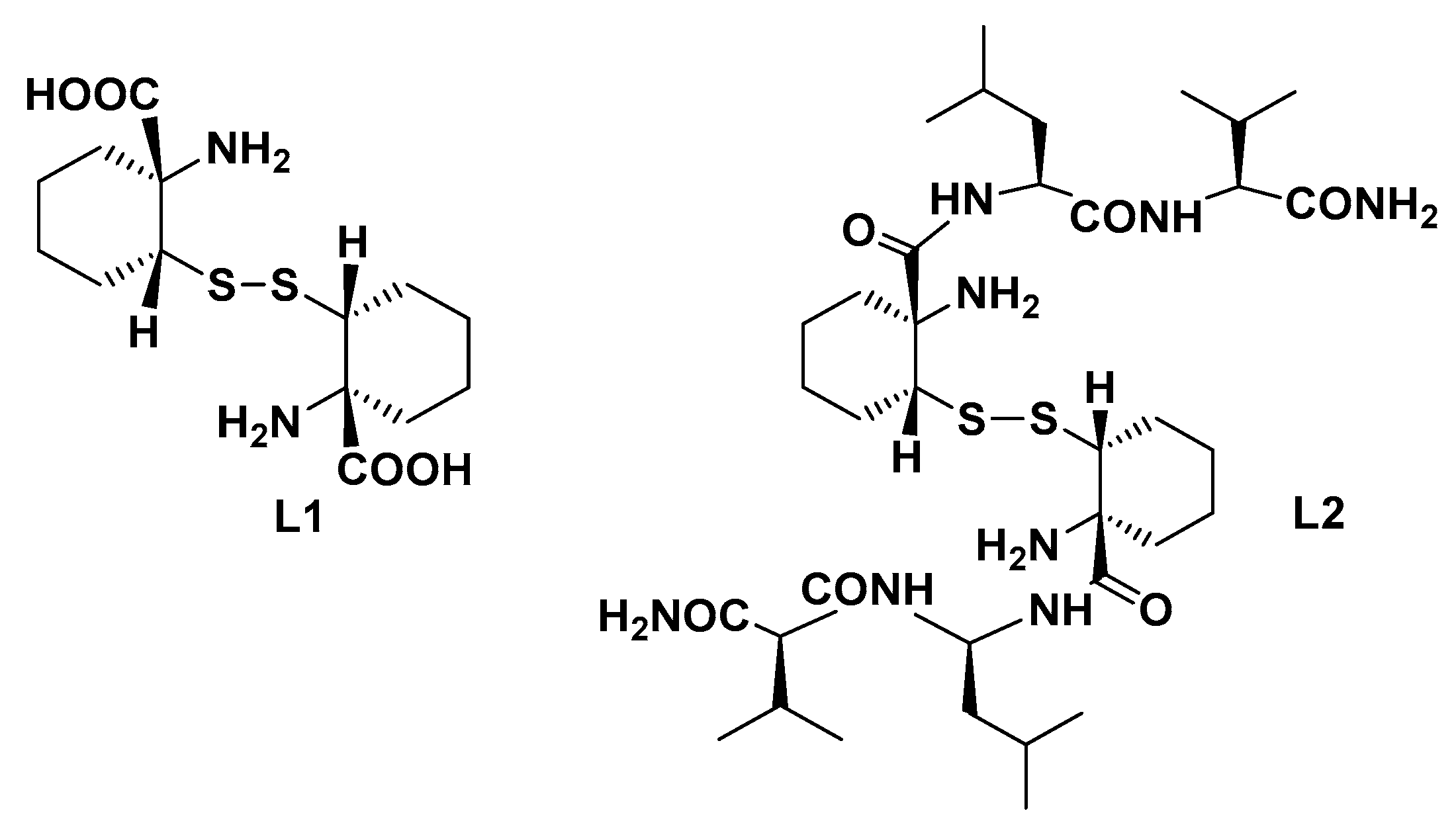
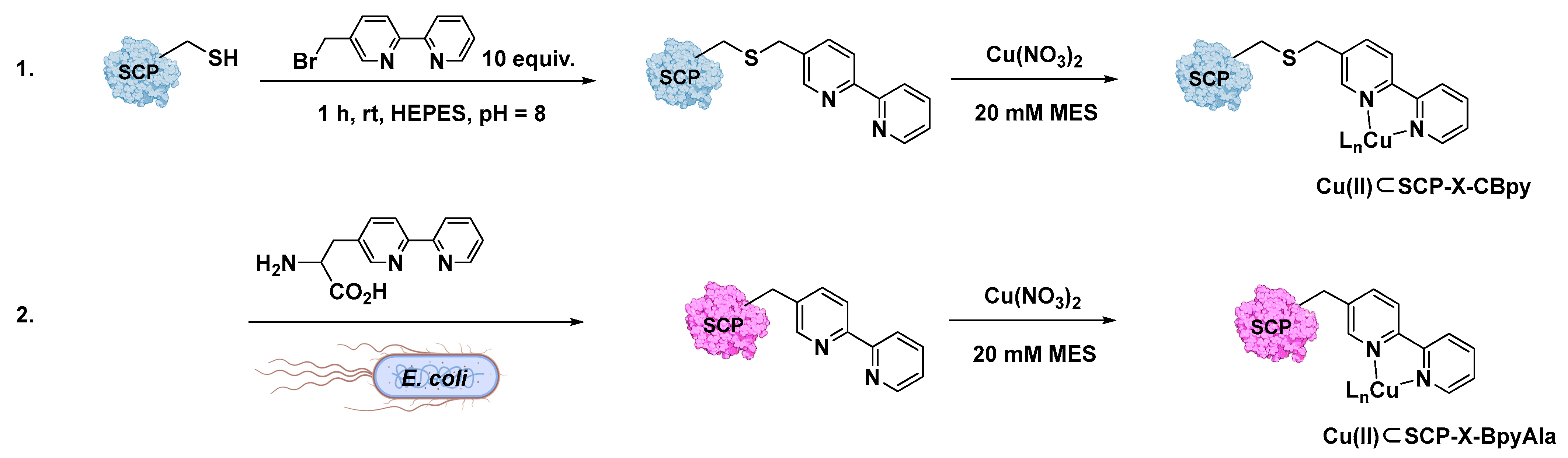
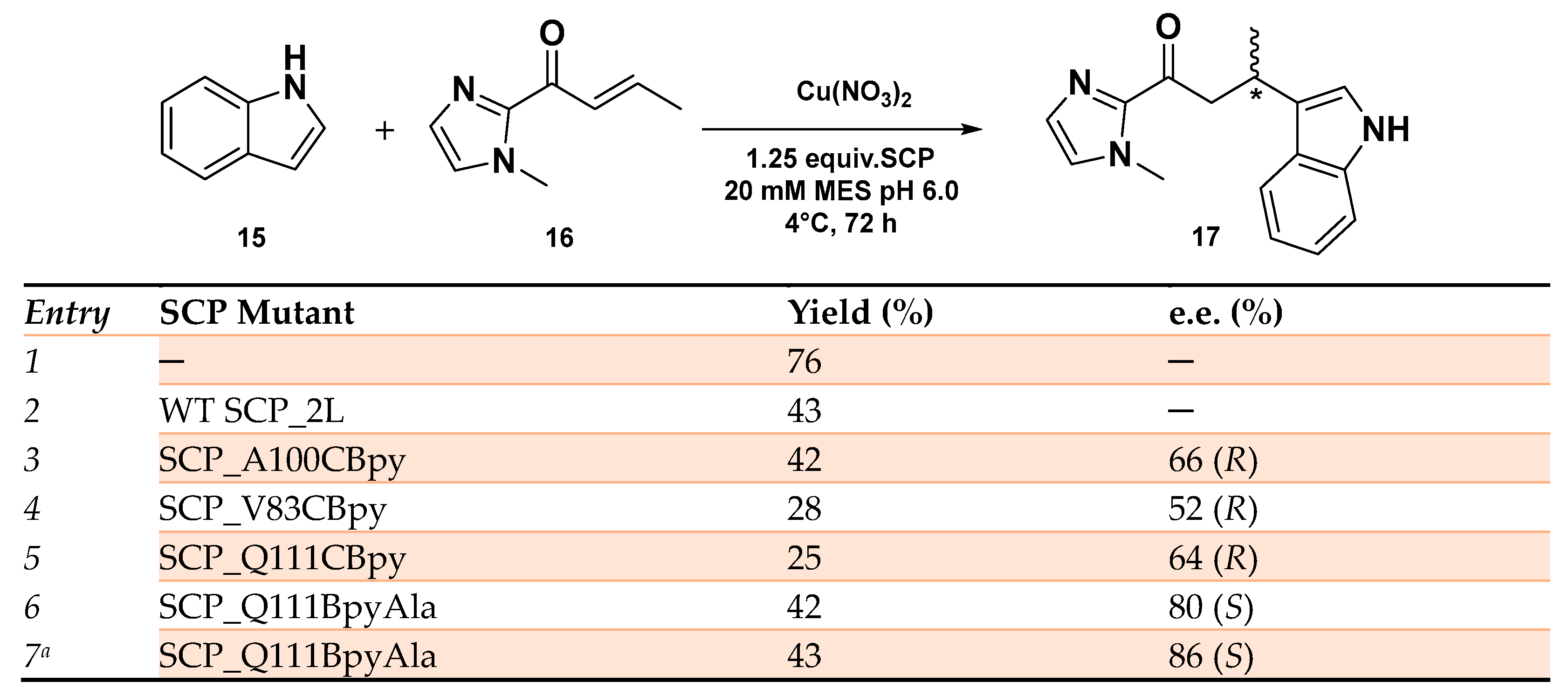
- Klemencic, E.; Brewster, R.C.; Ali, H.S.; Richardson, J.M.; Jarvis, A.G. Using BpyAla to generate copper artificial metalloenzymes: A catalytic and structural study. Catal. Sci. Technol. 2024, 14, 1622–1632. [Google Scholar] [CrossRef] [PubMed]
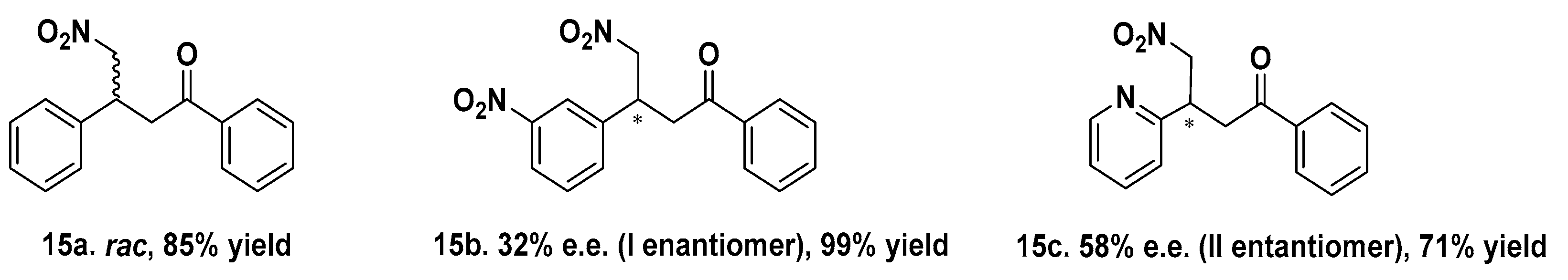
- Gutiérrez de Souza, C.; Bersellini, M.; Roelfes, G. Artificial Metalloenzymes based on TetR Proteins and Cu(II) for Enantioselective Friedel-Crafts Alkylation Reactions. ChemCatChem 2020, 12, 3190–3194. [Google Scholar] [CrossRef]
- Drienovská, I.; Scheele, R.A.; Gutiérrez de Souza, C.; Roelfes, G. A Hydroxyquinoline-Based Unnatural Amino Acid for the Design of Novel Artificial Metalloenzymes. ChemBioChem 2020, 21, 3077–3081. [Google Scholar] [CrossRef] [PubMed]
- Roelfes, G. Supramolecular Assembly of DNA—And Protein-Based Artificial Metalloenzymes. In Supramolecular Catalysis; Wiley-VCH GmbH: Weinheim, Germany, 2022; pp. 561–572. [Google Scholar]
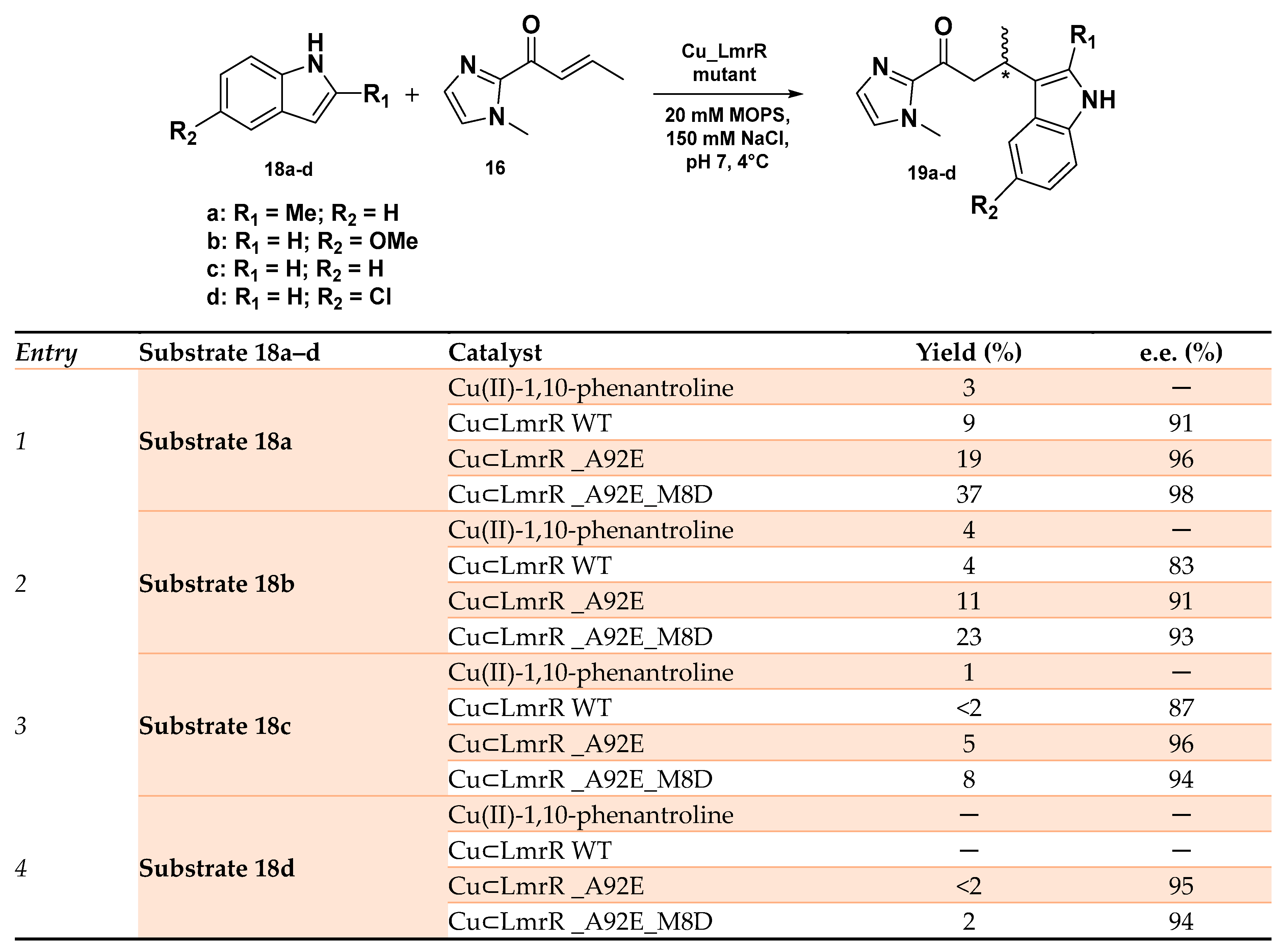
- Bos, J.; Browne, W.R.; Driessen, A.J.M.; Roelfes, G. Supramolecular Assembly of Artificial Metalloenzymes Based on the Dimeric Protein LmrR as Promiscuous Scaffold. J. Am. Chem. Soc. 2015, 137, 9796–9799. [Google Scholar] [CrossRef]
- Chordia, S.; Narasimhan, S.; Lucini Paioni, A.; Baldus, M.; Roelfes, G. In Vivo Assembly of Artificial Metalloenzymes and Application in Whole-Cell Biocatalysis. Angew. Chem. Int. Ed. 2021, 60, 5913–5920. [Google Scholar] [CrossRef]
- Saptal, V.B.; Ruta, V.; Bajada, M.A.; Vilé, G. Single-Atom Catalysis in Organic Synthesis. Angew. Chem. Int. Ed. 2023, 62, e202219306. [Google Scholar] [CrossRef]
- Kment, Š.; Bakandritsos, A.; Tantis, I.; Kmentová, H.; Zuo, Y.; Henrotte, O.; Naldoni, A.; Otyepka, M.; Varma, R.S.; Zbořil, R. Single Atom Catalysts Based on Earth-Abundant Metals for Energy-Related Applications. Chem. Rev. 2024. [Google Scholar] [CrossRef]
- Fang, J.; Chen, Q.; Li, Z.; Mao, J.; Li, Y. The synthesis of single-atom catalysts for heterogeneous catalysis. Chem. Commun. 2023, 59, 2854–2868. [Google Scholar] [CrossRef]
- Guo, Z.; Hong, J.; Song, N.; Liang, M. Single-Atom Nanozymes: From Precisely Engineering to Extensive Applications. Acc. Mater. Res. 2024, 5, 347–357. [Google Scholar] [CrossRef]
- Zhang, L.; Ren, Y.; Liu, W.; Wang, A.; Zhang, T. Single-atom catalyst: A rising star for green synthesis of fine chemicals. Natl. Sci. Rev. 2018, 5, 653–672. [Google Scholar] [CrossRef]
- Li, X.; Cao, Y.; Xiong, J.; Li, J.; Xiao, H.; Li, X.; Gou, Q.; Ge, J. Enzyme-metal-single-atom hybrid catalysts for one-pot chemoenzymatic reactions. Chin. J. Catal. 2023, 44, 139–145. [Google Scholar] [CrossRef]
- Zhu, J.; Zhang, Y.; Lu, D.; Zare, R.N.; Ge, J.; Liu, Z. Temperature-responsive enzyme–polymer nanoconjugates with enhanced catalytic activities in organic media. Chem. Commun. 2013, 49, 6090–6092. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Xu, P.; Ma, Y.; Yao, X.; Yao, Y.; Zong, M.; Li, X.; Lou, W. Recent advances in immobilized enzymes on nanocarriers. Chin. J. Catal. 2016, 37, 1814–1823. [Google Scholar] [CrossRef]
- Nikoshvili, L.Z.; Matveeva, V.G. Recent Progress in Pd-Catalyzed Tandem Processes. Catalysts 2023, 13, 1213. [Google Scholar] [CrossRef]
- Reissig, H.-U. Introduction to the Chemistry of Donor–Acceptor Cyclopropanes: A Historical and Personal Perspective. In Donor Acceptor Cyclopropanes in Organic Synthesis; Wiley-VCH GmbH: Weinheim, Germany, 2024; pp. 1–14. [Google Scholar]
- Talele, T.T. The “Cyclopropyl Fragment” is a Versatile Player that Frequently Appears in Preclinical/Clinical Drug Molecules. J. Med. Chem. 2016, 59, 8712–8756. [Google Scholar] [CrossRef]
- Sun, M.-R.; Li, H.-L.; Ba, M.-Y.; Cheng, W.; Zhu, H.-L.; Duan, Y.-T. Cyclopropyl Scaffold: A Generalist for Marketed Drugs. Mini Rev. Med. Chem. 2021, 21, 150–170. [Google Scholar] [CrossRef]
- Hoffman, G.R.; Olson, M.G.; Schoffstall, A.M.; Estévez, R.F.; Van den Eynde, V.; Gillman, P.K.; Stabio, M.E. Classics in Chemical Neuroscience: Selegiline, Isocarboxazid, Phenelzine, and Tranylcypromine. ACS Chem. Neurosci. 2023, 14, 4064–4075. [Google Scholar] [CrossRef]
- Li, H.; Cheng, J. 2-Phenylcyclopropylmethylamine (PCPMA) as a privileged scaffold for central nervous system drug design. Bioorganic Med. Chem. Lett. 2024, 101, 129654. [Google Scholar] [CrossRef]
- Tinoco, A.; Wei, Y.; Bacik, J.-P.; Carminati, D.M.; Moore, E.J.; Ando, N.; Zhang, Y.; Fasan, R. Origin of High Stereocontrol in Olefin Cyclopropanation Catalyzed by an Engineered Carbene Transferase. ACS Catal. 2019, 9, 1514–1524. [Google Scholar] [CrossRef]
- Chandgude, A.L.; Fasan, R. Highly Diastereo- and Enantioselective Synthesis of Nitrile-Substituted Cyclopropanes by Myoglobin-Mediated Carbene Transfer Catalysis. Angew. Chem. Int. Ed. 2018, 57, 15852–15856. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, P.; Sreenilayam, G.; Tyagi, V.; Fasan, R. Gram-Scale Synthesis of Chiral Cyclopropane-Containing Drugs and Drug Precursors with Engineered Myoglobin Catalysts Featuring Complementary Stereoselectivity. Angew. Chem. Int. Ed. 2016, 55, 16110–16114. [Google Scholar] [CrossRef] [PubMed]
- Carminati, D.M.; Fasan, R. Stereoselective Cyclopropanation of Electron-Deficient Olefins with a Cofactor Redesigned Carbene Transferase Featuring Radical Reactivity. ACS Catal. 2019, 9, 9683–9697. [Google Scholar] [CrossRef] [PubMed]
- Melngaile, R.; Videja, M.; Kuka, J.; Kinens, A.; Zacs, D.; Veliks, J. Synthetic Access to Fluorocyclopropylidenes. Org. Lett. 2023, 25, 2280–2284. [Google Scholar] [CrossRef]
- Zhang, G.; McCorvy, J.D.; Shen, S.; Cheng, J.; Roth, B.L.; Kozikowski, A.P. Design of fluorinated cyclopropane derivatives of 2-phenylcyclopropylmethylamine leading to identification of a selective serotonin 2C (5-HT2C) receptor agonist without 5-HT2B agonism. Eur. J. Med. Chem. 2019, 182, 111626. [Google Scholar] [CrossRef]
- Pons, A.; Delion, L.; Poisson, T.; Charette, A.B.; Jubault, P. Asymmetric Synthesis of Fluoro, Fluoromethyl, Difluoromethyl, and Trifluoromethylcyclopropanes. Acc. Chem. Res. 2021, 54, 2969–2990. [Google Scholar] [CrossRef]
- Yamani, K.; Pierre, H.; Archambeau, A.; Meyer, C.; Cossy, J. Asymmetric Transfer Hydrogenation of gem-Difluorocyclopropenyl Esters: Access to Enantioenriched gem-Difluorocyclopropanes. Angew. Chem. Int. Ed. 2020, 59, 18505–18509. [Google Scholar] [CrossRef]
- Pellissier, H. Recent developments in enantioselective zinc-catalyzed transformations. Coord. Chem. Rev. 2021, 439, 213926. [Google Scholar] [CrossRef]
- Schaus, L.; Das, A.; Knight, A.M.; Jimenez-Osés, G.; Houk, K.N.; Garcia-Borràs, M.; Arnold, F.H.; Huang, X. Protoglobin-Catalyzed Formation of cis-Trifluoromethyl-Substituted Cyclopropanes by Carbene Transfer. Angew. Chem. Int. Ed. 2023, 62, e202208936. [Google Scholar] [CrossRef]
- Ren, X.; Chandgude, A.L.; Carminati, D.M.; Shen, Z.; Khare, S.D.; Fasan, R. Highly stereoselective and enantiodivergent synthesis of cyclopropylphosphonates with engineered carbene transferases. Chem. Sci. 2022, 13, 8550–8556. [Google Scholar] [CrossRef]
- Villada, J.D.; Majhi, J.; Lehuédé, V.; Hendricks, M.E.; Neufeld, K.; Tona, V.; Fasan, R. Biocatalytic Strategy for the Highly Stereoselective Synthesis of Fluorinated Cyclopropanes. Angew. Chem. Int. Ed. 2024, 63, e202406779. [Google Scholar] [CrossRef] [PubMed]
- Sikandar, S.; Zahoor, A.F.; Ghaffar, A.; Anjum, M.N.; Noreen, R.; Irfan, A.; Munir, B.; Kotwica-Mojzych, K.; Mojzych, M. Unveiling the Chemistry and Synthetic Potential of Catalytic Cycloaddition Reaction of Allenes: A Review. Molecules 2023, 28, 704. [Google Scholar] [CrossRef] [PubMed]
- Gregg, T.M.; Farrugia, M.K.; Frost, J.R. Rhodium-mediated enantioselective cyclopropanation of allenes. Org. Lett. 2009, 11, 4434–4436. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, Y.; Cantin, T.; Decaens, J.; Couve-Bonnaire, S.; Charette, A.B.; Poisson, T.; Jubault, P. Catalytic Asymmetric Syntheses of Alkylidenecyclopropanes from Allenoates with Donor-Acceptor and Diacceptor Diazo Reagents. Chem.—A Eur. J. 2022, 28, e202201438. [Google Scholar] [CrossRef]
- Lindsay, V.N.G.; Fiset, D.; Gritsch, P.J.; Azzi, S.; Charette, A.B. Stereoselective Rh2(S-IBAZ)4-Catalyzed Cyclopropanation of Alkenes, Alkynes, and Allenes: Asymmetric Synthesis of Diacceptor Cyclopropylphosphonates and Alkylidenecyclopropanes. J. Am. Chem. Soc. 2013, 135, 1463–1470. [Google Scholar] [CrossRef]
- Bloomer, B.J.; Joyner, I.A.; Garcia-Borràs, M.; Hu, D.B.; Garçon, M.; Quest, A.; Ugarte Montero, C.; Yu, I.F.; Clark, D.S.; Hartwig, J.F. Enantio- and Diastereodivergent Cyclopropanation of Allenes by Directed Evolution of an Iridium-Containing Cytochrome. J. Am. Chem. Soc. 2024, 146, 1819–1824. [Google Scholar] [CrossRef]
- Maglio, O.; Chino, M.; D’Alonzo, D.; Leone, L.; Nastri, F.; Lombardi, A. Peptide-based Artificial Metalloenzymes by Design. In Peptide and Protein Engineering for Biotechnological and Therapeutic Applications; World Scientific Publishing Co.: Singapore, 2023; pp. 371–420. [Google Scholar]
- Zozulia, O.; Korendovych, I.V. Semi-Rationally Designed Short Peptides Self-Assemble and Bind Hemin to Promote Cyclopropanation. Angew. Chem. Int. Ed. 2020, 59, 8108–8112. [Google Scholar] [CrossRef]
- Villarino, L.; Splan, K.E.; Reddem, E.; Alonso-Cotchico, L.; Gutiérrez de Souza, C.; Lledós, A.; Maréchal, J.-D.; Thunnissen, A.-M.W.H.; Roelfes, G. An Artificial Heme Enzyme for Cyclopropanation Reactions. Angew. Chem. Int. Ed. 2018, 57, 7785–7789. [Google Scholar] [CrossRef]
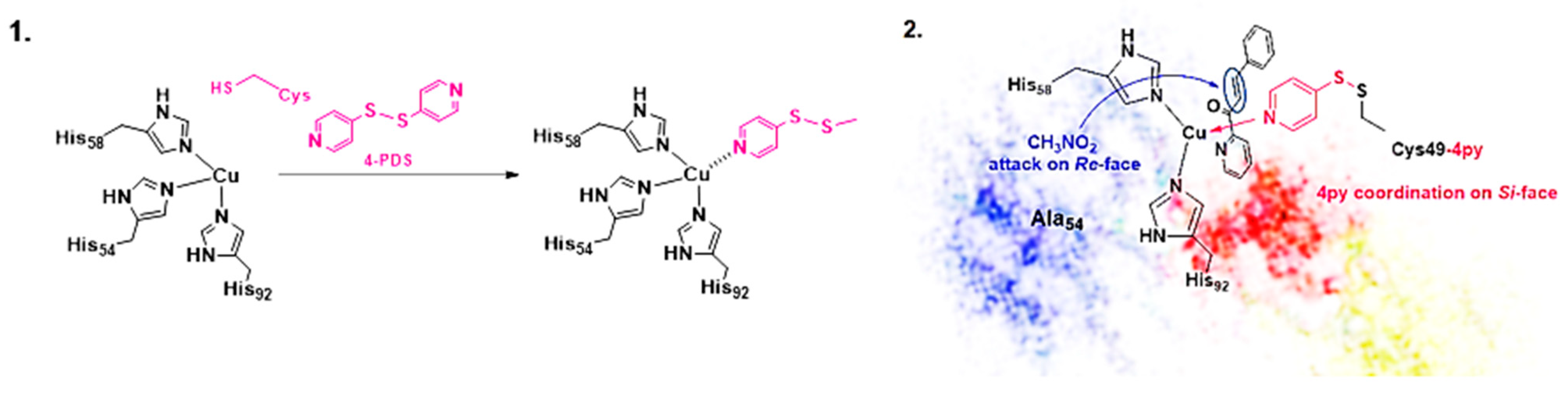
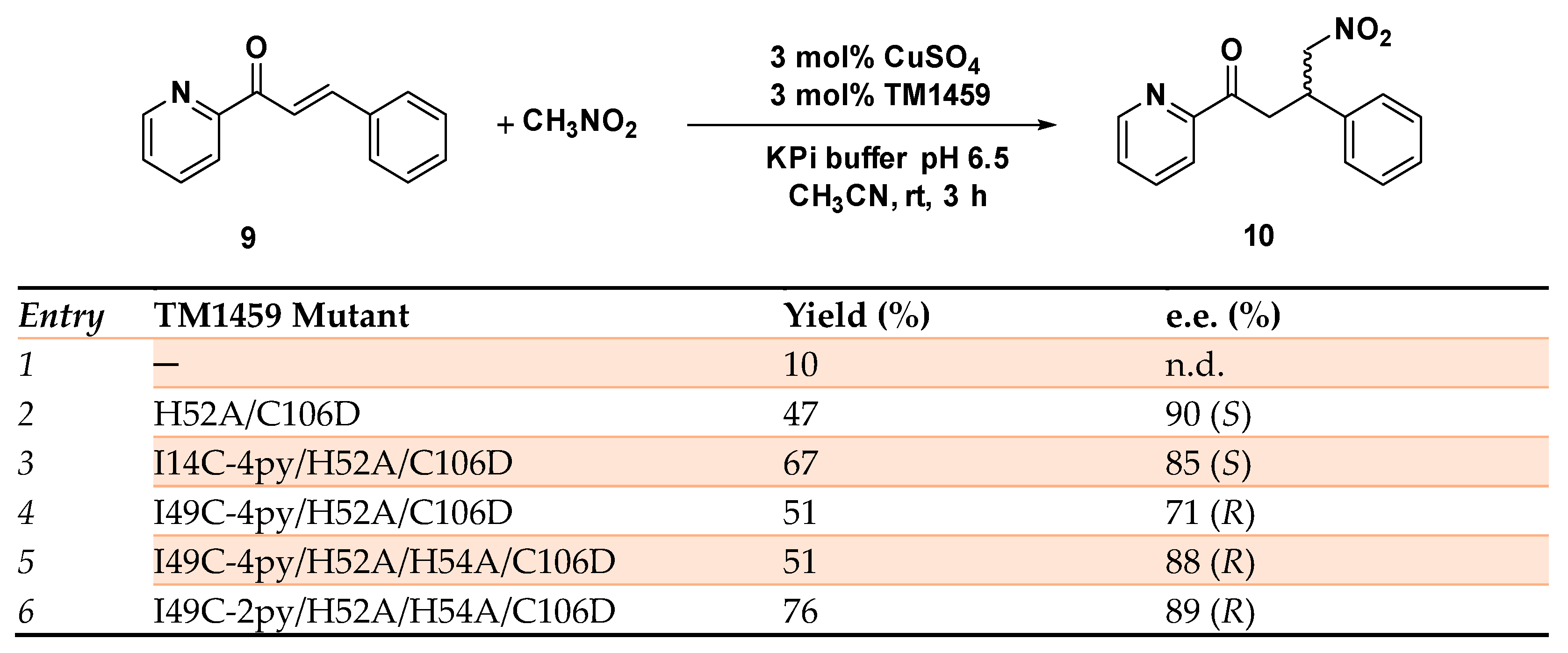
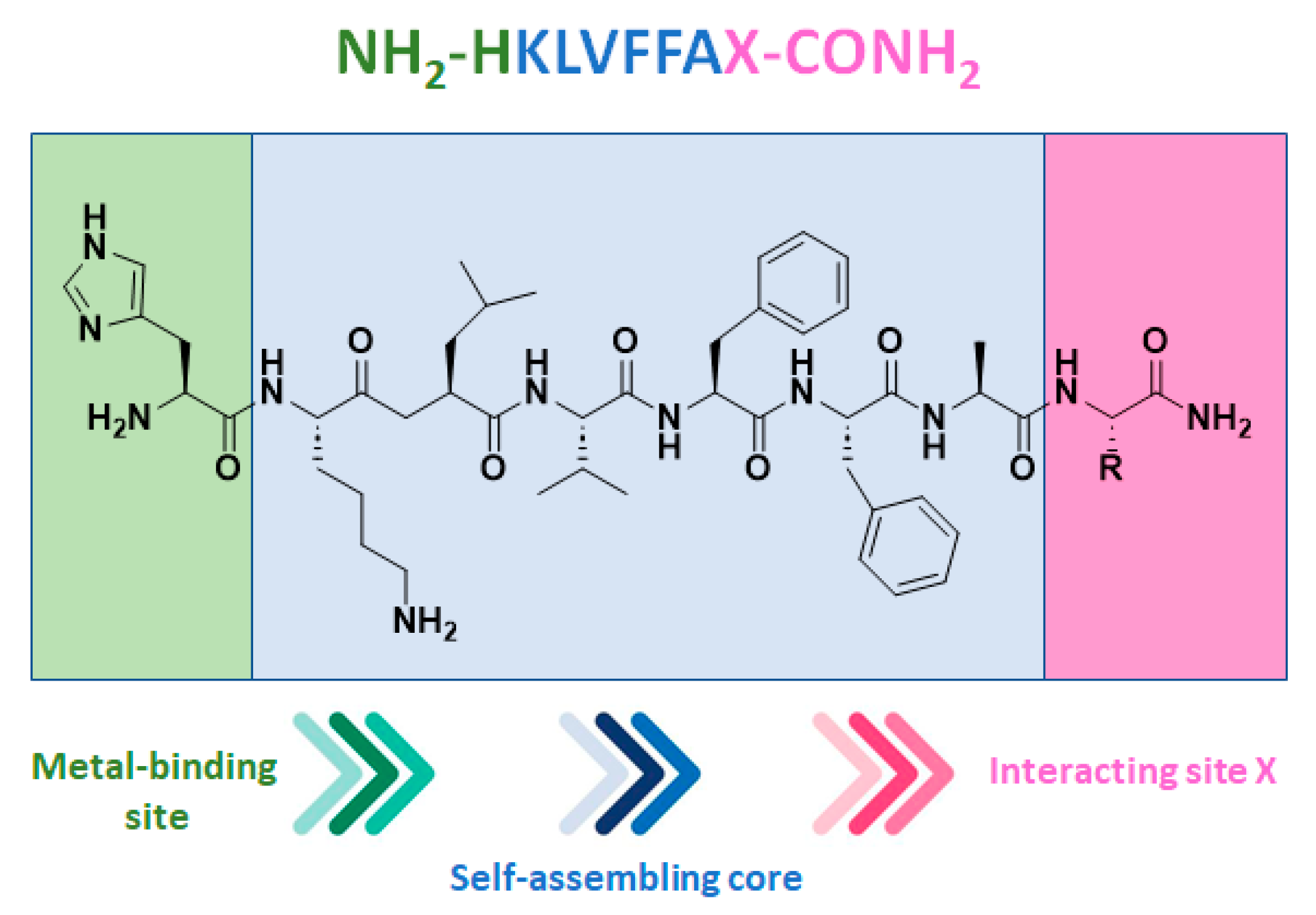
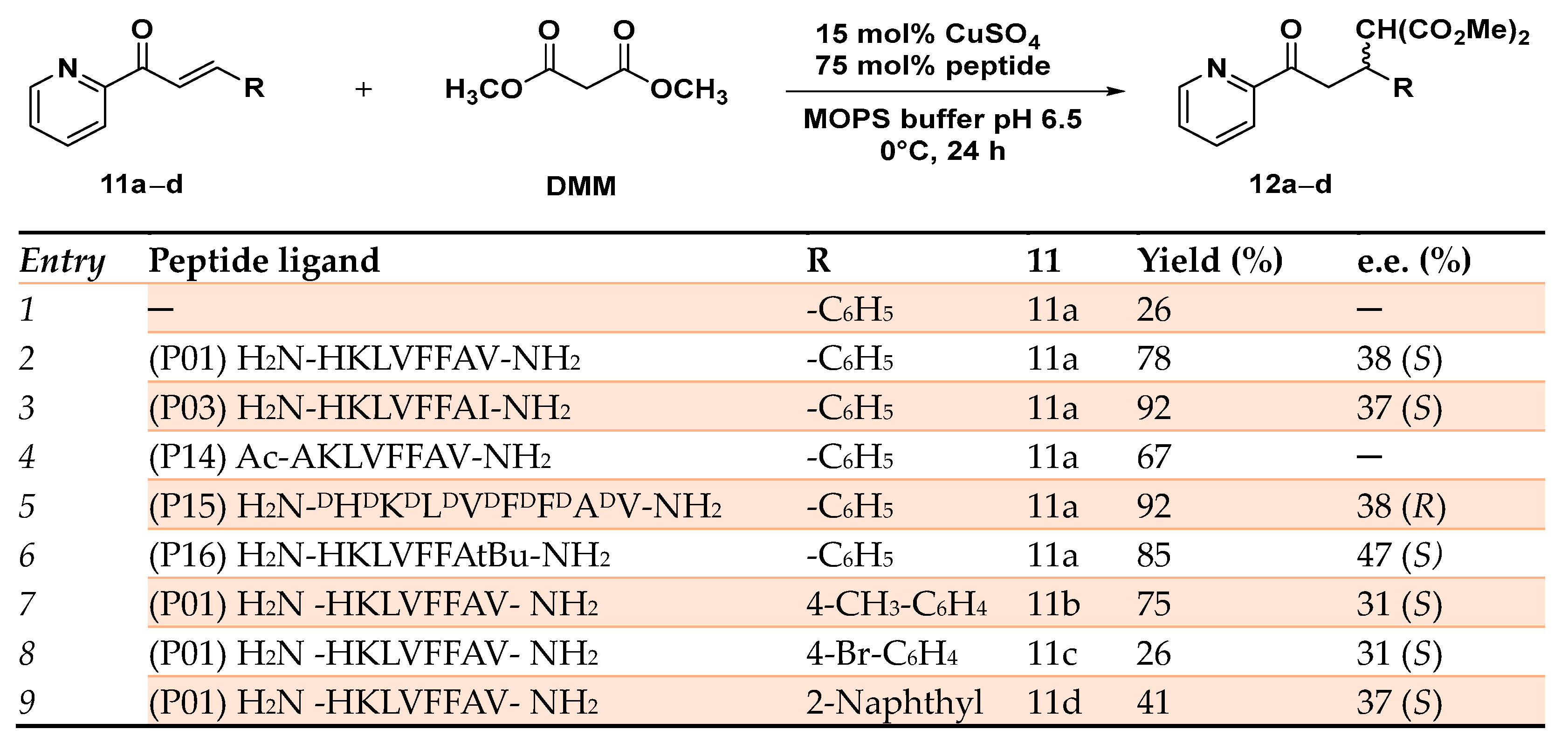
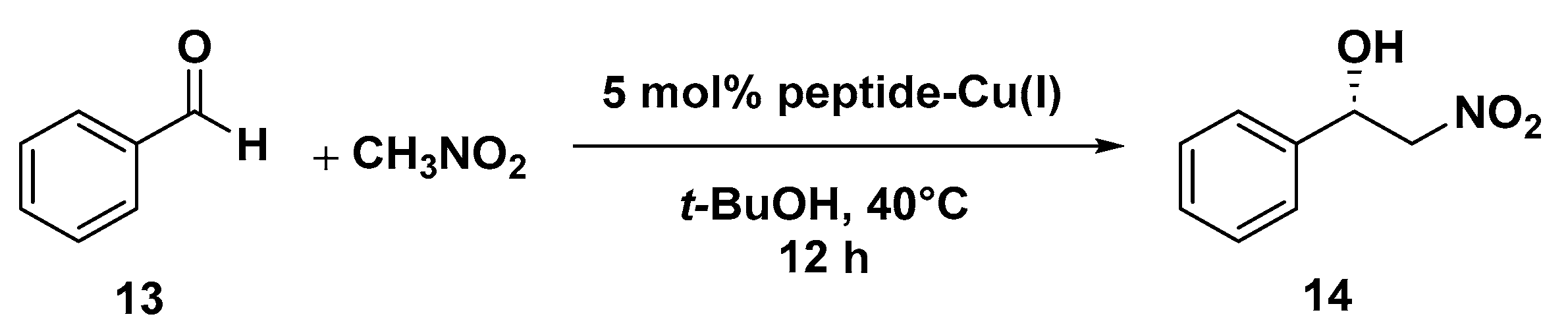

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rimoldi, I.; Coffetti, G.; Gandolfi, R.; Facchetti, G. Hybrid Metal Catalysts as Valuable Tools in Organic Synthesis: An Overview of the Recent Advances in Asymmetric C─C Bond Formation Reactions. Molecules 2024, 29, 5090. https://doi.org/10.3390/molecules29215090
Rimoldi I, Coffetti G, Gandolfi R, Facchetti G. Hybrid Metal Catalysts as Valuable Tools in Organic Synthesis: An Overview of the Recent Advances in Asymmetric C─C Bond Formation Reactions. Molecules. 2024; 29(21):5090. https://doi.org/10.3390/molecules29215090
Chicago/Turabian StyleRimoldi, Isabella, Giulia Coffetti, Raffaella Gandolfi, and Giorgio Facchetti. 2024. "Hybrid Metal Catalysts as Valuable Tools in Organic Synthesis: An Overview of the Recent Advances in Asymmetric C─C Bond Formation Reactions" Molecules 29, no. 21: 5090. https://doi.org/10.3390/molecules29215090
APA StyleRimoldi, I., Coffetti, G., Gandolfi, R., & Facchetti, G. (2024). Hybrid Metal Catalysts as Valuable Tools in Organic Synthesis: An Overview of the Recent Advances in Asymmetric C─C Bond Formation Reactions. Molecules, 29(21), 5090. https://doi.org/10.3390/molecules29215090