
Synthesis of Perfluoroalkylated Pyrazoles from α-Perfluoroalkenylated Aldehydes


Abstract
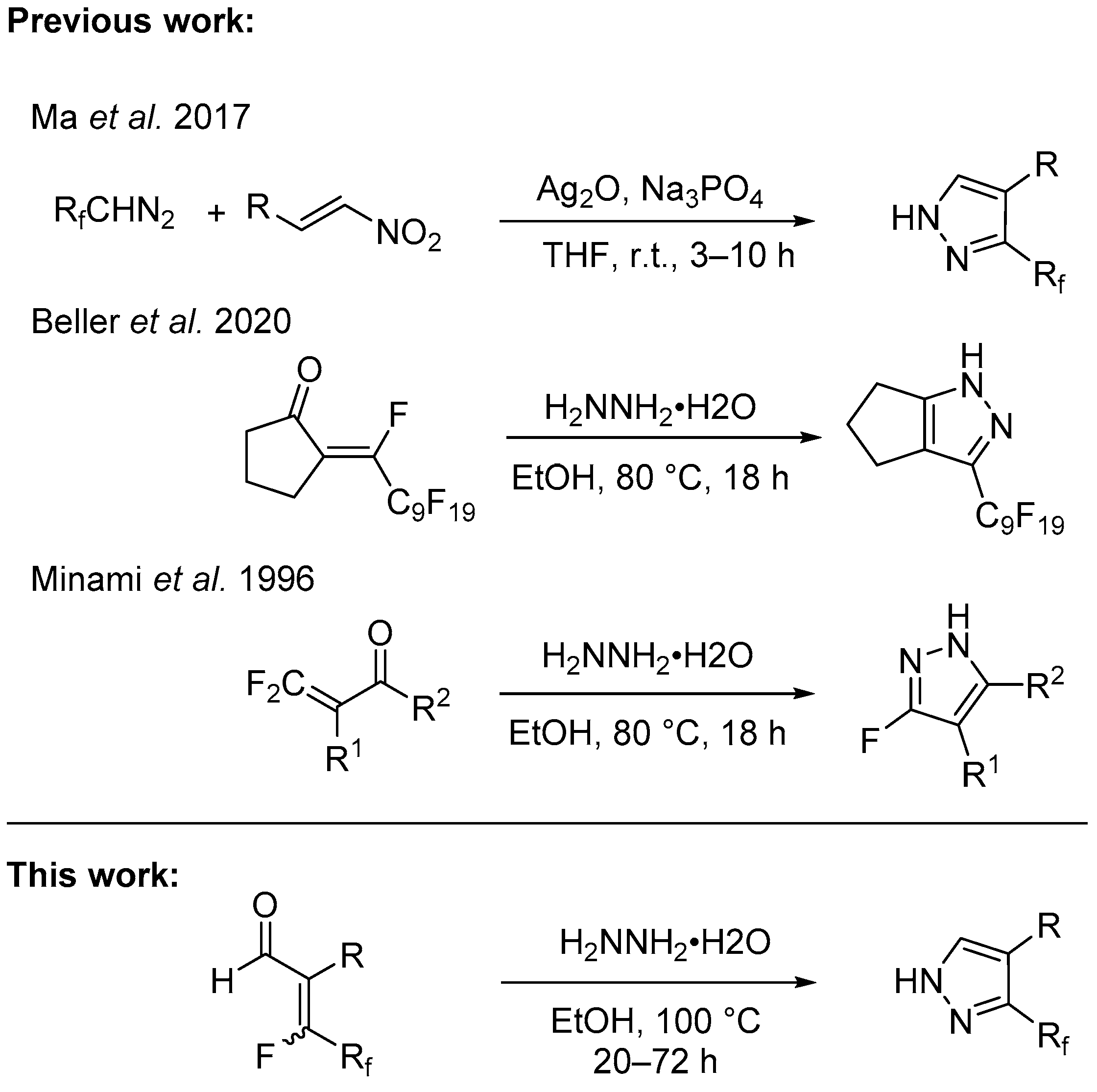
1. Introduction
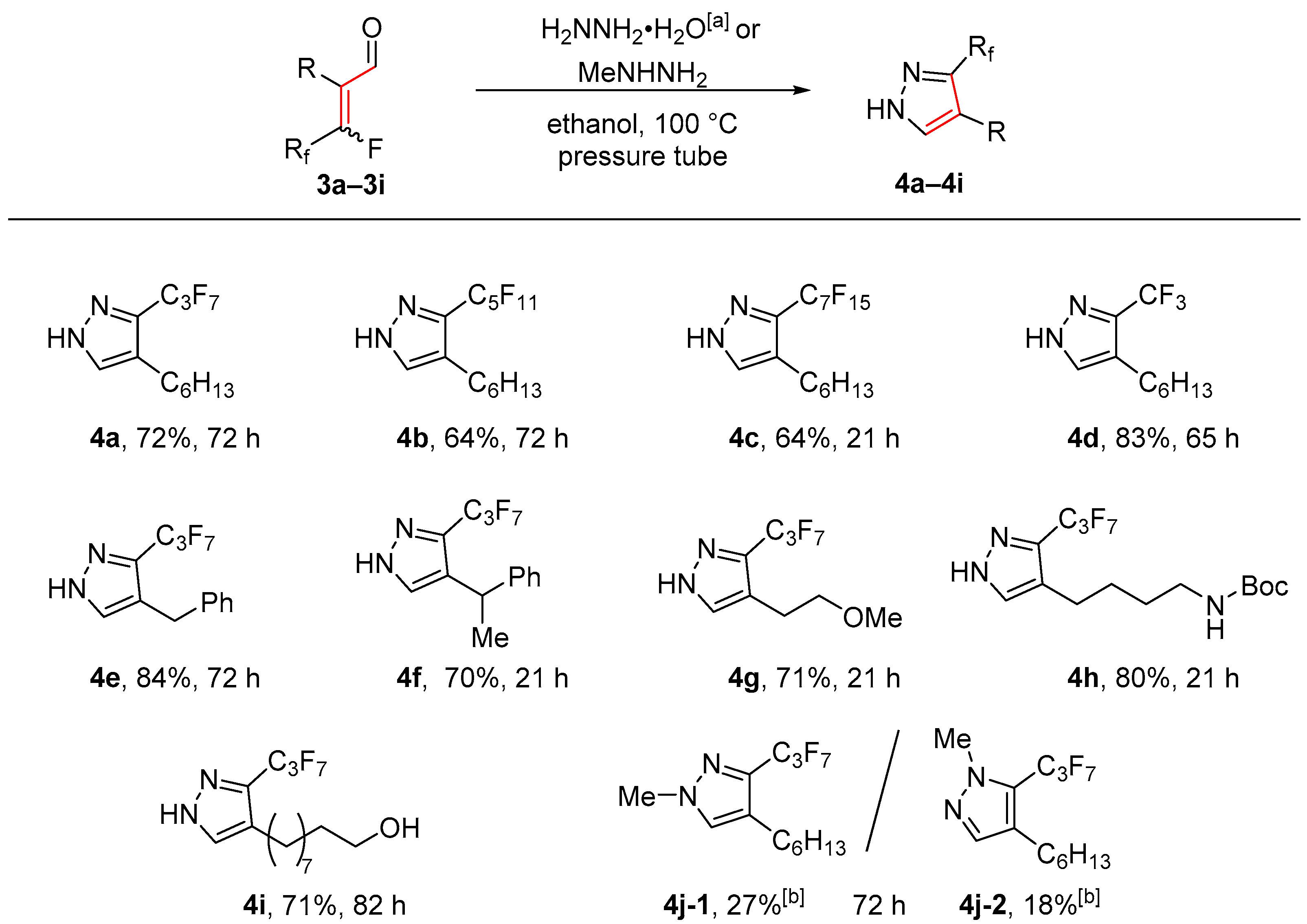
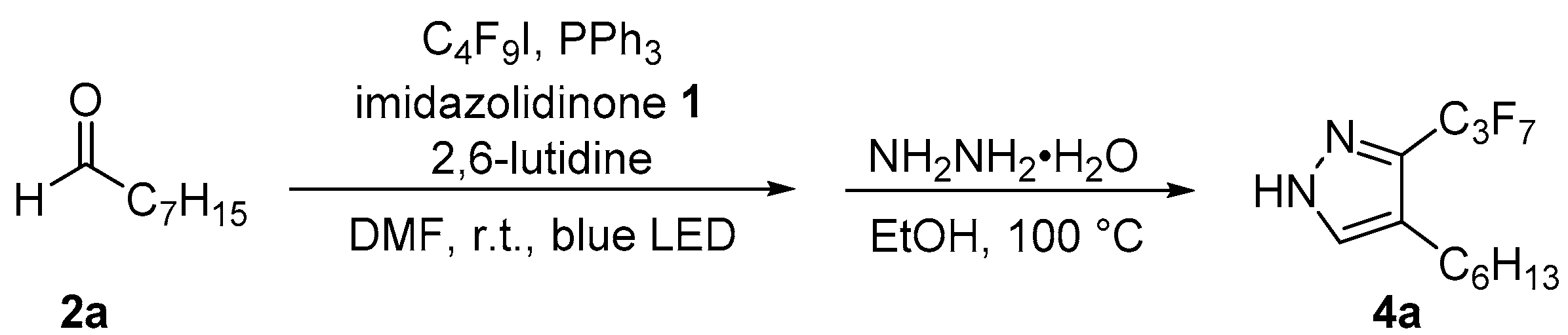
2. Results and Discussion
3. Experimental Section
3.1. General Information
3.2. Synthesis of Starting Materials
- General procedure I for the photocatalytic perfluoroalkenylation of aldehydes
- 2-(perfluoroethylidene)-octanal (3d). Following general procedure I, octanal (2a) (0.128 g, 1.00 mmol, 1.00 equiv), (S)-2,2,3,5-tetramethylimidazolidin-4-on (1a) (16.4 mg, 0.115 mmol 0.15 equiv), triphenylphosphine (26.2 mg, 0.100 mmol, 0.10 equiv), pentafluoro-1-iodoethane (0.17 mL, 1.4 mmol, 1.4 equiv), and 2,6-lutidine (0.14 mL, 1.2 mmol, 1.2 equiv) were mixed with dimethylformamide (0.5 mL) and irritated for 21 h. Purification by flash chromatography (n-pentane/diethyl ether 95:5) gave a mixture of (E)- and (Z)-2-(perfluoroethylidene)-octanal (0.143 g, 0.632 mmol, 63%, E/Z; 60:40) as a colorless liquid.
- 1H NMR (300 MHz, CDCl3) δ = 10.19 (s, 0.4H), 10.06 (s, 0.6H), 2.43–2.27 (m, 2H), 1.43–1.24 (m, 8H), 0.88 (t, 3H). 19F NMR (282 MHz, CDCl3) δ = −63.52 (d, J = 7.8 Hz), −67.66 (d, J = 6.6 Hz), −110.52 (q, J = 7.9 Hz), −129.26 (q, J = 6.6 Hz). 13C NMR (75 MHz, CDCl3) δ 188.09–187.24 (m), 160.00–116.39 (m), 77.41, 76.98, 76.56, 34.11, 31.35, 31.28, 29.12, 28.99, 27.90, 22.70, 22.66, 22.44, 22.32, 21.86, 13.99, 13.92. IR: [cm−1] = 2960, 2933, 2863, 2367, 2341, 1698, 1669, 1335, 1204, 1156, 1094.
- 2-(perfluorobutylidene)-3-methyl-3-phenylpropanal (3f). Following general procedure I, 3-phenylbutyraldehyde (2f) (0.223 mL, 1.50 mmol, 1.00 equiv), (S)-2,2,3,5-tetramethylimidazolidin-4-on (1a) (27.4 mg, 0.193 mmol, 0.13 equiv), triphenylphosphine (47.5 mg, 0.181 mmol, 0.12 equiv), nonafluoro-1-iodobutane (0.535 mL, 3.11 mmol, 2.07 equiv), and 2,6-lutidine (0.23 mL, 2.0 mmol, 1.3 equiv) were mixed with dimethylformamide (1 mL) and irritated for 20 h. Purification by flash chromatography (n-pentane/diethyl ether 99.4:0.6) gave a mixture of (E)- and (Z)-2-(perfluorobutylidene)-3-methyl-3-phenylpropanal (0.090 g, 0.26 mmol, 17%, E/Z; 86:14) as a colorless liquid.
- 1H NMR (300 MHz, CDCl3) δ = 10.04 (d, J = 1.9 Hz, 0.1H), 9.94 (td, J = 1.7 Hz, 0.9, 0.9H), 7.37–7.27 (m, 3H), 7.27–7.17 (m, 2H), 4.46 (q, J = 7.3 Hz, 0.9H), 4.13 (d, J = 7.5 Hz, 0.1H), 1.70–1.58 (m, 3H). 19F NMR (282 MHz, CDCl3) δ = −80.27–−80.51 (m, 3F), −106.33 (m, 0.9F), −111.97 (m, 1.8F), −112.69–−112.80 (m, 0.01F), −113.62–−113.87 (m, 0.09F), −114.32–−114.51 (m, 0.08F), −115.44 (m, 0.01F), −122.97 (m, z0.03F), −126.81–−126.95 (m, 0.2F), −127.39 (dd, J = 6.6, 2.9 Hz, 1.7F). 13C NMR (151 MHz, CDCl3) δ = 187.65 (m), 187.44–187.17 (m), 156.55–155.77 (m), 154.62–154.04 (m) 141.02, 140.27, 137.32, 137.24, 135.45, 135.41, 133.81, 133.68, 128.68, 128.54, 128.49, 128.45, 128.43, 127.29, 127.18, 126.90, 126.82, 119.07–107.59 (m), 35.06, 34.14, 17.19, 17.16. IR: [cm−1] = 3059, 2930, 1698, 1654, 1435, 1354, 1265, 1232, 1189, 1127.
3.3. Synthesis of Perfluoroalkylated Pyrazoles
- General procedure II for the cyclization of the α-perfluoroalkenylated aldehydes to the corresponding pyrazols (4a–4j)
- 4-hexyl-3-perfluoropropyl-1H-pyrazol (4a). Following general procedure II, 2-(perfluorobutylidene)-octanal (3a) (0.169 g, 0.518 mmol, 1.00 equiv), hydrazine monohydrate (0.095 mL, 2.0 mmol, 3.9 equiv), and ethanol (2 mL) were mixed and stirred at 100 °C for 72 h. Purification by flash chromatography (n-pentane/diethyl ether 7:3) gave the desired product (120 mg, 0.373 mmol, 72%) as a colorless oil.
- 1H NMR (300 MHz, CDCl3) δ 13.19 (s, 1H), 7.47 (s, 1H), 2.57 (t, J = 7.8 Hz, 2H), 1.67–1.51 (m, 1H), 1.45–1.21 (m, 2H), 0.96–0.84 (m, 3H). 19F NMR (282 MHz, CDCl3) δ −80.25 (t, J = 9.6 Hz, 3F), −109.81 (q, J = 9.7 Hz, 2F), −126.75 (q, J = 7.4 Hz, 2F). 13C NMR (151 MHz, CDCl3) δ = 137.95 (t, J = 27.0 Hz.), 129.31, 122.85, 121.66–106.69 (m), 31.72, 30.55, 29.14, 23.35, 22.71, 14.11. IR: [cm−1] = 3177, 2962, 2935, 2863, 2360, 2341, 1347, 1230, 1212, 1116. HRMS-ESI: calcd for C12H15F7N2 [M + H+] m/z 321.1196, found 321.1197.; Rf = 0.3 in n-pentane/diethyl ether 7:3.
- 4-hexyl-3-perfluoropentyl-1H-pyrazol (4b). Following general procedure II, 2-(perfluorohexylidene)octanal (3b) (0.212 g, 0.498 mmol, 1.00 equiv), hydrazine monohydrate (0.090 mL, 1.9 mmol, 3.8 equiv), and ethanol (2 mL) were mixed and stirred at 100 °C for 72 h. Purification by flash chromatography (n-pentane/diethyl ether 7:3) gave the desired product (0.135 g, 0.321 mmol, 64%) as a colorless oil.
- 1H NMR (300 MHz, CDCl3) δ = 13.16 (s, 1H, H-1), 7.46 (s, 1H), 2.58 (t, J = 7.8 Hz, 2H), 1.67–1.51 (m, 2H), 1.45–1.21 (m, 6H,), 0.97–0.84 (m, 3H). 19F NMR (282 MHz, CDCl3) δ = −80.83 (tt, J = 10.0, 2.7 Hz, 3F), −108.77–−109.06 (m, 2F), −122.28–−122.50 (m, 2F), −122.49–−122.72 (m, 2F), −126.08–−126.34 (m, 2F). 13C NMR (151 MHz, CDCl3) δ = 138.24–137.73 (m), 129.21, 122.86, 120.85–105.82 (m), 30.45, 29.10, 23.31, 22.68, 14.12. IR: [cm−1] = 3177, 2962, 2934, 2863, 1469, 1361, 1239, 1205, 1145, 1106. HRMS-ESI: calcd for C14H15F11N2 [M + H+] m/z 421.1132, found 421.1133. Rf = 0.25 in n-pentane/diethyl ether 9:1.
- 4-hexyl-3-perfluoroheptyl-1H-pyrazol (4c). Following general procedure II, 2-(perfluorooctylidene)octanal (3c) (0.263 g, 0.500 mmol, 1.00 equiv), hydrazine monohydrate (0.090 mL, 1.9 mmol, 3.8 equiv), and ethanol (2 mL) were mixed and stirred at 100 °C for 21 h. Purification by flash chromatography (n-pentane/diethyl ether 7:3) gave the desired product (0.167 g, 0.322 mmol, 64%) as a colorless solid.
- 1H NMR (300 MHz, CDCl3) δ = 12.86 (s, 1H), 7.46 (d, J = 1.6 Hz, 1H), 2.57 (t, J = 7.8 Hz, 2H), 1.67–1.51 (m, 2H), 1.45–1.21 (m, 6H), 0.95–0.83 (m, 3H). 19F NMR (282 MHz, CDCl3) δ = −80.83 (tt, J = 9.9, 2.4 Hz, 3F), −108.74–−109.00 (m, 2F), −121.45–−121.77 (m, 2F), −121.95–−122.29 (m, 4F), −122.56–−122.87 (m, 2F), −126.15 (dddt, J = 19.3, 10.9, 7.3, 4.1 Hz, 2F). 13C NMR (151 MHz, CDCl3) δ = 137.77 (t, J = 27.5 Hz), 129.06, 122.68, 120.68–105.88 (m), 31.53, 30.26, 28.94, 23.14, 22.50, 13.90. IR: [cm−1] = 3178, 3116, 2962, 2936, 2865, 1471, 1366, 1241, 1210, 1150. HRMS-ESI: calcd for C16H15F15N2 [M + H+] m/z 521.1068, found 521.1063. Mp. 35–36 °C. Rf = 0.3 in n-pentane/diethyl ether 4:1.
- 4-hexyl-3-(trifluoromethyl)-1H-pyrazole (4d). Following general procedure II, 2-(perfluoroethylidene)octanal (3d) (0.104 g, 0.460 mmol, 1.00 equiv), hydrazine monohydrate (0.085 mL, 1.8 mmol, 3.9 equiv), and ethanol (1.9 mL) were mixed and stirred at 100 °C for 65 h. Purification by flash chromatography (n-pentane/diethyl ether 8:2) gave the desired product (0.084 g, 0.38 mmol, 83%) as a colorless oil.
- 1H NMR (300 MHz, CDCl3) δ = 13.62 (s, 1H), 7.49 (q, J = 1.0 Hz, 1H), 2.60 (t, J = 7.8 Hz, 2H), 1.67–1.56 (m, 2H), 1.41–1.27 (m, 6H), 0.96–0.86 (m, 3H). 19F NMR (282 MHz, CDCl3) δ = −60.77. 13C NMR (75 MHz, CDCl3) δ = 139.74 (q, J = 36.0 Hz), 129.24, 123.03 (q, J = 267.17), 120.66, 31.70, 30.52, 29.09, 23.17, 22.73, 14.18. IR: [cm−1] = 3177, 2959, 2861, 1490, 1377, 1281, 1158, 1130, 1062, 958. HRMS-ESI: calcd for C10H15F3N2 [M + H+] m/z 221.1260, found 221.1262. Rf = 0.25 in n-hexane/ethyl acetate 8:2.
- 4-benzyl-3-perfluoropropyl-1H-pyrazol (4e). Following general procedure II, 2-(perfluorobutylidene)-3-phenylpropanal (3e) (0.153 g, 0.461 mmol, 1.00 equiv), hydrazine monohydrate (0.085 mL, 1.7 mmol, 3.7 equiv), and ethanol (2 mL) were mixed and stirred at 100 °C for 72 h. Purification by flash chromatography (n-pentane/diethyl ether 8:2) gave the desired product (0.127 g, 0.388 mmol, 84%) as a colorless oil.
- 1H NMR (300 MHz, CDCl3) δ = 12.79 (s, 1H), 7.37–7.13 (m, 6H), 3.92 (s, 2H). 19F NMR (282 MHz, CDCl3) δ = −80.21 (t, J = 9.6 Hz, 3F), −109.80 (q, J = 9.7 Hz, 2F), −126.69 (d, J = 7.7 Hz, 2F). 13C NMR (151 MHz, CDCl3) δ = 139.61, 137.86 (t, J = 27.5 Hz), 130.57, 128.76, 128.70, 126.68, 121.79, 121.36–106.68 (m), 29.65. IR: [cm−1] = 3177, 3063, 2962, 1497, 1348, 1231, 1211, 1184, 1115, 1034. HRMS-ESI: calcd for C13H9F7N2 [M + H+] m/z 327.0727, found 327.0730. Rf = 0.3 in n-pentane/diethyl ether 4:1.
- 3-(perfluorpropyl)-4-(1-phenylethyl)-1H-pyrazol (4f). Following general procedure II, 2-(perfluorobutylidene)-3-methylen-3-phenylpropanal (3f) (0.103 g, 0.310 mmol, 1.00 equiv), hydrazine monohydrate (0.055 mL, 1.1 mmol, 3.5 equiv), and ethanol (1.4 mL) were mixed and stirred at 100 °C for 21 h. Purification by flash chromatography (n-pentane/diethyl ether 7:3) gave the desired product (73.8 mg, 0.217 mmol, 70%) as a colorless oil.
- 1H NMR (300 MHz, CDCl3) δ = 12.84 (s, 1H), 7.50 (d, J = 1.4 Hz, 1H), 7.34–7.16 (m, 5H), 4.26 (q, J = 7.2 Hz, 1H), 1.60 (d, J = 7.2 Hz, 3H). 19F NMR (282 MHz, CDCl3) δ = −80.17 (t, J = 9.7 Hz, 3F), −108.76–−109.12 (m, 2F), −126.47 (d, J = 6.6 Hz, 2F). 13C NMR (151 MHz, CDCl3) δ = 145.56, 137.30 (t, J = 27.6 Hz), 129.15, 128.61, 127.72, 127.04, 126.49, 121.70–106.26 (m), 34.62, 23.76. IR: [cm−1] = 3176, 2967, 1494, 1454, 1349, 1228, 1210, 1186, 1116, 1024. HRMS-ESI: calcd for C14H11F7N2 [M + H+] m/z 341.0883, found 341.0885.
- 4-(2-methoxyethyl)-3-(perfluorpropyl)-1H-pyrazol (4g). Following general procedure II, 2-(perfluorobutylidene)-4-methoxybutanal (3g) (0.164 g, 0.545 mmol, 1.00 equiv), hydrazine monohydrate (0.10 mL, 2.2 mmol, 4.0 equiv), and ethanol (2.2 mL) were mixed and stirred at 100 °C for 21 h. Purification by flash chromatography (n-pentane/diethyl ether 7:3) gave the desired product (0.114 g, 0.388 mmol, 71%) as a colorless oil.
- 1H NMR (300 MHz, CDCl3) δ = 13.04 (s, 1H), 7.63–7.56 (m, 1H), 3.57 (t, J = 6.5 Hz, 2H), 3.37 (s, 3H), 2.91–2.81 (m, 2H). 19F NMR (282 MHz, CDCl3) δ = −80.23 (t, J = 9.7 Hz, 3F), −109.82 (q, J = 9.7 Hz, 2.1F), −126.67–−126.88 (m, 2F). 13C NMR (151 MHz, CDCl3) δ = 137.93 (t, J = 27.2 Hz), 130.13, 118.82, 120.83–106.26 (m), 72.10, 58.50, 23.71. IR: [cm−1] = 3184, 2963, 2361, 2355, 2341, 1468, 1347, 1232, 1117, 1038. HRMS-ESI: calcd for C9H9F7N2 [M + H+] m/z 295.0676, found 295.0679. Rf = 0.16 in n-pentane/diethyl ether 4:1.
- tert-butyl (4-(3-(perfluorpropyl)-1H-pyrazol-4-yl)butyl)carbamate (4h). Following general procedure II, tert-butyl-5-(perfluorobutylidene)-(6-oxohexyl)carbamate (3h) (0.201 g, 0.487 mmol, 1.00 equiv), hydrazine monohydrate (0.090 mL, 1.9 mmol, 3.9 equiv), and ethanol (2.5 mL) were mixed and stirred at 100 °C for 21 h. Purification by flash chromatography (n-pentane/diethyl ether 1:1) gave the desired product (0.158 g, 0.388 mmol, 80%) as a colorless oil.
- 1H NMR (300 MHz, CDCl3) δ = 12.86 (s, 1H), 7.46 (d, J = 1.6 Hz, 1H), 4.57 (s, 1H), 3.15 (q, J = 6.4 Hz, 2H), 2.58 (t, J = 7.3 Hz, 2H), 1.57 (dddd, J = 16.8, 15.0, 8.3, 3.4 Hz, 3H), 1.44 (s, 10H). 19F NMR (282 MHz, CDCl3) δ = −80.21 (t, J = 9.6 Hz, 3F), −109.67 (p, J = 8.5 Hz, 2F), −126.75 (d, J = 7.5 Hz, 2F). 13C NMR (151 MHz, CDCl3) δ = 156.24, 137.91 (t, J = 51.08), 129.21, 122.09, 121.62–106.15 (m), 79.38, 40.42, 29.92, 28.51, 27.83, 23.02. IR: [cm−1] = 3302, 3190, 2943, 1692, 1519, 1457, 1368, 1228, 1180, 1115. HRMS-ESI: calcd for C15H20F7N3NaO2 [M + Na+] m/z 430.1336, found 430.1135. Rf = 0.16 in n-pentane/diethyl ether 1:1.
- 7-(3-(perfluorpropyl)-1H-pyrazol-4-yl)heptan-1-ol (4i). Following general procedure II, 9-hydroxy-2-(perfluorobutylidene)nonanal (3i) (0.179 g, 0.504 mmol, 1.00 equiv), hydrazine monohydrate (0.090 mL, 1.9 mmol, 3.8 equiv), and ethanol (2 mL) were mixed and stirred at 100 °C for 82 h. Purification by flash chromatography (n-pentane/diethyl ether 3:7) gave the desired product (0.125 g, 0.357 mmol, 71%) as a colorless oil.
- 1H NMR (300 MHz, CDCl3) δ = 12.99 (s, 1H, H-1), 7.44 (s, 1H, H-2), 3.65 (t, J = 6.6 Hz, 2H, H-12), 2.55 (t, J = 7.8 Hz, 2H, H-6), 1.66–1.49 (m, 4H, CH2), 1.46–1.27 (m, 6H, CH2). 19F NMR (282 MHz, CDCl3) δ = −80.26 (t, J = 9.6 Hz, 3F), −109.75 (q, J = 9.7 Hz, 2.1F), −126.74–−126.81 (m, 2F). 13C NMR (151 MHz, CDCl3) δ = 137.75 (t, J = 27.5 Hz), 129.15, 122.61, 121.75–106.86 (m), 63.01, 32.75, 30.49, 29.37, 29.25, 25.78, 23.27. IR: [cm−1] = 3183, 2935, 2862, 1467, 1347, 1227, 1209, 1184, 1116, 1037. HRMS-ESI: calcd for C13H17F7N2O [M + H+] m/z 351.1302, found 351.1298. Rf = 0.1 in n-pentane/diethyl ether 3:7.
- 4-hexyl-1-methyl-3(5)-(perfluoropropyl)-1H-pyrazol (4j). General procedure II was employed, but hydrazine hydrate was exchanged by methylhydrazine. 2-(Perfluorobutylidene)octanal (3a) (0.169 g, 0.518 mmol, 1.00 equiv), methylhydrazine (0.098 mL, 1.9 mmol, 3.7 equiv), and ethanol (2 mL) were mixed and stirred at 100 °C for 72 h. Purification by flash chromatography (n-pentane/diethyl ether; 98:2, 95:5, 7:3) gave 4-hexyl-1-methyl-3-(perfluoropropyl)-1H-pyrazol (4j-1) (45 mg, 0.14 mmol, 27%) and 4-hexyl-1-methyl-5-(perfluoropropyl)-1H-pyrazol (4j-2) (30 mg, 0.090 mmol, 18%) as a slightly brown oil.
- 4-Hexyl-1-methyl-3-(perfluoropropyl)-1H-pyrazol (4j-1); 1H NMR (300 MHz, CDCl3) δ = 7.38 (d, J = 1.3 Hz, 1H), 3.94 (t, J = 1.8 Hz 8, 3H), 2.55–2.45 (m, 2H), 1.61–1.49 (m, 2H), 1.35–1.25 (m, 6H), 0.91–0.82 (m, 4H). 19F NMR (282 MHz, CDCl3) δ = −80.16 (t, J = 10.0 Hz, 3F), −107.68–−107.87 (m, 2F), −126.12–−126.28 (m, 2F). 13C NMR (75 MHz, CDCl3) δ = 138.86, 126.74, 125.64–125.18 (m), 39.50, 31.57, 30.49, 29.02, 23.67, 22.56, 22.33, 14.02. IR: [cm−1] = 2959, 2932, 2862, 1469, 1347, 1228, 1182, 1134, 1115, 1023. HRMS-ESI: calcd for C13H18F7N2 [M + H+] m/z 335.1353, found 335.1358. Rf = 0.8 in n-pentane/diethyl ether 9:1.
- 4-Hexyl-1-methyl-5-(perfluoropropyl)-1H-pyrazol (4j-2); 1H NMR (300 MHz, CDCl3) δ = 7.22 (s, 1H), 3.91 (s, 3H), 2.50 (t, J = 7.8 Hz, 2H), 1.57–1.46 (m, 2H), 1.37–1.22 (m, 6H), 0.92–0.85 (m, 3H). 19F NMR (282 MHz, CDCl3) δ = −80.21 (t, J = 9.7 Hz, 3F), −109.29 (q, J = 9.6 Hz, 2F), −126.56–−126.63 (m, 2F). 13C NMR (75 MHz, CDCl3) δ = 137.59 (t, J = 28.0 Hz), 130.38, 123.42, 120.72–104.88 (m), 39.48 (d, J = 3.1 Hz), 34.12, 31.58, 30.47, 29.00, 23.29, 22.57, 14.01. IR: [cm−1] = 2960, 2932, 2862, 1462, 1390, 1346, 1229, 1203, 1138, 1115. HRMS-ESI: calcd for C13H18F7N2 [M + H+] m/z 335.1353, found 335.1348. Rf = 0.4 in n-pentane/diethyl ether 9:1.
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fustero, S.; Sánchez-Roselló, M.; Barrio, P.; Simón-Fuentes, A. From 2000 to mid-2010: A fruitful decade for the synthesis of pyrazoles. Chem. Rev. 2011, 111, 6984–7034. [Google Scholar] [CrossRef] [PubMed]
- Ansari, A.; Ali, A.; Asif, M.; Shamsuzzaman, S. Review: Biologically active pyrazole derivatives. New J. Chem. 2017, 41, 16–41. [Google Scholar] [CrossRef]
- Steinbach, G.; Lynch, P.M.; Phillips, R.K.; Wallace, M.H.; Hawk, E.; Gordon, G.B.; Wakabayashi, N.; Saunders, B.; Shen, Y.; Fujimura, T.; et al. The effect of celecoxib, a cyclooxygenase-2 inhibitor, in familial adenomatous polyposis. N. Engl. J. Med. 2000, 342, 26. [Google Scholar] [CrossRef] [PubMed]
- Uslaner, J.M.; Parmentier-Batteur, S.; Flick, R.B.; Surles, N.O.; Lam, J.S.H.; McNaughton, C.H.; Jacobson, M.A.; Hutson, P.H. Dose-dependent effect of CDPPB, the mGluR5 positive allosteric modulator, on recognition memory is associated with GluR1 and CREB phosphorylation in the prefrontal cortex and hippocampus. Neuropharmacology 2009, 57, 531–538. [Google Scholar] [CrossRef] [PubMed]
- Hampp, C.; Hartzema, A.G.; Kauf, T.L. Cost-utility analysis of rimonabant in the treatment of obesity. Value Health 2008, 11, 389–399. [Google Scholar] [CrossRef]
- Vinick, F.J.; Heym, J.H. Chapter 1 Antipsychotic Agents. In Annual Reports in Medicinal Chemistry; Bailey, D.M., Ed.; Academic Press: Orlando, FL, USA, 1987; Volume 22, pp. 1–10. [Google Scholar]
- Kameyama, T.; Nabeshima, T. Effects of 1,3-diphenyl-5-(2-dimethylaminopropionamide)-pyrazoledifenamizole on a conditioned avoidance response. Neuropharmacology 1978, 17, 249–256. [Google Scholar] [CrossRef]
- Luttinger, D.; Hlasta, D.J. Chapter 3 Antidepressant Agents. In Annual Reports in Medicinal Chemistry; Bailey, D.M., Ed.; Academic Press: Orlando, FL, USA, 1987; Volume 22, pp. 21–30. [Google Scholar]
- O’Hagan, D. Understanding organofluorine chemistry. An introduction to the C–F bond. Chem. Soc. Rev. 2008, 37, 308–319. [Google Scholar] [CrossRef]
- Müller, K.; Faeh, C.; Diederich, F. Fluorine in pharmaceuticals: Looking beyond intuition. Science 2007, 317, 1881–1886. [Google Scholar] [CrossRef]
- Hagmann, W.K. The many roles for fluorine in medicinal chemistry. J. Med. Chem. 2008, 51, 4359–4369. [Google Scholar] [CrossRef]
- Purser, S.; Moore, P.R.; Swallow, S.; Gouverneur, V. Fluorine in medicinal chemistry. Chem. Soc. Rev. 2008, 37, 320–330. [Google Scholar] [CrossRef]
- Gillis, E.P.; Eastman, K.J.; Hill, M.D.; Donnelly, D.J.; Meanwell, N.A. Applications of Fluorine in Medicinal Chemistry. J. Med. Chem. 2015, 58, 8315–8359. [Google Scholar] [CrossRef] [PubMed]
- Mykhailiuk, P.K. Fluorinated Pyrazoles: From Synthesis to Applications. Chem. Rev. 2021, 121, 1670–1715. [Google Scholar] [CrossRef]
- Lamberth, C.; Dinges, J. (Eds.) Bioactive Heterocyclic Compound Classes: Agrochemicals; John Wiley distributor: Chichester, UK; Wiley-VCH: Weinheim, Germany, 2012; ISBN 3527664440. [Google Scholar]
- Kane, S.P. Celecoxib—Drug Usage Statistics, ClinCalc DrugStats Database. Available online: https://clincalc.com/DrugStats/Drugs/Celecoxib (accessed on 7 June 2024).
- Betcke, I.; Götzinger, A.C.; Kornet, M.M.; Müller, T.J.J. Multicomponent syntheses of pyrazoles via (3 + 2)-cyclocondensation and (3 + 2)-cycloaddition key steps. Beilstein J. Org. Chem. 2024, 20, 2024–2077. [Google Scholar] [CrossRef] [PubMed]
- Kino, T.; Nagase, Y.; Ohtsuka, Y.; Yamamoto, K.; Uraguchi, D.; Tokuhisa, K.; Yamakawa, T. Trifluoromethylation of various aromatic compounds by CF3I in the presence of Fe(II) compound, H2O2 and dimethylsulfoxide. J. Fluor. Chem. 2010, 131, 98–105. [Google Scholar] [CrossRef]
- Sloop, J.C.; Jackson, J.L.; Schmidt, R.D. Microwave-mediated pyrazole fluorinations using selectfluor®. Heteroat. Chem. 2009, 20, 341–345. [Google Scholar] [CrossRef]
- Fustero, S.; Román, R.; Sanz-Cervera, J.F.; Simón-Fuentes, A.; Cuñat, A.C.; Villanova, S.; Murguía, M. Improved regioselectivity in pyrazole formation through the use of fluorinated alcohols as solvents: Synthesis and biological activity of fluorinated tebufenpyrad analogs. J. Org. Chem. 2008, 73, 3523–3529. [Google Scholar] [CrossRef]
- Mykhailiuk, P.K. In Situ generation of difluoromethyl diazomethane for 3 + 2 cycloadditions with alkynes. Angew. Chem. Int. Ed. Engl. 2015, 54, 6558–6561. [Google Scholar] [CrossRef]
- Mertens, L.; Hock, K.J.; Koenigs, R.M. Fluoroalkyl-Substituted Diazomethanes and Their Application in a General Synthesis of Pyrazoles and Pyrazolines. Chem. Eur. J. 2016, 22, 9542–9545. [Google Scholar] [CrossRef]
- Chen, Z.; Zheng, Y.; Ma, J.-A. Use of a Traceless Activating and Directing Group for the Construction of Trifluoromethylpyrazoles: One-Pot Transformation of Nitroolefins and Trifluorodiazoethane. Angew. Chem. 2017, 129, 4640–4645. [Google Scholar] [CrossRef]
- Ichikawa, J.; Kobayashi, M.; Noda, Y.; Yokota, N.; Amano, K.; Minami, T. Regiocontrolled Syntheses of 3- or 5-Fluorinated Pyrazoles from 2,2-Difluorovinyl Ketones. J. Org. Chem. 1996, 61, 2763–2769. [Google Scholar] [CrossRef]
- Ye, F.; Zhang, S.; Wei, Z.; Weniger, F.; Spannenberg, A.; Taeschler, C.; Ellinger, S.; Jiao, H.; Neumann, H.; Beller, M. Versatile Fluorinated Building Blocks by Stereoselective (Per)fluoroalkenylation of Ketones. Eur. J. Org. Chem. 2020, 2020, 70–81. [Google Scholar] [CrossRef]
- Wulkesch, C.; Czekelius, C. Straightforward Synthesis of Fluorinated Enals via Photocatalytic α-Perfluoroalkenylation of Aldehydes. J. Org. Chem. 2021, 86, 7425–7438. [Google Scholar] [CrossRef] [PubMed]
- Bracker, M.; Helmecke, L.; Kleinschmidt, M.; Czekelius, C.; Marian, C.M. Visible Light-Induced Homolytic Cleavage of Perfluoroalkyl Iodides Mediated by Phosphines. Molecules 2020, 25, 1606. [Google Scholar] [CrossRef] [PubMed]
- Jarończyk, M.; Dobrowolski, J.C.; Mazurek, A.P. Theoretical studies on tautomerism and IR spectra of pyrazole derivatives. J. Mol. Struct. 2004, 673, 17–28. [Google Scholar] [CrossRef]
- Bouillon, J.-P.; Didier, B.; Dondy, B.; Doussot, P.; Plantier-Royon, R.; Portella, C. Efficient Synthesis of 4-Fluoro-5-(perfluoroalkyl)pyrazoles from Organofluorosilicon Building Blocks. Eur. J. Org. Chem. 2001, 2001, 187–192. [Google Scholar] [CrossRef]
- Helmecke, L.; Spittler, M.; Baumgarten, K.; Czekelius, C. Metal-Free Activation of C-I Bonds and Perfluoroalkylation of Alkenes with Visible Light Using Phosphine Catalysts. Org. Lett. 2019, 21, 7823–7827. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
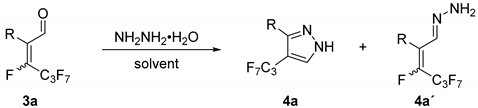 | |||||
| Entry | Solvent | Hydrazine [Equiv] | Temperature [°C] | Time [h] | Conversion of 4a′ to 4a [%] |
|---|---|---|---|---|---|
| 1 | ethanol | 1.3 | 80 | 21 | 31 |
| 2 | ethanol | 2.1 | 100 | 19 | 65 |
| 3 | ethanol | 4.9 | 100 | 21 | 77 |
| 4 | ethanol | 2.1 | 100 | 264 | 96 |
| 5 | DMF | 2.9 | 120 | 21 | n.a. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bunnemann, L.; Wulkesch, C.; Voigt, V.C.; Czekelius, C. Synthesis of Perfluoroalkylated Pyrazoles from α-Perfluoroalkenylated Aldehydes. Molecules 2024, 29, 5034. https://doi.org/10.3390/molecules29215034
Bunnemann L, Wulkesch C, Voigt VC, Czekelius C. Synthesis of Perfluoroalkylated Pyrazoles from α-Perfluoroalkenylated Aldehydes. Molecules. 2024; 29(21):5034. https://doi.org/10.3390/molecules29215034
Chicago/Turabian StyleBunnemann, Lennart, Christian Wulkesch, Victoria Carina Voigt, and Constantin Czekelius. 2024. "Synthesis of Perfluoroalkylated Pyrazoles from α-Perfluoroalkenylated Aldehydes" Molecules 29, no. 21: 5034. https://doi.org/10.3390/molecules29215034
APA StyleBunnemann, L., Wulkesch, C., Voigt, V. C., & Czekelius, C. (2024). Synthesis of Perfluoroalkylated Pyrazoles from α-Perfluoroalkenylated Aldehydes. Molecules, 29(21), 5034. https://doi.org/10.3390/molecules29215034