
Computational Multiscale Study of the Interaction Between the PDMS Polymer and Sunscreen-Related Pollutant Molecules



Abstract
1. Introduction
2. Results and Discussion
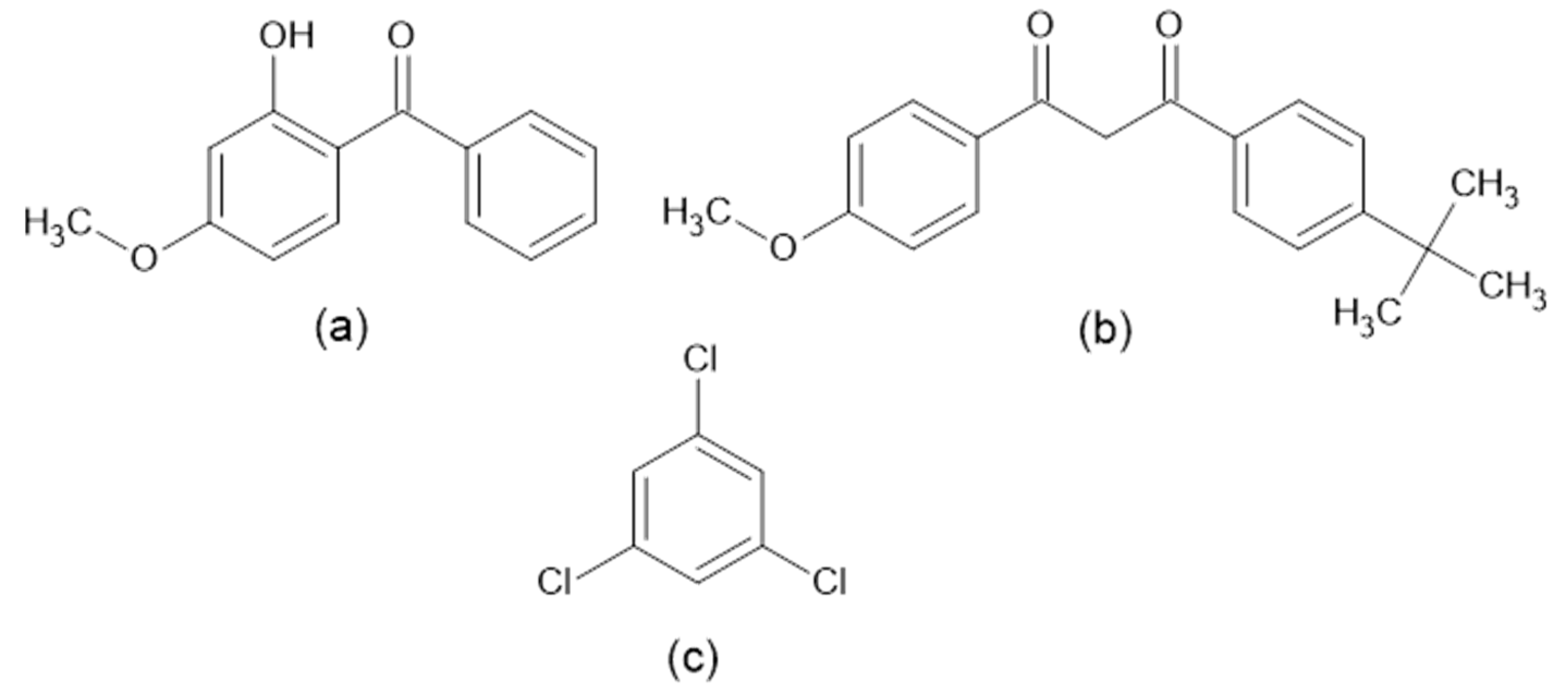
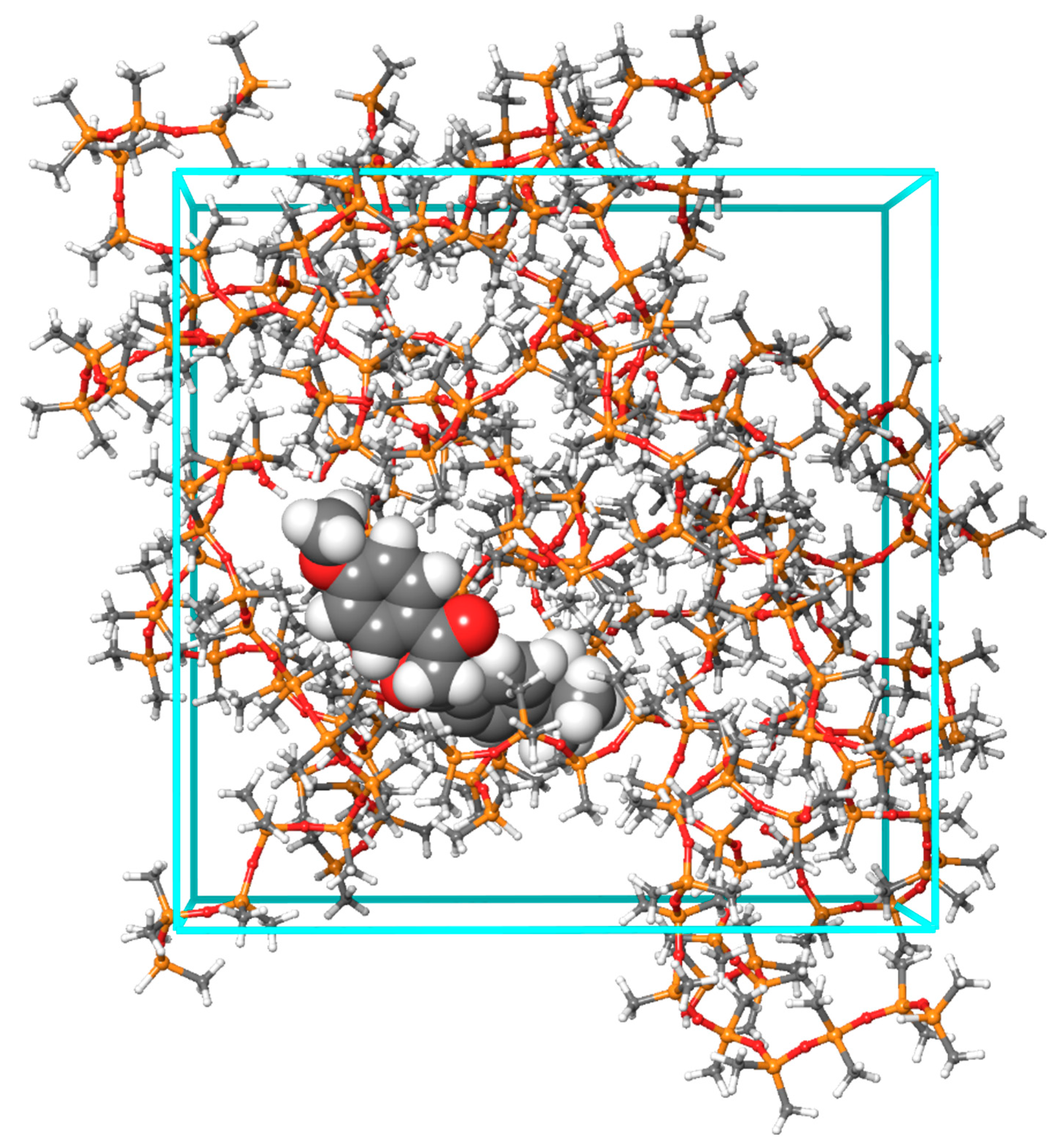
2.1. Molecular Structures and Computational Workflow
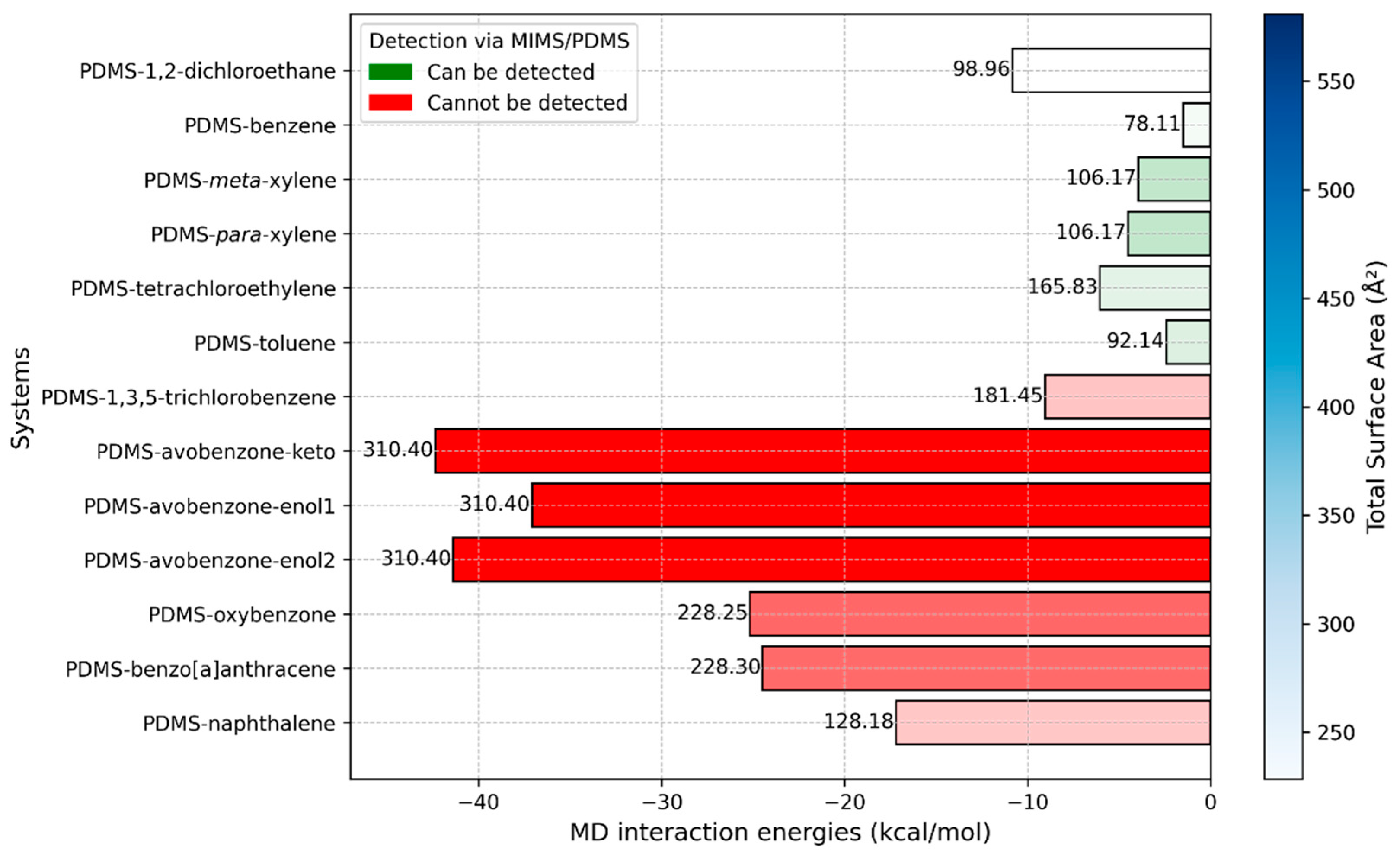
2.2. MD Simulations—Interaction Energies
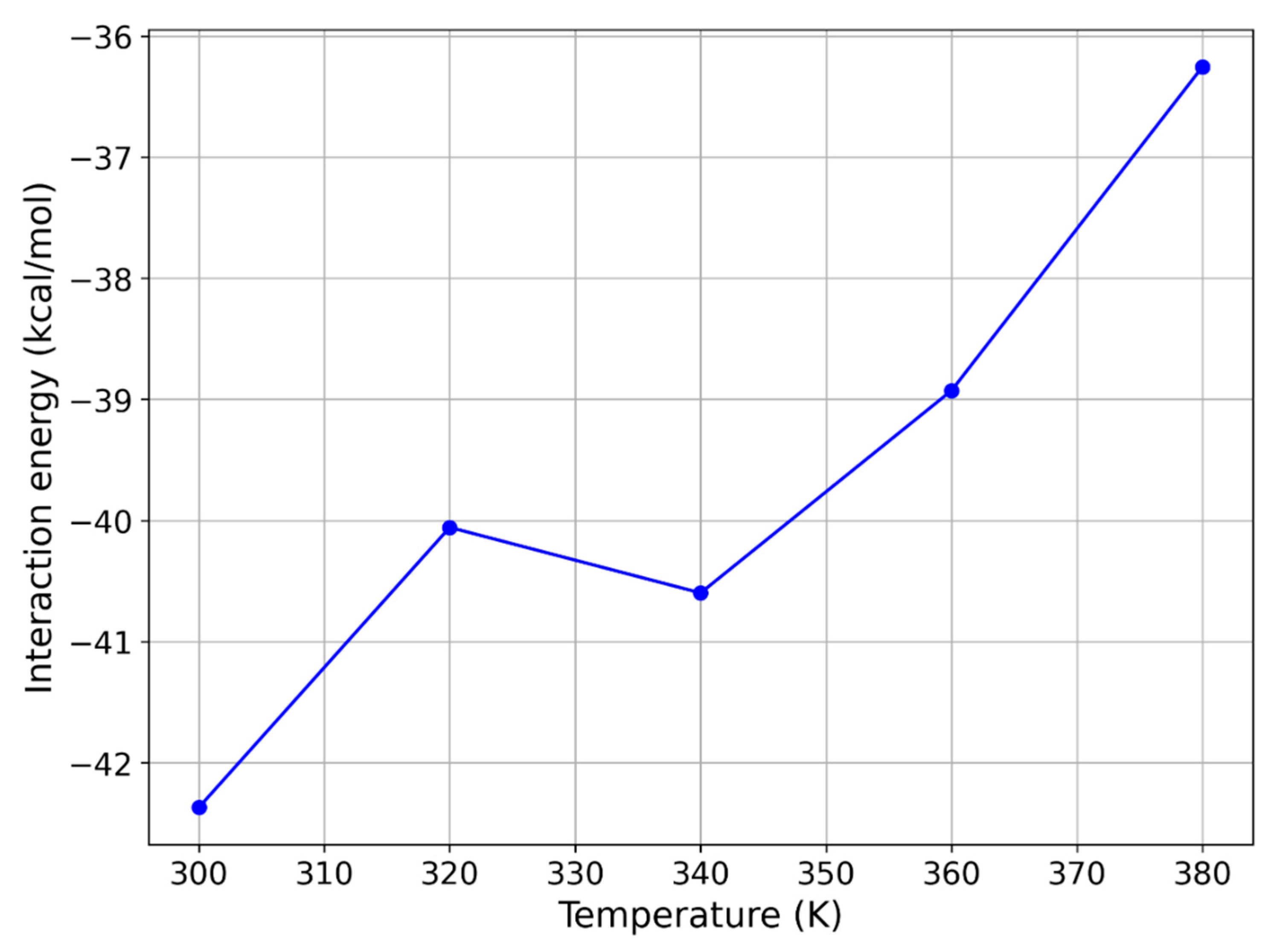
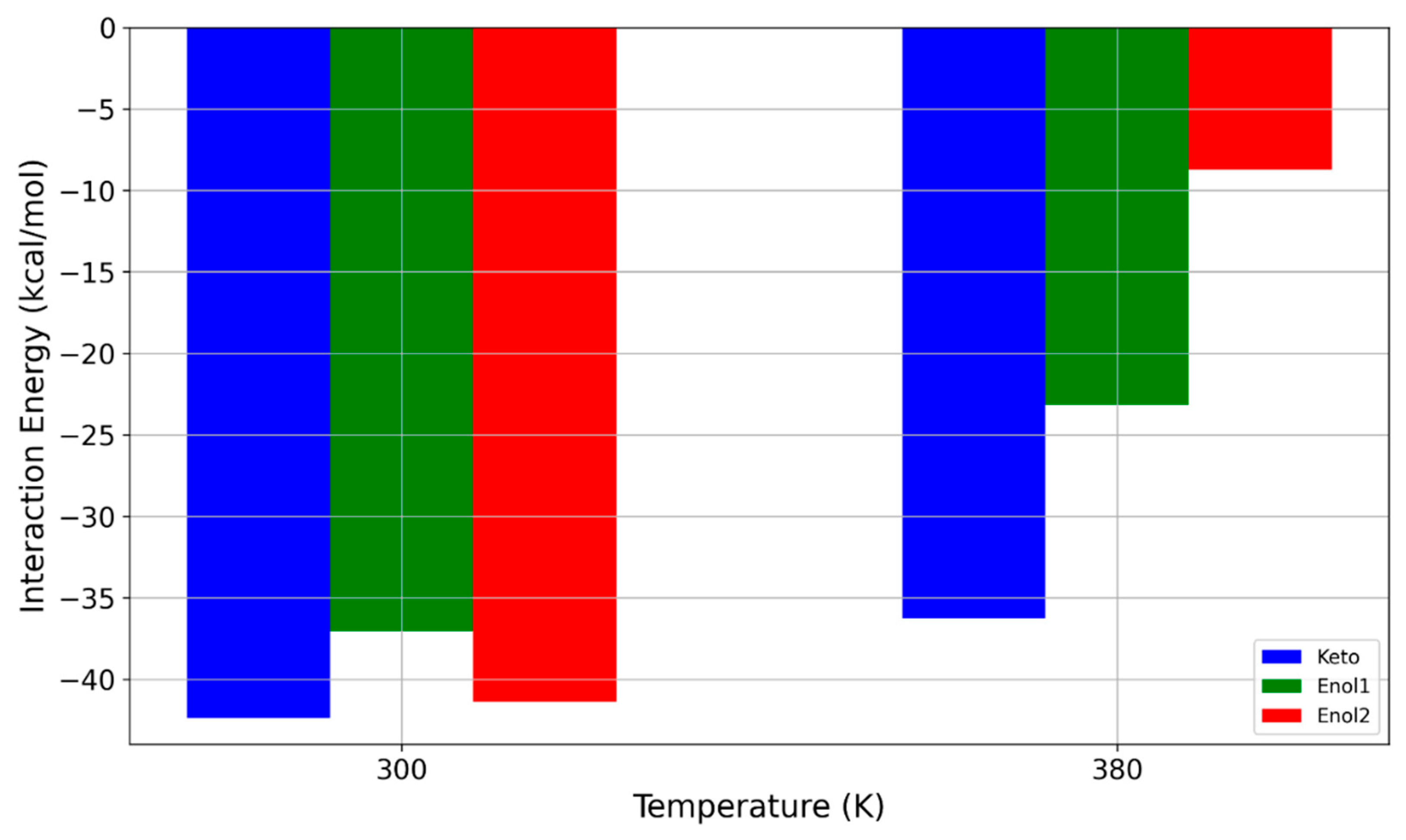
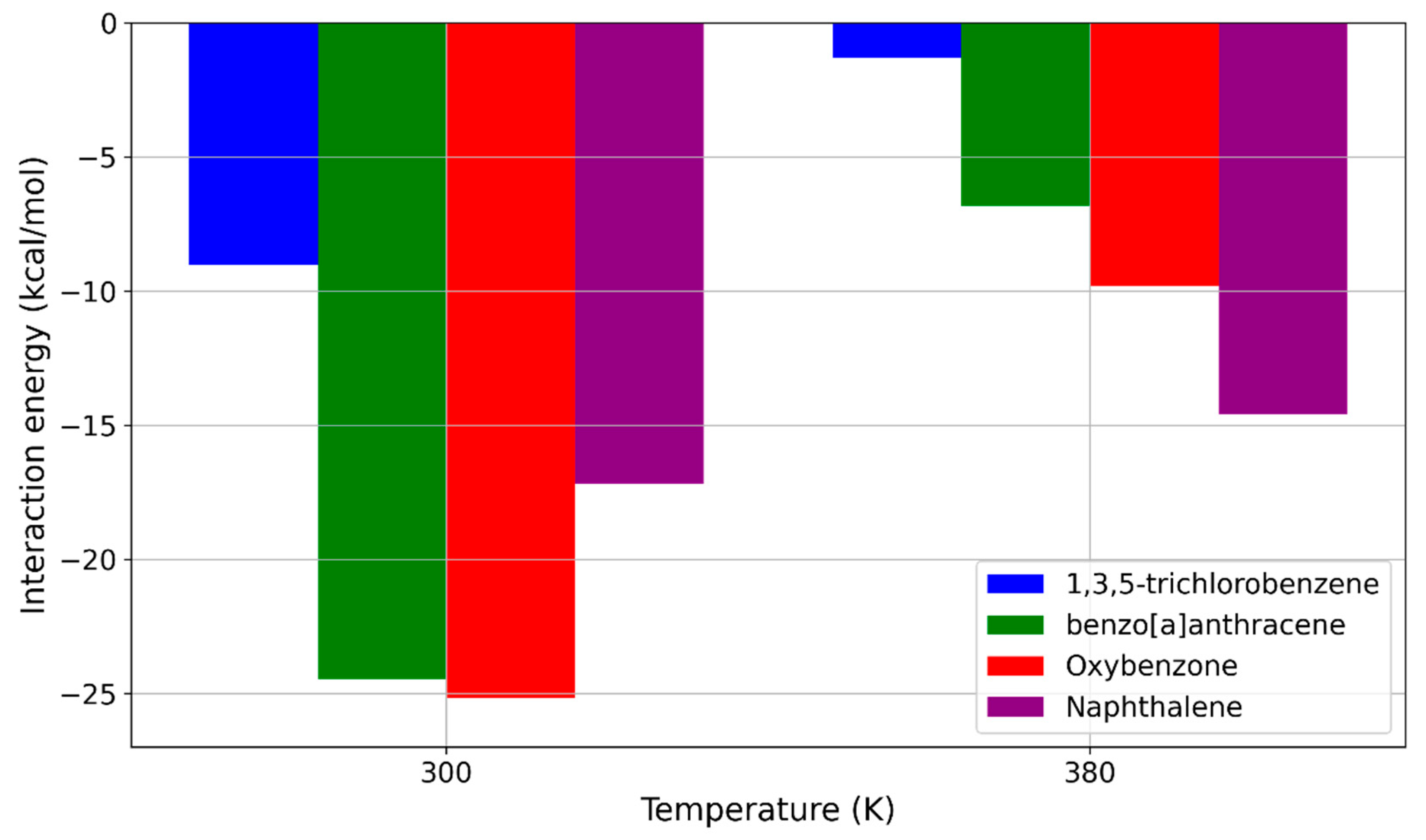
2.3. MD Simulations—Temperature Effects on the Interaction Strength
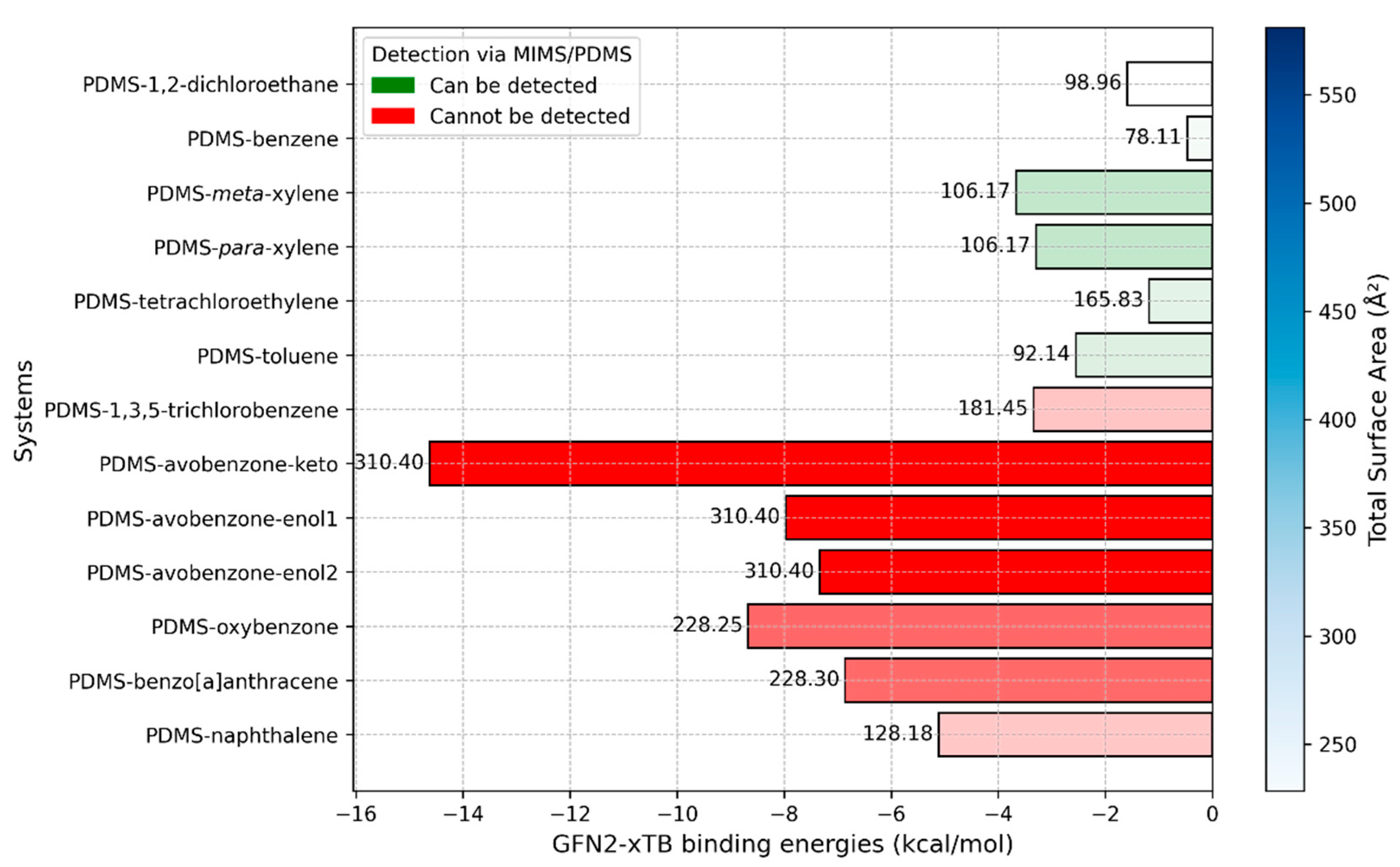
2.4. GFN2-xTB Ground State Geometries and Binding Energies
2.5. Noncovalent Interactions Between PDMS and Avobenzone Forms
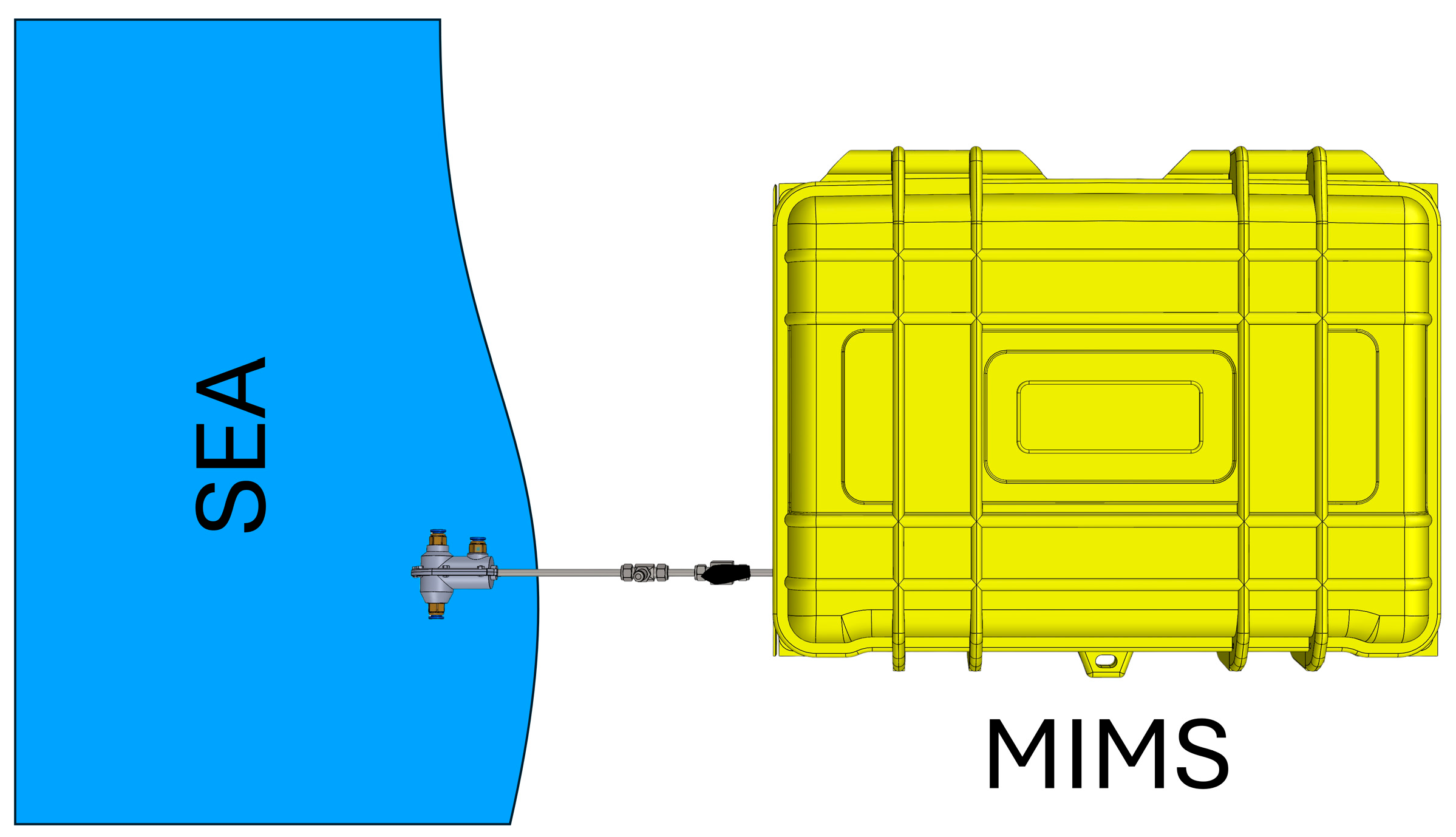
3. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mallah, M.A.; Changxing, L.; Mallah, M.A.; Noreen, S.; Liu, Y.; Saeed, M.; Xi, H.; Ahmed, B.; Feng, F.; Mirjat, A.A.; et al. Polycyclic Aromatic Hydrocarbon and Its Effects on Human Health: An Overeview. Chemosphere 2022, 296, 133948. [Google Scholar] [CrossRef]
- Zhao, X.; Gao, J.; Zhai, L.; Yu, X.; Xiao, Y. Recent Evidence on Polycyclic Aromatic Hydrocarbon Exposure. Healthcare 2023, 11, 1958. [Google Scholar] [CrossRef]
- World Health Organization (WHO). Human Health Effects of Polycyclic Aromatic Hydrocarbons as Ambient Air Pollutants—Report of the Working Group on Polycyclic Aromatic Hydrocarbons of the Joint Task Force on the Health Aspects of Air Pollution. Available online: https://www.who.int/europe/publications/i/item/9789289056533 (accessed on 28 August 2024).
- Trebše, P.; Polyakova, O.V.; Baranova, M.; Kralj, M.B.; Dolenc, D.; Sarakha, M.; Kutin, A.; Lebedev, A.T. Transformation of Avobenzone in Conditions of Aquatic Chlorination and UV-Irradiation. Water Res. 2016, 101, 95–102. [Google Scholar] [CrossRef]
- Autier, P.; Doré, J.-F.; Schifflers, E.; Cesarini, J.-P.; Bollaerts, A.; Koelmel, K.F.; Gefeller, O.; Liabeuf, A.; Lejeune, F.; Lienard, D.; et al. Melanoma and Use of Sunscreens: An EORTC Case-Control Study in Germany, Belgium and France. Int. J. Cancer 1995, 61, 749–755. [Google Scholar] [CrossRef]
- Hanson, K.M.; Gratton, E.; Bardeen, C.J. Sunscreen Enhancement of UV-Induced Reactive Oxygen Species in the Skin. Free Radic. Biol. Med. 2006, 41, 1205–1212. [Google Scholar] [CrossRef]
- Garland, C.F.; Garland, F.C.; Gorham, E.D. Rising Trends in Melanoma an Hypothesis Concerning Sunscreen Effectiveness. Ann. Epidemiol. 1993, 3, 103–110. [Google Scholar] [CrossRef]
- Sahoo, D.K.; Pooja; Jena, S.; Mohanty, P.; Biswal, H.S.; Gowd, K.H. Probing the Photostability of Avobenzone with N-Acetylcysteine Using UV Spectroscopy, Computational Studies and Integration into Aloe Vera Gel. J. Photochem. Photobiol. A Chem. 2024, 447, 115196. [Google Scholar] [CrossRef]
- Trichlorobenzenes|ToxFAQsTM|ATSDR. Available online: https://wwwn.cdc.gov/TSP/ToxFAQs/ToxFAQsDetails.aspx?faqid=1169&toxid=255 (accessed on 29 August 2024).
- HEALTH EFFECTS. Toxicological Profile for Trichlorobenzenes; Agency for Toxic Substances and Disease Registry (US): Atlanta, GA, USA, 2014. Available online: https://www.atsdr.cdc.gov/toxprofiles/tp199-p.pdf (accessed on 29 August 2024).
- Implementing Decision—2022/1307-EN—EUR-Lex. Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX%3A32022D1307&qid=1658824912292 (accessed on 29 August 2024).
- Directive 2013/39/EU of the European Parliament and of the Council of 12 August 2013 Amending Directives 2000/60/EC and 2008/105/EC as Regards Priority Substances in the Field of Water policyText with EEA Relevance. Available online: https://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2013:226:0001:0017:en:PDF (accessed on 29 August 2024).
- Jeffery, J.; Carradus, M.; Songin, K.; Pettit, M.; Pettit, K.; Wright, C. Optimized Method for Determination of 16 FDA Polycyclic Aromatic Hydrocarbons (PAHs) in Mainstream Cigarette Smoke by Gas Chromatography–Mass Spectrometry. Chem. Cent. J. 2018, 12, 27. [Google Scholar] [CrossRef]
- Vistnes, H.; Sossalla, N.A.; Røsvik, A.; Gonzalez, S.V.; Zhang, J.; Meyn, T.; Asimakopoulos, A.G. The Determination of Polycyclic Aromatic Hydrocarbons (PAHs) with HPLC-DAD-FLD and GC-MS Techniques in the Dissolved and Particulate Phase of Road-Tunnel Wash Water: A Case Study for Cross-Array Comparisons and Applications. Toxics 2022, 10, 399. [Google Scholar] [CrossRef]
- Peruchi, L.M.; Rath, S. Development and Application of a HPLC Method for Eight Sunscreen Agents in Suncare Products. Int. J. Cosmet. Sci. 2012, 34, 226–233. [Google Scholar] [CrossRef]
- Chang, N.I.; Yoo, M.Y.; Lee, S.H. Determination of Fourteen Sunscreen Agents in Cosmetics Using High-Performance Liquid Chromatography. Int. J. Cosmet. Sci. 2015, 37, 175–180. [Google Scholar] [CrossRef]
- Hoch, G.; Kok, B. A Mass Spectrometer Inlet System for Sampling Gases Dissolved in Liquid Phases. Arch. Biochem. Biophys. 1963, 101, 160–170. [Google Scholar] [CrossRef]
- Ketola, R.A.; Lauritsen, F.R. Membrane Inlet Mass Spectrometry (MIMS) in Historical Perspective. In The Encyclopedia of Mass Spectrometry; Elsevier: Amsterdam, The Netherlands, 2016; pp. 143–148. ISBN 978-0-08-043848-1. [Google Scholar]
- Aleksić Née Jakšić, M.; Simeon, A.; Vujić, D.; Giannoukos, S.; Brkić, B. Food and Lifestyle Impact on Breath VOCs Using Portable Mass Spectrometer—Pilot Study across European Countries. J. Breath Res. 2023, 17, 143–148. [Google Scholar] [CrossRef]
- Armaković, S.; Vujić, D.; Brkić, B. A Computational Study of Polydimethylsiloxane Derivatives as a Semi-Permeable Membrane for in-Field Identification of Naphthenic Acids in Water Using Portable Mass Spectrometry. J. Mol. Liq. 2022, 351, 118657. [Google Scholar] [CrossRef]
- Giannoukos, S.; Brkić, B.; Taylor, S.; France, N. Membrane Inlet Mass Spectrometry for Homeland Security and Forensic Applications. J. Am. Soc. Mass Spectrom. 2015, 26, 231–239. [Google Scholar] [CrossRef]
- Aleksić Née Jakšić, M.; Simeon, A.; Vujić, D.; Giannoukos, S.; Brkić, B. Membrane Inlet Mass Spectrometry Method for Food Intake Impact Assessment on Specific Volatile Organic Compounds in Exhaled Breath. Anal. Bioanal. Chem. 2022, 414, 6077–6091. [Google Scholar] [CrossRef]
- Aneesh Kumar, R.; Jamelah Al-Otaibi, S.; Sheena Mary, Y.; Shyma Mary, Y.; Acharjee, N.; Thomas, R.; Raveendran Pillai, R.; Leena, T.L. Surface Adsorption of Adenine on Pristine and B/N/O/P-Doped Coronene as Biosensing Substrate for DNA Detection- DFT Study. J. Mol. Liq. 2023, 393, 123546. [Google Scholar] [CrossRef]
- Al-Otaibi, J.S.; Mary, Y.S.; Mary, Y.S.; Thomas, R. Evidences of Noncovalent Interactions between Indole and Dichloromethane under Different Solvent Conditions. J. Mol. Model. 2023, 29, 246. [Google Scholar] [CrossRef]
- Pooventhiran, T.; Thomas, R.; Bhattacharyya, U.; Sowrirajan, S.; Irfan, A.; Rao, D.J. Structural Aspects, Reactivity Analysis, Wavefunction Based Properties, Cluster Formation with Helicene and Subsequent Detection from Surface Enhancement in Raman Spectra of Triclabendazole Studies Using First Principle Simulations. Vietnam J. Chem. 2021, 59, 887–901. [Google Scholar] [CrossRef]
- Apaolaza, A.; Richard, D.; Tejerina, M.R. Experimental and Ab Initio Study of the Structural and Optical Properties of ZnO Coatings: Performance of the DFT+ U Approach. Process. Appl. Ceram. 2020, 14, 362–371. [Google Scholar] [CrossRef]
- Afroz Bakht, M.; Alharthi, A.I.; Thangaiyan, P.; Ahmad, A.; Ali, I.; Thomas, R. Interaction of Serotonin and Histamine with Water and Ethanol: Evidence from Theoretical Investigations. Comput. Theor. Chem. 2023, 1228, 114299. [Google Scholar] [CrossRef]
- Pooventhiran, T.; Bhattacharyya, U.; Rao, D.J.; Chandramohan, V.; Karunakar, P.; Irfan, A.; Mary, Y.S.; Thomas, R. Detailed Spectra, Electronic Properties, Qualitative Non-Covalent Interaction Analysis, Solvatochromism, Docking and Molecular Dynamics Simulations in Different Solvent Atmosphere of Cenobamate. Struct. Chem. 2020, 31, 2475–2485. [Google Scholar] [CrossRef]
- Tonel, M.Z.; González-Durruthy, M.; Zanella, I.; Fagan, S.B. Interactions of Graphene Derivatives with Glutamate-Neurotransmitter: A Parallel First Principles—Docking Investigation. J. Mol. Graph. Model. 2019, 88, 121–127. [Google Scholar] [CrossRef]
- de Oliveira, P.V.; Zanella, I.; Bulhões, L.O.S.; Fagan, S.B. Adsorption of 17 β-Estradiol in Graphene Oxide through the Competing Methanol Co-Solvent: Experimental and Computational Analysis. J. Mol. Liq. 2021, 321, 114738. [Google Scholar] [CrossRef]
- da Silva Bruckmann, F.; Zuchetto, T.; Ledur, C.M.; dos Santos, C.L.; da Silva, W.L.; Binotto Fagan, S.; Zanella da Silva, I.; Bohn Rhoden, C.R. Methylphenidate Adsorption onto Graphene Derivatives: Theory and Experiment. New J. Chem. 2022, 46, 4283–4291. [Google Scholar] [CrossRef]
- Martínez, J.M.; Martínez, L. Packing Optimization for Automated Generation of Complex System’s Initial Configurations for Molecular Dynamics and Docking. J. Comput. Chem. 2003, 24, 819–825. [Google Scholar] [CrossRef]
- Martínez, L.; Andrade, R.; Birgin, E.G.; Martínez, J.M. PACKMOL: A Package for Building Initial Configurations for Molecular Dynamics Simulations. J. Comput. Chem. 2009, 30, 2157–2164. [Google Scholar] [CrossRef]
- Armaković, S.; Armaković, S.J. Atomistica.Online—Web Application for Generating Input Files for ORCA Molecular Modelling Package Made with the Anvil Platform. Mol. Simul. 2023, 49, 117–123. [Google Scholar] [CrossRef]
- Armaković, S.; Armaković, S.J. Online and Desktop Graphical User Interfaces for Xtb Programme from Atomistica.Online Platform. Mol. Simul. 2024, 50, 560–570. [Google Scholar] [CrossRef]
- Armaković, S.; Ilić, D.; Brkić, B. Design of Novel Membranes for the Efficient Separation of Bee Alarm Pheromones in Portable Membrane Inlet Mass Spectrometric Systems. Int. J. Mol. Sci. 2024, 25, 8599. [Google Scholar] [CrossRef]
- Otero-de-la-Roza, A.; Johnson, E.R.; Contreras-Garcia, J. Revealing Non-Covalent Interactions in Solids: NCI Plots Revisited. Phys. Chem. Chem. Phys. 2012, 14, 12165–12172. [Google Scholar] [CrossRef]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef]
- Contreras-García, J.; Johnson, E.R.; Keinan, S.; Chaudret, R.; Piquemal, J.-P.; Beratan, D.N.; Yang, W. NCIPLOT: A Program for Plotting Noncovalent Interaction Regions. J. Chem. Theory Comput. 2011, 7, 625–632. [Google Scholar] [CrossRef]
- Ehlert, S.; Stahn, M.; Spicher, S.; Grimme, S. Robust and Efficient Implicit Solvation Model for Fast Semiempirical Methods. J. Chem. Theory Comput. 2021, 17, 4250–4261. [Google Scholar] [CrossRef]
- Bannwarth, C.; Caldeweyher, E.; Ehlert, S.; Hansen, A.; Pracht, P.; Seibert, J.; Spicher, S.; Grimme, S. Extended Tight-Binding Quantum Chemistry Methods. WIREs Comput. Mol. Sci. 2021, 11, e1493. [Google Scholar] [CrossRef]
- Bannwarth, C.; Ehlert, S.; Grimme, S. GFN2-xTB—An Accurate and Broadly Parametrized Self-Consistent Tight-Binding Quantum Chemical Method with Multipole Electrostatics and Density-Dependent Dispersion Contributions. J. Chem. Theory Comput. 2019, 15, 1652–1671. [Google Scholar] [CrossRef]
- Pracht, P.; Caldeweyher, E.; Ehlert, S.; Grimme, S. A Robust Non-Self-Consistent Tight-Binding Quantum Chemistry Method for Large Molecules 2019. ChemRxiv 2019. [Google Scholar] [CrossRef]
- Grimme, S.; Bannwarth, C.; Shushkov, P. A Robust and Accurate Tight-Binding Quantum Chemical Method for Structures, Vibrational Frequencies, and Noncovalent Interactions of Large Molecular Systems Parametrized for All Spd-Block Elements (Z = 1–86). J. Chem. Theory Comput. 2017, 13, 1989–2009. [Google Scholar] [CrossRef]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785. [Google Scholar] [CrossRef]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate Spin-Dependent Electron Liquid Correlation Energies for Local Spin Density Calculations: A Critical Analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. Density Functionals with Broad Applicability in Chemistry. Acc. Chem. Res. 2008, 41, 157–167. [Google Scholar] [CrossRef]
- Valero, R.; Costa, R.; de P. R. Moreira, I.; Truhlar, D.G.; Illas, F. Performance of the M06 Family of Exchange-Correlation Functionals for Predicting Magnetic Coupling in Organic and Inorganic Molecules. J. Chem. Phys. 2008, 128, 114103. [Google Scholar] [CrossRef]
- Jacquemin, D.; Perpète, E.A.; Ciofini, I.; Adamo, C.; Valero, R.; Zhao, Y.; Truhlar, D.G. On the Performances of the M06 Family of Density Functionals for Electronic Excitation Energies. J. Chem. Theory Comput. 2010, 6, 2071–2085. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 Other Functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Cao, Y.; Halls, M.D.; Vadicherla, T.R.; Friesner, R.A. Pseudospectral Implementations of Long-Range Corrected Density Functional Theory. J. Comput. Chem. 2021, 42, 2089–2102. [Google Scholar] [CrossRef]
- Jacobson, L.D.; Bochevarov, A.D.; Watson, M.A.; Hughes, T.F.; Rinaldo, D.; Ehrlich, S.; Steinbrecher, T.B.; Vaitheeswaran, S.; Philipp, D.M.; Halls, M.D. Automated Transition State Search and Its Application to Diverse Types of Organic Reactions. J. Chem. Theory Comput. 2017, 13, 5780–5797. [Google Scholar] [CrossRef]
- Cao, Y.; Hughes, T.; Giesen, D.; Halls, M.D.; Goldberg, A.; Vadicherla, T.R.; Sastry, M.; Patel, B.; Sherman, W.; Weisman, A.L.; et al. Highly Efficient Implementation of Pseudospectral Time-Dependent Density-Functional Theory for the Calculation of Excitation Energies of Large Molecules. J. Comput. Chem. 2016, 37, 1425–1441. [Google Scholar] [CrossRef]
- Bochevarov, A.D.; Harder, E.; Hughes, T.F.; Greenwood, J.R.; Braden, D.A.; Philipp, D.M.; Rinaldo, D.; Halls, M.D.; Zhang, J.; Friesner, R.A. Jaguar: A High-Performance Quantum Chemistry Software Program with Strengths in Life and Materials Sciences. Int. J. Quantum Chem. 2013, 113, 2110–2142. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Detectable by Current MIMS | Undetectable by Current MIMS |
|---|---|
| 1,2-dichloroethane | 1,3,5-trichlorobenzene |
| Benzene | Avobenzone and his keto and enol forms |
| Xylene (meta and para) | Benzo[a]anthracene |
| Toluene | Naphthalene |
| Tetrachloroethylene | Oxybenzone |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Armaković, S.; Vujić, Đ.; Brkić, B. Computational Multiscale Study of the Interaction Between the PDMS Polymer and Sunscreen-Related Pollutant Molecules. Molecules 2024, 29, 4908. https://doi.org/10.3390/molecules29204908
Armaković S, Vujić Đ, Brkić B. Computational Multiscale Study of the Interaction Between the PDMS Polymer and Sunscreen-Related Pollutant Molecules. Molecules. 2024; 29(20):4908. https://doi.org/10.3390/molecules29204908
Chicago/Turabian StyleArmaković, Stevan, Đorđe Vujić, and Boris Brkić. 2024. "Computational Multiscale Study of the Interaction Between the PDMS Polymer and Sunscreen-Related Pollutant Molecules" Molecules 29, no. 20: 4908. https://doi.org/10.3390/molecules29204908
APA StyleArmaković, S., Vujić, Đ., & Brkić, B. (2024). Computational Multiscale Study of the Interaction Between the PDMS Polymer and Sunscreen-Related Pollutant Molecules. Molecules, 29(20), 4908. https://doi.org/10.3390/molecules29204908