
Photoinduced Transformations with Diverse Maleimide Scaffolds


Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Light-Mediated Photoreactions of Maleimides
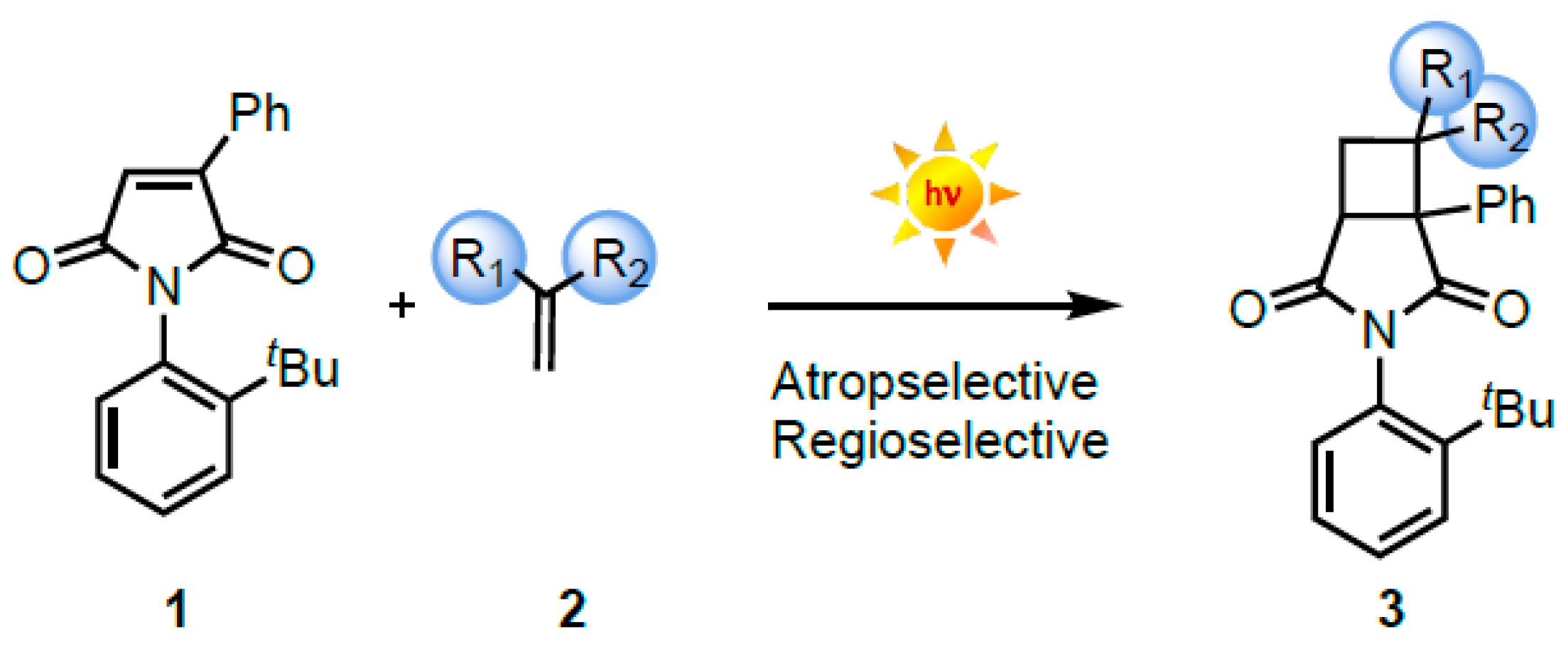
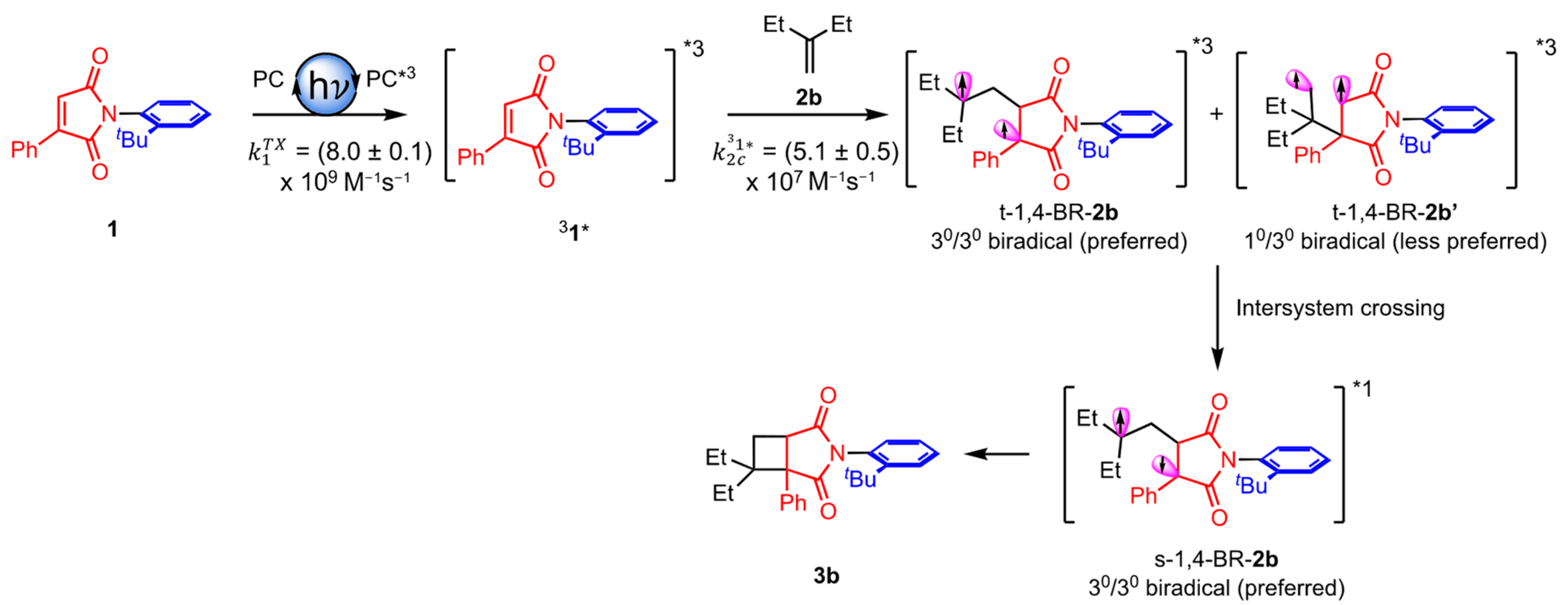
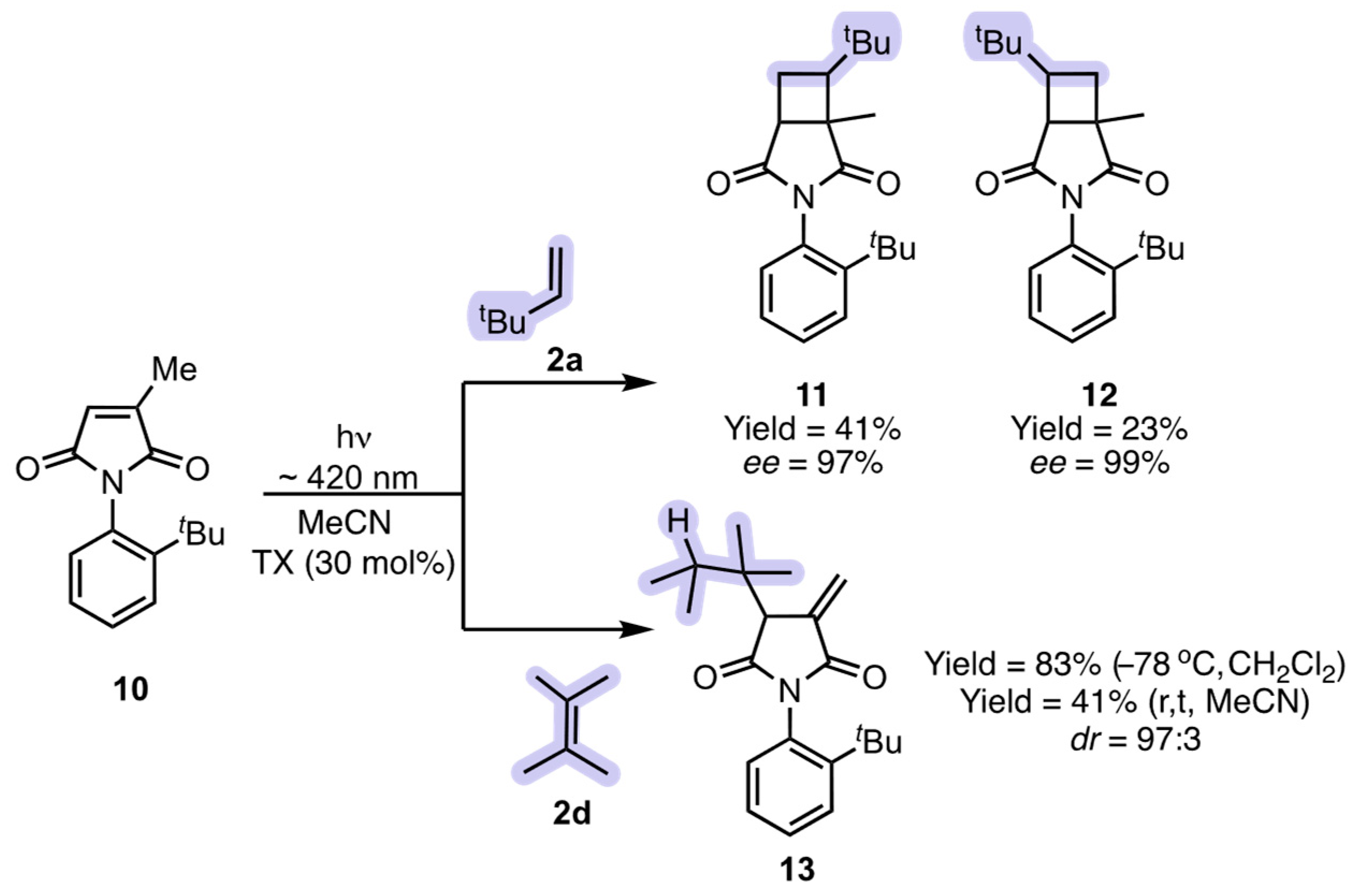
2.1. Atrop- and Regioselective [2 + 2] Photoycloaddition
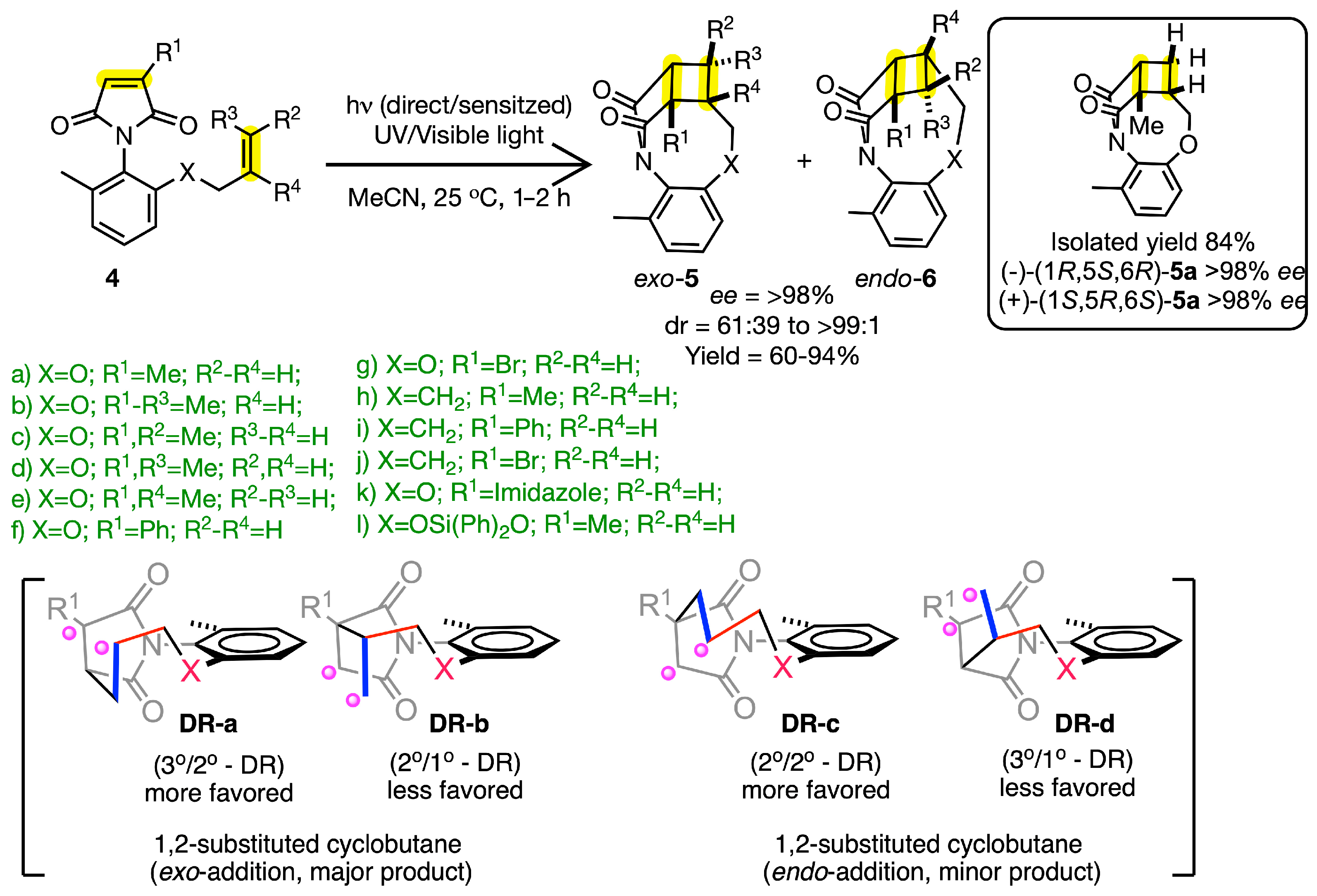
2.2. Atrop-Selective Intramolecular [2 + 2] Photocycloaddition of Maleimides
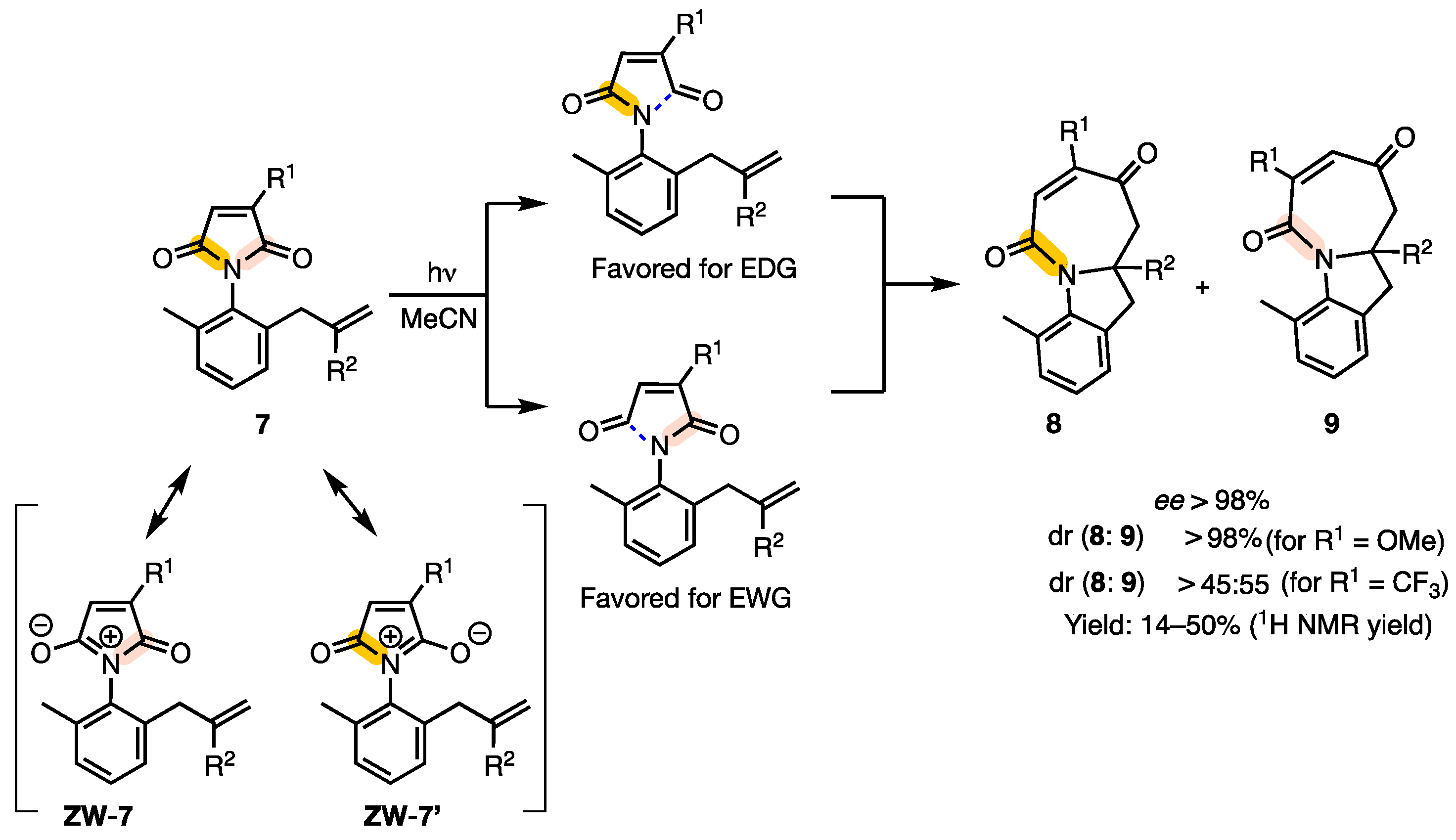
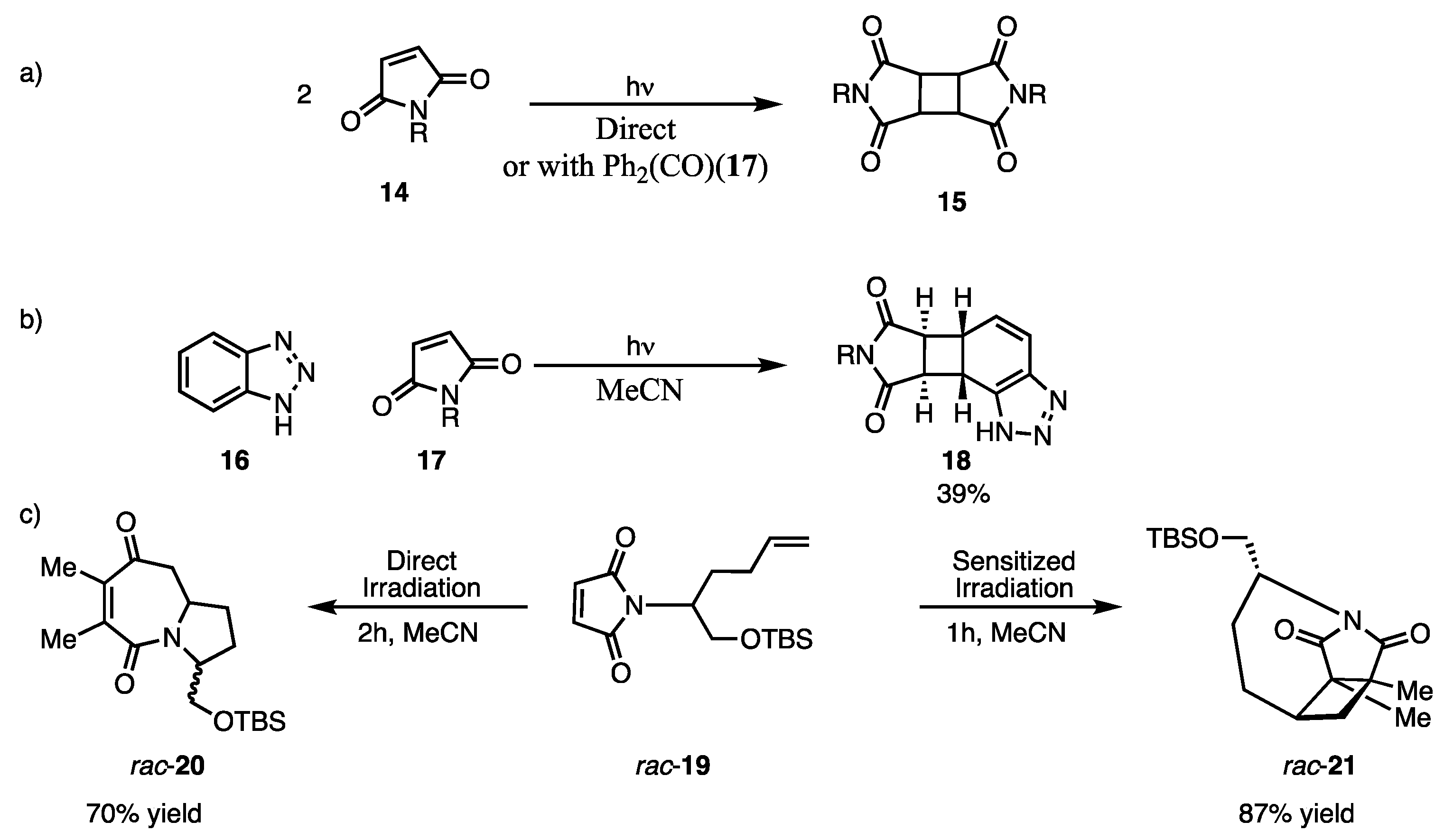
2.3. [5 + 2] Photocycloaddition of Maleimides
2.4. Photo-ene Reaction of Maleimides
2.5. Diversity in Photoreactions of Maleimides—An Overview
3. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Poplata, S.; Tröster, A.; Zou, Y.-Q.; Bach, T. Recent Advances in the Synthesis of Cyclobutanes by Olefin [2 + 2] Photocycloaddition Reactions. Chem. Rev. 2016, 116, 9748–9815. [Google Scholar] [CrossRef] [PubMed]
- Bos, P.H.; Antalek, M.T.; Porco, J.A.; Stephenson, C.R.J. Tandem Dienone Photorearrangement–Cycloaddition for the Rapid Generation of Molecular Complexity. J. Am. Chem. Soc. 2013, 135, 17978–17982. [Google Scholar] [CrossRef] [PubMed]
- Ahuja, S.; Jockusch, S.; Ugrinov, A.; Sivaguru, J. Energy Transfer Catalysis by Visible Light: Atrop– and Regio–Selective Intermolecular [2 + 2]—Photocycloaddition of Maleimide with Alkenes. Eur. J. Org. Chem. 2020, 2020, 1478–1481. [Google Scholar] [CrossRef]
- Skubi, K.L.; Blum, T.R.; Yoon, T.P. Dual Catalysis Strategies in Photochemical Synthesis. Chem. Rev. 2016, 116, 10035–10074. [Google Scholar] [CrossRef]
- Ye, X.; Peng, L.; Bao, X.; Tan, C.-H.; Wang, H. Recent developments in highly efficient construction of P–stereogenic centers. Green Synth. Catal. 2021, 2, 6–18. [Google Scholar] [CrossRef]
- Vallavoju, N.; Selvakumar, S.; Jockusch, S.; Sibi, M.P.; Sivaguru, J. Enantioselective Organo–Photocatalysis Mediated by Atropisomeric Thiourea Derivatives. Angew. Chem. Int. Ed. 2014, 53, 5604–5608. [Google Scholar] [CrossRef]
- CRC Handbook of Organic Photochemistry and Photobiology; CRC Press: Boca Raton, FL, USA, 1995; p. 1636.
- Liebermann, C. Ueber Polythymochinon. Ber. Dtsch. Chem. Ges. 1877, 10, 2177–2179. [Google Scholar] [CrossRef]
- Rabinovich, D.; Schmidt, G.M.J. Topochemistry. Part XV. The solid–state photochemistry of p–quinones. J. Chem. Soc. B 1967, 144–149. [Google Scholar] [CrossRef]
- Bertram, J.; Kürsten, R. Ueber das Vorkommen des Orthocumaraldehyd–methyläthers im Cassiaöl. J. Prakt. Chem. 1895, 51, 316–325. [Google Scholar] [CrossRef]
- Riiber, C.N. Das directe Ueberführen der Zimmtsäure in α–Truxillsäure. Ber. Dtsch. Chem. Ges. 1902, 35, 2908–2909. [Google Scholar] [CrossRef]
- Roth, H.D. The Beginnings of Organic Photochemistry. Angew. Chem. Int. Ed. 1989, 28, 1193–1207. [Google Scholar] [CrossRef]
- Sivaguru, J.; Bach, T.; Ramamurthy, V. Keeping the name clean: [2 + 2] photocycloaddition. Photochem. Photobiol. Sci. 2022, 21, 1333–1340. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, D.; Bera, N.; Ghosh, S. [2 + 2] Photochemical Cycloaddition in Organic Synthesis. Eur. J. Org. Chem. 2019, 2020, 1310–1326. [Google Scholar] [CrossRef]
- Vallavoju, N.; Sivaguru, J. Supramolecular photocatalysis: Combining confinement and non–covalent interactions to control light initiated reactions. Chem. Soc. Rev. 2014, 43, 4084. [Google Scholar] [CrossRef] [PubMed]
- Sivaguru, J.; Natarajan, A.; Kaanumalle, L.S.; Shailaja, J.; Uppili, S.; Joy, A.; Ramamurthy, V. Asymmetric Photoreactions within Zeolites: Role of Confinement and Alkali Metal Ions. Acc. Chem. Res. 2003, 36, 509–521. [Google Scholar] [CrossRef] [PubMed]
- Ramamurthy, V. Photochemistry within a Water–Soluble Organic Capsule. Acc. Chem. Res. 2015, 48, 2904–2917. [Google Scholar] [CrossRef]
- Brimioulle, R.; Lenhart, D.; Maturi, M.M.; Bach, T. Enantioselective Catalysis of Photochemical Reactions. Angew. Chem. Int. Ed. 2015, 54, 3872–3890. [Google Scholar] [CrossRef]
- Blum, T.R.; Miller, Z.D.; Bates, D.M.; Guzei, I.A.; Yoon, T.P. Enantioselective photochemistry through Lewis acid–catalyzed triplet energy transfer. Science 2016, 354, 1391–1395. [Google Scholar] [CrossRef]
- Kumarasamy, E.; Raghunathan, R.; Sibi, M.P.; Sivaguru, J. Nonbiaryl and Heterobiaryl Atropisomers: Molecular Templates with Promise for Atropselective Chemical Transformations. Chem. Rev. 2015, 115, 11239–11300. [Google Scholar] [CrossRef]
- Kumarasamy, E.; Ayitou, A.J.-L.; Vallavoju, N.; Raghunathan, R.; Iyer, A.; Clay, A.; Kandappa, S.K.; Sivaguru, J. Tale of Twisted Molecules. Atropselective Photoreactions: Taming Light Induced Asymmetric Transformations through Non–biaryl Atropisomers. Acc. Chem. Res. 2016, 49, 2713–2724. [Google Scholar] [CrossRef]
- Ahuja, S.; Raghunathan, R.; Kumarasamy, E.; Jockusch, S.; Sivaguru, J. Realizing the Photoene Reaction with Alkenes under Visible Light Irradiation and Bypassing the Favored [2 + 2]–Photocycloaddition. J. Am. Chem. Soc. 2018, 140, 13185–13189. [Google Scholar] [CrossRef]
- Kumarasamy, E.; Raghunathan, R.; Jockusch, S.; Ugrinov, A.; Sivaguru, J. Tailoring Atropisomeric Maleimides for Stereospecific [2 + 2] Photocycloaddition—Photochemical and Photophysical Investigations Leading to Visible–Light Photocatalysis. J. Am. Chem. Soc. 2014, 136, 8729–8737. [Google Scholar] [CrossRef] [PubMed]
- Raghunathan, R.; Kumarasamy, E.; Jockusch, S.; Ugrinov, A.; Sivaguru, J. Engaging electronic effects for atropselective [5 + 2]–photocycloaddition of maleimides. Chem. Commun. 2016, 52, 8305–8308. [Google Scholar] [CrossRef] [PubMed]
- Ahuja, S.; Baburaj, S.; Valloli, L.K.; Rakhimov, S.A.; Manal, K.; Kushwaha, A.; Jockusch, S.; Forbes, M.D.E.; Sivaguru, J. Photochemical [2+ 4]–Dimerization Reaction from the Excited State. Angew. Chem. Int. Ed. 2023, 63, e202316662. [Google Scholar] [CrossRef] [PubMed]
- Shim, S.-C.; Bong, P.-H. Photocyclodimerization of Maleimide. Bull. Kor. Chem. Soc. 1982, 3, 115–119. [Google Scholar]
- Roscini, C.; Cubbage, K.L.; Berry, M.; Orr–Ewing, A.J.; Booker–Milburn, K.I. Reaction Control in Synthetic Organic Photochemistry: Switching between [5 + 2] and [2 + 2] Modes of Cycloaddition. Angew. Chem. Int. Ed. 2009, 48, 8716–8720. [Google Scholar] [CrossRef]
- Booker-Milburn, K.I.; Anson, C.E.; Clissold, C.; Costin, N.J.; Dainty, R.F.; Murray, M.; Patel, D.; Sharpe, A. Intramolecular Photocycloaddition ofN–Alkenyl Substituted Maleimides: A Potential Tool for the Rapid Construction of Perhydroazaazulene Alkaloids. Eur. J. Org. Chem. 2001, 2001, 1473–1482. [Google Scholar] [CrossRef]
- Skolia, E.; Kokotos, C.G. Photochemical [2 + 2] Cycloaddition of Alkenes with Maleimides: Highlighting the Differences between N–Alkyl vs N–Aryl Maleimides. ACS Org. Inorg. Au 2023, 3, 96–103. [Google Scholar] [CrossRef]
- Clark, S.C.; Hoyle, C.E.; Jönsson, S.; Morel, F.; Decker, C. Photopolymerization of acrylates using N–aliphaticmaleimides as photoinitiators. Polymer 1999, 40, 5063–5072. [Google Scholar] [CrossRef]
- Kandappa, S.K.; Ahuja, S.; Singathi, R.; Valloli, L.K.; Baburaj, S.; Parthiban, J.; Sivaguru, J. Using Restricted Bond Rotations to Enforce Excited–State Behavior of Organic Molecules. Synlett 2022, 33, 1123–1134. [Google Scholar]
- Goti, G.; Manal, K.; Sivaguru, J.; Dell’Amico, L. The impact of UV light on synthetic photochemistry and photocatalysis. Nat. Chem. 2024, 16, 684–692. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parthiban, J.; Garg, D.; Sivaguru, J. Photoinduced Transformations with Diverse Maleimide Scaffolds. Molecules 2024, 29, 4895. https://doi.org/10.3390/molecules29204895
Parthiban J, Garg D, Sivaguru J. Photoinduced Transformations with Diverse Maleimide Scaffolds. Molecules. 2024; 29(20):4895. https://doi.org/10.3390/molecules29204895
Chicago/Turabian StyleParthiban, Jayachandran, Dipti Garg, and Jayaraman Sivaguru. 2024. "Photoinduced Transformations with Diverse Maleimide Scaffolds" Molecules 29, no. 20: 4895. https://doi.org/10.3390/molecules29204895
APA StyleParthiban, J., Garg, D., & Sivaguru, J. (2024). Photoinduced Transformations with Diverse Maleimide Scaffolds. Molecules, 29(20), 4895. https://doi.org/10.3390/molecules29204895