
Identification of Novel PPARγ Partial Agonists Based on Virtual Screening Strategy: In Silico and In Vitro Experimental Validation


Abstract

1. Introduction
2. Results
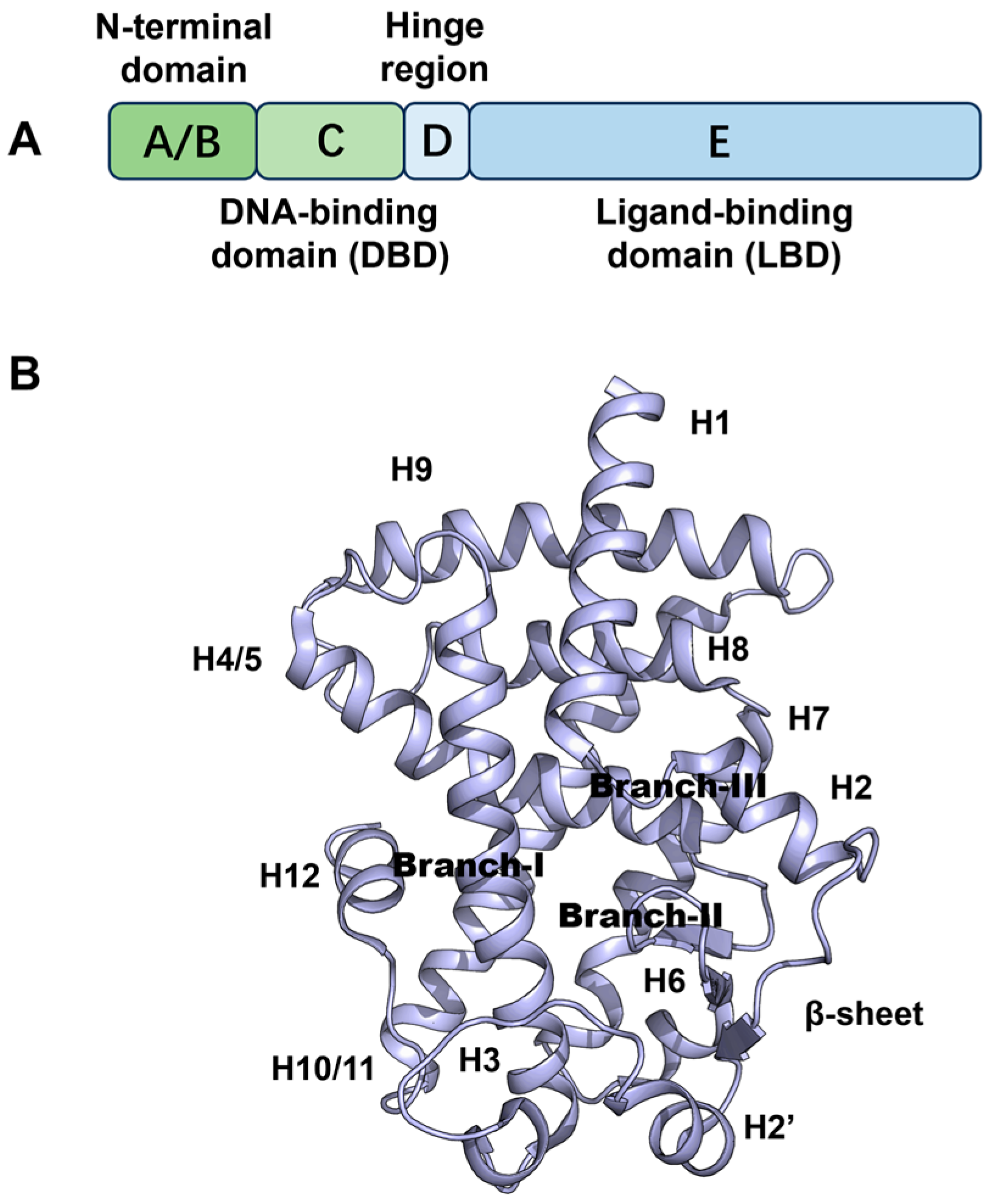
2.1. Docking Validation, Virtual Screening and Molecular Docking
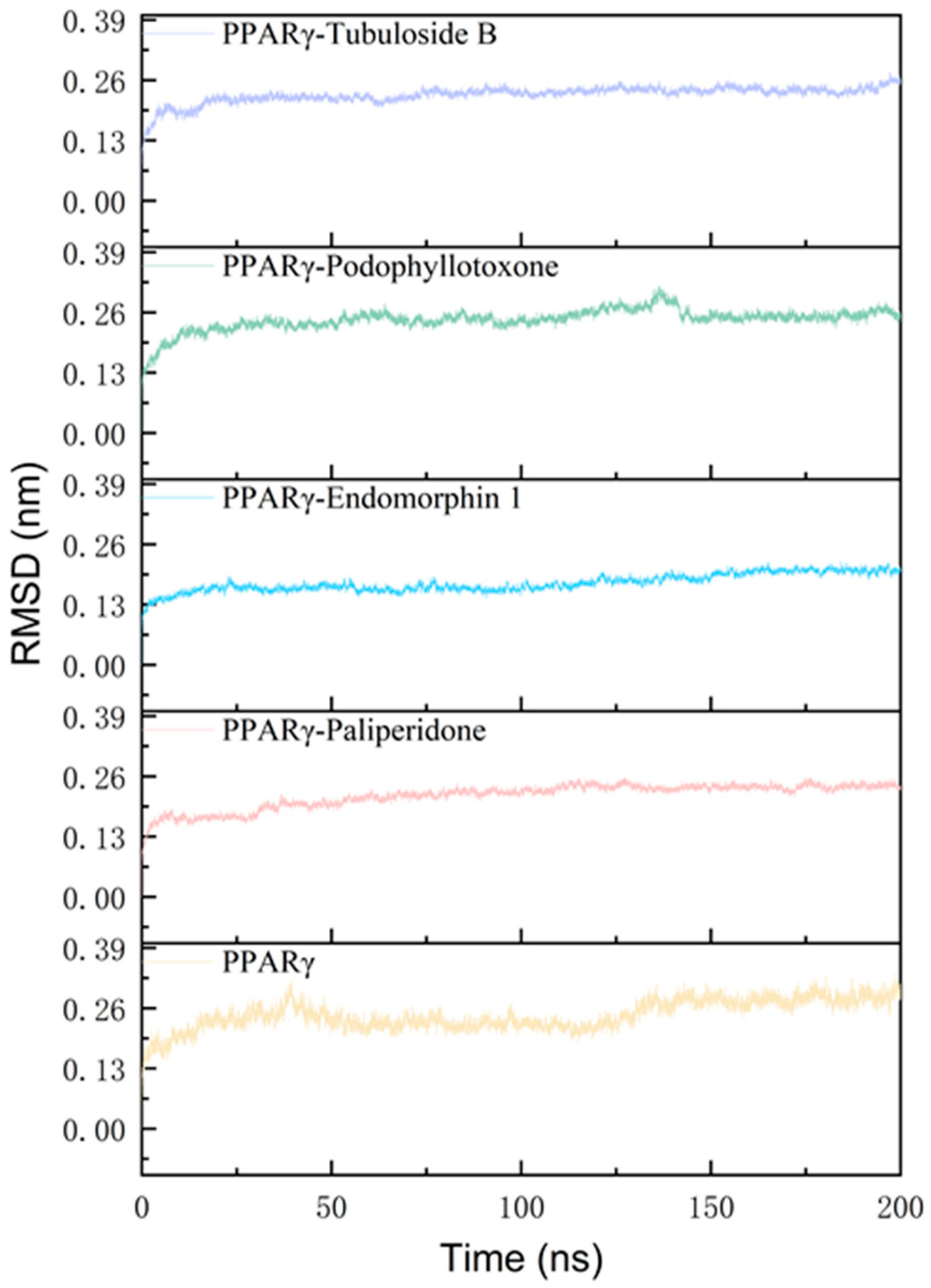
2.2. Stable Assessment of MD Simulation
2.3. Analysis of the Binding Energy
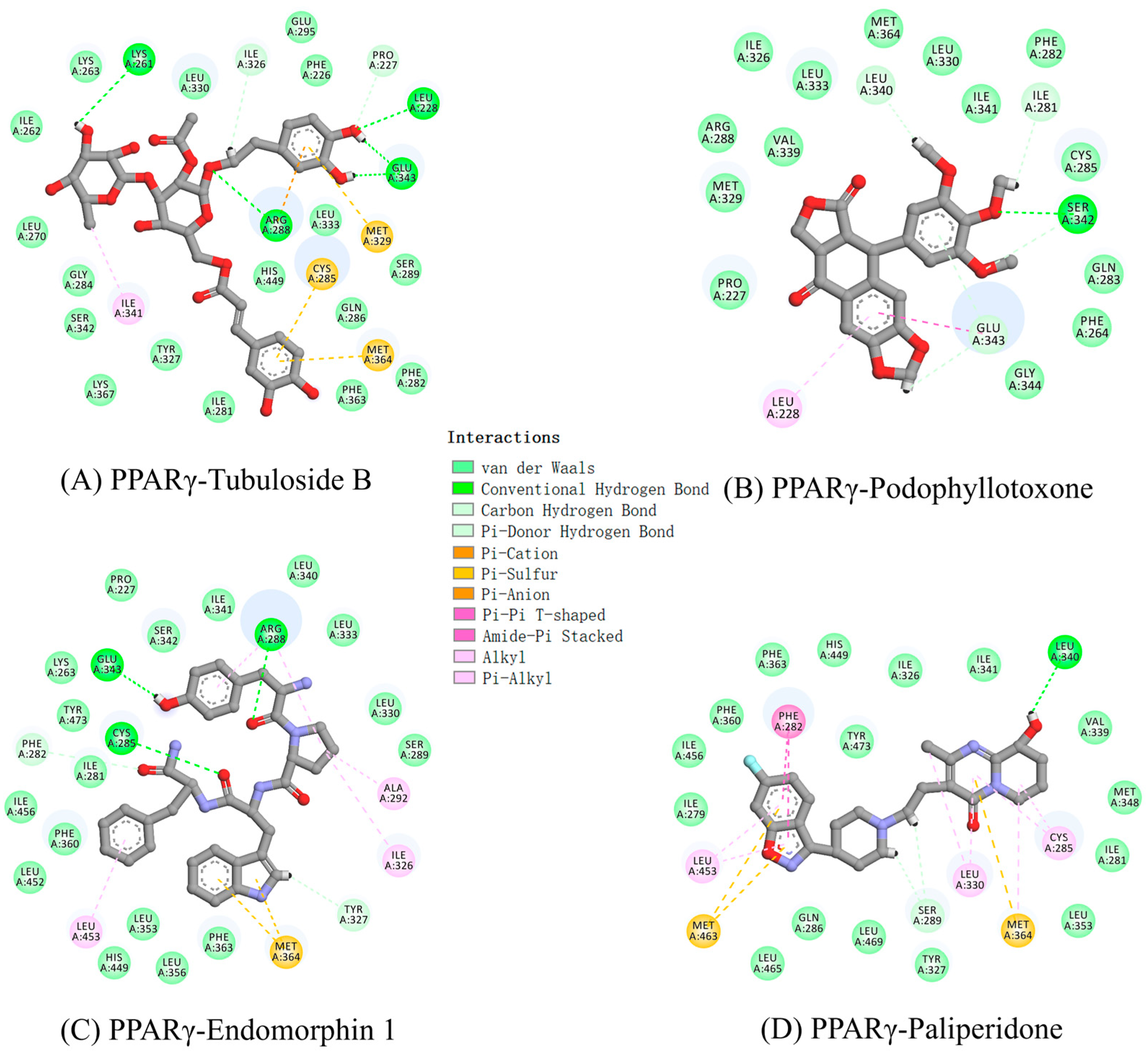
2.4. The Receptor and Ligand Interactions Analysis
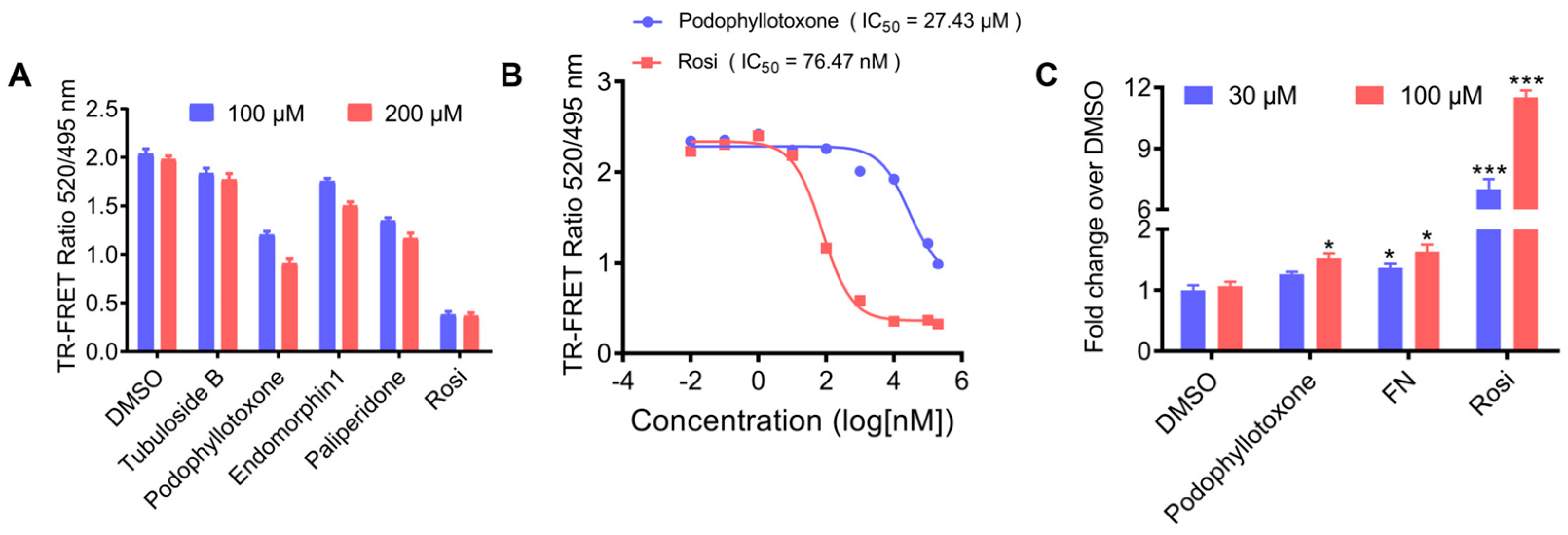
2.5. TR-FRET Competitive Binding Assay and PPARγ Transactivation Assay
3. Materials and Methods
3.1. Receptor and Ligand Preparations
3.2. Docking Validation, Virtual Screening and Molecular Docking
3.3. Molecular Dynamics Simulation
3.4. Calculation of the Binding Energy
3.5. Materials
3.6. TR-FRET Competitive Binding Assay
3.7. PPARγ Transactivation Assay
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ong, K.L.; Stafford, L.K.; McLaughlin, S.A.; Boyko, E.J.; Vollset, S.E.; Smith, A.E.; Dalton, B.E.; Duprey, J.; Cruz, J.A.; Hagins, H.; et al. Global, regional, and national burden of diabetes from 1990 to 2021, with projections of prevalence to 2050: A systematic analysis for the Global Burden of Disease Study 2021. Lancet 2023, 402, 203–234. [Google Scholar] [CrossRef] [PubMed]
- Demir, S.; Nawroth, P.P.; Herzig, S.; Ekim Üstünel, B. Emerging Targets in Type 2 Diabetes and Diabetic Complications. Adv. Sci. 2021, 8, 2100275. [Google Scholar] [CrossRef] [PubMed]
- Rangwala, S.M.; Lazar, M.A. Peroxisome proliferator-activated receptor γ in diabetes and metabolism. Trends Pharmacol. Sci. 2004, 25, 331–336. [Google Scholar] [CrossRef]
- Janani, C.; Ranjitha Kumari, B.D. PPAR gamma gene—A review. Diabetes Metab. Syndr. Clin. Res. Rev. 2015, 9, 46–50. [Google Scholar] [CrossRef]
- Chigurupati, S.; Dhanaraj, S.A.; Balakumar, P. A step ahead of PPARγ full agonists to PPARγ partial agonists: Therapeutic perspectives in the management of diabetic insulin resistance. Eur. J. Pharmacol. 2015, 755, 50–57. [Google Scholar] [CrossRef]
- Ahmadian, M.; Suh, J.M.; Hah, N.; Liddle, C.; Atkins, A.R.; Downes, M.; Evans, R.M. PPARγ signaling and metabolism: The good, the bad and the future. Nat. Med. 2013, 19, 557–566. [Google Scholar] [CrossRef]
- Kroker, A.J.; Bruning, J.B. Review of the Structural and Dynamic Mechanisms of PPARγ Partial Agonism. PPAR Res. 2015, 2015, 816856. [Google Scholar] [CrossRef]
- Chan, L.S.A.; Wells, R.A.; Shi, X.-M. Cross-Talk between PPARs and the Partners of RXR: A Molecular Perspective. PPAR Res. 2009, 2009, 925309. [Google Scholar] [CrossRef]
- Lebovitz, H.E. Thiazolidinediones: The Forgotten Diabetes Medications. Curr. Diabetes Rep. 2019, 19, 151. [Google Scholar] [CrossRef]
- Choi, J.H.; Banks, A.S.; Estall, J.L.; Kajimura, S.; Boström, P.; Laznik, D.; Ruas, J.L.; Chalmers, M.J.; Kamenecka, T.M.; Blüher, M.; et al. Anti-diabetic drugs inhibit obesity-linked phosphorylation of PPARγ by Cdk5. Nature 2010, 466, 451–456. [Google Scholar] [CrossRef]
- Rennings, A.J.M.; Smits, P.; Stewart, M.W.; Tack, C.J. Fluid Retention and Vascular Effects of Rosiglitazone in Obese, Insulin-Resistant, Nondiabetic Subjects. Diabetes Care 2006, 29, 581–587. [Google Scholar] [CrossRef]
- Guan, Y.; Hao, C.; Cha, D.R.; Rao, R.; Lu, W.; Kohan, D.E.; Magnuson, M.A.; Redha, R.; Zhang, Y.; Breyer, M.D. Thiazolidinediones expand body fluid volume through PPARγ stimulation of ENaC-mediated renal salt absorption. Nat. Med. 2005, 11, 861–866. [Google Scholar] [CrossRef]
- Rosen, C.J. Revisiting the Rosiglitazone Story—Lessons Learned. N. Engl. J. Med. 2010, 363, 803–806. [Google Scholar] [CrossRef]
- Weidner, C.; de Groot, J.C.; Prasad, A.; Freiwald, A.; Quedenau, C.; Kliem, M.; Witzke, A.; Kodelja, V.; Han, C.-T.; Giegold, S.; et al. Amorfrutins are potent antidiabetic dietary natural products. Proc. Natl. Acad. Sci. USA 2012, 109, 7257–7262. [Google Scholar] [CrossRef]
- Bruning, J.B.; Chalmers, M.J.; Prasad, S.; Busby, S.A.; Kamenecka, T.M.; He, Y.; Nettles, K.W.; Griffin, P.R. Partial Agonists Activate PPARγ Using a Helix 12 Independent Mechanism. Structure 2007, 15, 1258–1271. [Google Scholar] [CrossRef]
- Frkic, R.L.; Pederick, J.L.; Horsfall, A.J.; Jovcevski, B.; Crame, E.E.; Kowalczyk, W.; Pukala, T.L.; Chang, M.R.; Zheng, J.; Blayo, A.-L.; et al. PPARγ Corepression Involves Alternate Ligand Conformation and Inflation of H12 Ensembles. ACS Chem. Biol. 2023, 18, 1115–1123. [Google Scholar] [CrossRef]
- Frkic, R.L.; Marshall, A.C.; Blayo, A.-L.; Pukala, T.L.; Kamenecka, T.M.; Griffin, P.R.; Bruning, J.B. PPARγ in Complex with an Antagonist and Inverse Agonist: A Tumble and Trap Mechanism of the Activation Helix. iScience 2018, 5, 69–79. [Google Scholar] [CrossRef]
- Chen, F.; Ma, L.; Cai, G.; Tang, J.; Wang, Y.; Liu, Q.; Liu, X.; Hou, N.; Zhou, Z.; Yi, W. Identification of a novel PPARγ modulator with good anti-diabetic therapeutic index via structure-based screening, optimization and biological validation. Biomed. Pharmacother. 2022, 154, 113653. [Google Scholar] [CrossRef]
- Omoboyowa, D.A.; Singh, G.; Fatoki, J.O.; Oyeneyin, O.E. Computational investigation of phytochemicals from Abrus precatorius seeds as modulators of peroxisome proliferator-activated receptor gamma (PPARγ). J. Biomol. Struct. Dyn. 2022, 41, 5568–5582. [Google Scholar] [CrossRef]
- Chu, Y.; Gui, S.; Zheng, Y.; Zhao, J.; Zhao, Y.; Li, Y.; Chen, X. The natural compounds, Magnolol or Honokiol, promote adipose tissue browning and resist obesity through modulating PPARα/γ activity. Eur. J. Pharmacol. 2024, 969, 176438. [Google Scholar] [CrossRef]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef] [PubMed]
- Mandal, S.; Faizan, S.; Raghavendra, N.M.; Kumar, B.R.P. Molecular dynamics articulated multilevel virtual screening protocol to discover novel dual PPAR α/γ agonists for anti-diabetic and metabolic applications. Mol. Divers. 2022, 27, 2605–2631. [Google Scholar] [CrossRef] [PubMed]
- Pathak, R.K.; Gupta, A.; Shukla, R.; Baunthiyal, M. Identification of new drug-like compounds from millets as Xanthine oxidoreductase inhibitors for treatment of Hyperuricemia: A molecular docking and simulation study. Comput. Biol. Chem. 2018, 76, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Zhang, Y.; Jiang, W.; Zhang, H. Virtual Screening of FDA-Approved Drugs for Enhanced Binding with Mitochondrial Aldehyde Dehydrogenase. Molecules 2022, 27, 8773. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Chen, J.; Zhang, P.; Zheng, N.; Ma, L.; Zhang, Y.; Zhang, H. Repurposing Drugs for Inhibition against ALDH2 via a 2D/3D Ligand-Based Similarity Search and Molecular Simulation. Molecules 2023, 28, 7325. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Tang, J.; Cai, G.; Chen, F.; Liu, Q.; Zhou, Z.; Zhang, S.; Liu, X.; Hou, N.; Yi, W. Structure-based screening and biological validation of the anti-thrombotic drug-dicoumarol as a novel and potent PPARγ-modulating ligand. Bioorganic Chem. 2022, 129, 106191. [Google Scholar] [CrossRef]
- Capelli, D.; Cerchia, C.; Montanari, R.; Loiodice, F.; Tortorella, P.; Laghezza, A.; Cervoni, L.; Pochetti, G.; Lavecchia, A. Structural basis for PPAR partial or full activation revealed by a novel ligand binding mode. Sci. Rep. 2016, 6, 34792. [Google Scholar] [CrossRef]
- Laghezza, A.; Montanari, R.; Lavecchia, A.; Piemontese, L.; Pochetti, G.; Iacobazzi, V.; Infantino, V.; Capelli, D.; De Bellis, M.; Liantonio, A.; et al. On the Metabolically Active Form of Metaglidasen: Improved Synthesis and Investigation of Its Peculiar Activity on Peroxisome Proliferator-Activated Receptors and Skeletal Muscles. ChemMedChem 2015, 10, 555–565. [Google Scholar] [CrossRef]
- de Groot, J.C.; Weidner, C.; Krausze, J.; Kawamoto, K.; Schroeder, F.C.; Sauer, S.; Büssow, K. Structural Characterization of Amorfrutins Bound to the Peroxisome Proliferator-Activated Receptor γ. J. Med. Chem. 2013, 56, 1535–1543. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, M.; Gamo, K.; Oyama, T.; Miyachi, H. Peroxisome proliferator-activated receptor gamma (PPARγ) has multiple binding points that accommodate ligands in various conformations: Structurally similar PPARγ partial agonists bind to PPARγ LBD in different conformations. Bioorganic Med. Chem. Lett. 2015, 25, 2758–2762. [Google Scholar] [CrossRef] [PubMed]
- Ahsan, W. The Journey of Thiazolidinediones as Modulators of PPARs for the Management of Diabetes: A Current Perspective. Curr. Pharm. Des. 2019, 25, 2540–2554. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Lian, Y.; Tang, J.; Chen, F.; Gao, H.; Zhou, Z.; Hou, N.; Yi, W. Identification of the anti-fungal drug fenticonazole nitrate as a novel PPARγ-modulating ligand with good therapeutic index: Structure-based screening and biological validation. Pharmacol. Res. 2021, 173, 105860. [Google Scholar] [CrossRef]
- Burley, S.K.; Bhikadiya, C.; Bi, C.; Bittrich, S.; Chao, H.; Chen, L.; Craig, P.A.; Crichlow, G.V.; Dalenberg, K.; Duarte, J.M.; et al. RCSB Protein Data Bank (RCSB.org): Delivery of experimentally-determined PDB structures alongside one million computed structure models of proteins from artificial intelligence/machine learning. Nucleic Acids Res. 2023, 51, 488–508. [Google Scholar] [CrossRef]
- Páll, S.; Zhmurov, A.; Bauer, P.; Abraham, M.; Lundborg, M.; Gray, A.; Hess, B.; Lindahl, E. Heterogeneous parallelization and acceleration of molecular dynamics simulations in GROMACS. J. Chem. Phys. 2020, 153, 134110. [Google Scholar] [CrossRef]
- Neese, F. Software update: The ORCA program system—Version 5.0. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2022, 12, e1606. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2011, 33, 580–592. [Google Scholar] [CrossRef]
- Lindorff-Larsen, K.; Piana, S.; Palmo, K.; Maragakis, P.; Klepeis, J.L.; Dror, R.O.; Shaw, D.E. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 2010, 78, 1950–1958. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [PubMed]
- Bernetti, M.; Bussi, G. Pressure control using stochastic cell rescaling. J. Chem. Phys. 2020, 153, 114107. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Valdés-Tresanco, M.S.; Valdés-Tresanco, M.E.; Valiente, P.A.; Moreno, E. gmx_MMPBSA: A New Tool to Perform End-State Free Energy Calculations with GROMACS. J. Chem. Theory Comput. 2021, 17, 6281–6291. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Tang, J.; Chen, F.; Liu, Q.; Huang, J.; Liu, X.; Zhou, Z.; Yi, W. Structure-based screening, optimization and biological evaluation of novel chrysin-based derivatives as selective PPARγ modulators for the treatment of T2DM and hepatic steatosis. Eur. J. Med. Chem. 2024, 276, 116728. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Zhou, X.E.; Shi, J.; Zhou, Z.; Zhao, G.; Zhang, X.; Sun, Y.; Suino-Powell, K.; Ma, L.; Gao, H.; et al. Identification and structural insight of an effective PPARγ modulator with improved therapeutic index for anti-diabetic drug discovery. Chem. Sci. 2020, 11, 2260–2268. [Google Scholar] [CrossRef]
- Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.Z.H.; Hou, T. End-Point Binding Free Energy Calculation with MM/PBSA and MM/GBSA: Strategies and Applications in Drug Design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | CAS | Molecular Structure | Docking Score (kcal/mol) | Hydrogen Bond | Hydrophobic Interaction |
|---|---|---|---|---|---|
| Tubuloside B | 112516-04-8 | 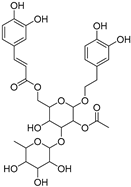 | −10.07 | Leu228, Gln286, Arg288, Ser342, Glu343 | Phe282, Arg288, Ala292, Met329, Leu333 |
| Podophyllotoxone | 477-49-6 | 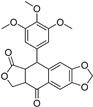 | −10.04 | Ser342 | Arg288, Ala292, Ile326, Leu330, Ile341 |
| Endomorphin 1 | 189388-22-5 |  | −10.68 | Gly284, Cys285 Arg288 | Leu228, Met329, Leu333, Arg288, Phe287, Lys263, Ile341, Cys285, Met364, Val339, Met348, Leu353, Ile326 |
| Paliperidone | 144598-75-4 | 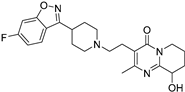 | −10.44 | Ser342 | Leu228, Leu333, Ile341, Arg288, Ala292, Ile326, Leu330, Cys285, His449, Phe363, Tyr327 |
| Compound | ∆EvdW | ∆Eelec | ∆EMM | ∆Gpolar | ∆Gnonpolar | ∆Gsol | ∆Ebind |
|---|---|---|---|---|---|---|---|
| Tubuloside B | −69.12 ± 0.43 | −79.92 ± 1.61 | −149.04 ± 1.62 | 111.05 ± 1.25 | −7.19 ± 0.02 | 103.86 ± 1.24 | −45.18 ± 0.70 |
| Podophyllotoxone | −53.00 ± 0.38 | −21.67 ± 0.49 | −74.67 ± 0.61 | 49.23 ± 0.41 | −4.71 ± 0.01 | 44.52 ± 0.41 | −30.15 ± 0.38 |
| Endomorphin 1 | −66.03 ± 0.38 | −54.33 ± 1.39 | −120.35 ± 1.49 | 88.34 ± 1.26 | −6.58 ± 0.02 | 81.49 ± 1.25 | −38.86 ± 0.54 |
| Paliperidone | −55.47 ± 0.29 | −18.96 ± 0.53 | −74.43 ± 0.60 | 51.56 ± 0.53 | −5.21 ± 0.01 | 46.35 ± 0.52 | −28.08 ± 0.34 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lian, Y.-E.; Wang, M.; Ma, L.; Yi, W.; Liao, S.; Gao, H.; Zhou, Z. Identification of Novel PPARγ Partial Agonists Based on Virtual Screening Strategy: In Silico and In Vitro Experimental Validation. Molecules 2024, 29, 4881. https://doi.org/10.3390/molecules29204881
Lian Y-E, Wang M, Ma L, Yi W, Liao S, Gao H, Zhou Z. Identification of Novel PPARγ Partial Agonists Based on Virtual Screening Strategy: In Silico and In Vitro Experimental Validation. Molecules. 2024; 29(20):4881. https://doi.org/10.3390/molecules29204881
Chicago/Turabian StyleLian, Yu-E, Mei Wang, Lei Ma, Wei Yi, Siyan Liao, Hui Gao, and Zhi Zhou. 2024. "Identification of Novel PPARγ Partial Agonists Based on Virtual Screening Strategy: In Silico and In Vitro Experimental Validation" Molecules 29, no. 20: 4881. https://doi.org/10.3390/molecules29204881
APA StyleLian, Y.-E., Wang, M., Ma, L., Yi, W., Liao, S., Gao, H., & Zhou, Z. (2024). Identification of Novel PPARγ Partial Agonists Based on Virtual Screening Strategy: In Silico and In Vitro Experimental Validation. Molecules, 29(20), 4881. https://doi.org/10.3390/molecules29204881