Abstract
The coordination chemistry of 2-pyridyl ketoximes continues to attract the interest of many inorganic chemistry groups around the world for a variety of reasons. Cadmium(II) complexes of such ligands have provided models of solvent extraction of this toxic metal ion from aqueous environments using 2-pyridyl ketoxime extractants. Di-2-pyridyl ketone oxime (dpkoxH) is a unique member of this family of ligands because its substituent on the oxime carbon bears another potential donor site, i.e., a second 2-pyridyl group. The goal of this study was to investigate the reactions of cadmium(II) halides and dpkoxH in order to assess the structural role (if any) of the halogeno ligand and compare the products with their zinc(II) analogs. The synthetic studies provided access to complexes {[CdCl2(dpkoxH)∙2H2O]}n (1∙2H2O), {[CdBr2(dpkoxH)]}n (2) and {[CdI2(dpkoxH)]}n (3) in 50–60% yields. The structures of the complexes were determined by single-crystal X-ray crystallography. The compounds consist of structurally similar 1D zigzag chains, but only 2 and 3 are strictly isomorphous. Neighboring CdII atoms are alternately doubly bridged by halogeno and dpkoxH ligands, the latter adopting the η1:η1:η1:μ (or 2.0111 using Harris notation) coordination mode. A terminal halogeno group completes distorted octahedral coordination at each metal ion, and the coordination sphere of the CdII atoms is {CdII(η1 − X)(μ − X)2(Npyridyl)2(Noxime)} (X = Cl, Br, I). The trans-donor–atom pairs in 1∙2H2O are Clterminal/Noxime and two Clbridging/Npyridyl; on the contrary, these donor–atom pairs are Xterminal/Npyridyl, Xbridging/Noxime, and Xbridging/Npyridyl (X = Br, I). There are intrachain H-bonding interactions in the structures. The packing of the chains in 1∙2H2O is achieved via π-π stacking interactions, while the 3D architecture of the isomorphous 2 and 3 is built via C-H∙∙∙Cg (Cg is the centroid of one pyridyl ring) and π-π overlaps. The molecular structures of 1∙2H2O and 2 are different compared with their [ZnX2(dpkoxH)] (X = Cl, Br) analogs. The polymeric compounds were characterized by IR and Raman spectroscopies in the solid state, and the data were interpreted in terms of the known molecular structures. The solid-state structures of the complexes are not retained in DMSO, as proven via NMR (1H, 13C, and 113Cd NMR) spectroscopy and molar conductivity data. The complexes completely release the coordinated dpkoxH molecule, and the dominant species in solution seem to be [Cd(DMSO)6]2+ in the case of the chloro and bromo complexes and [CdI2(DMSO)4].
1. Introduction
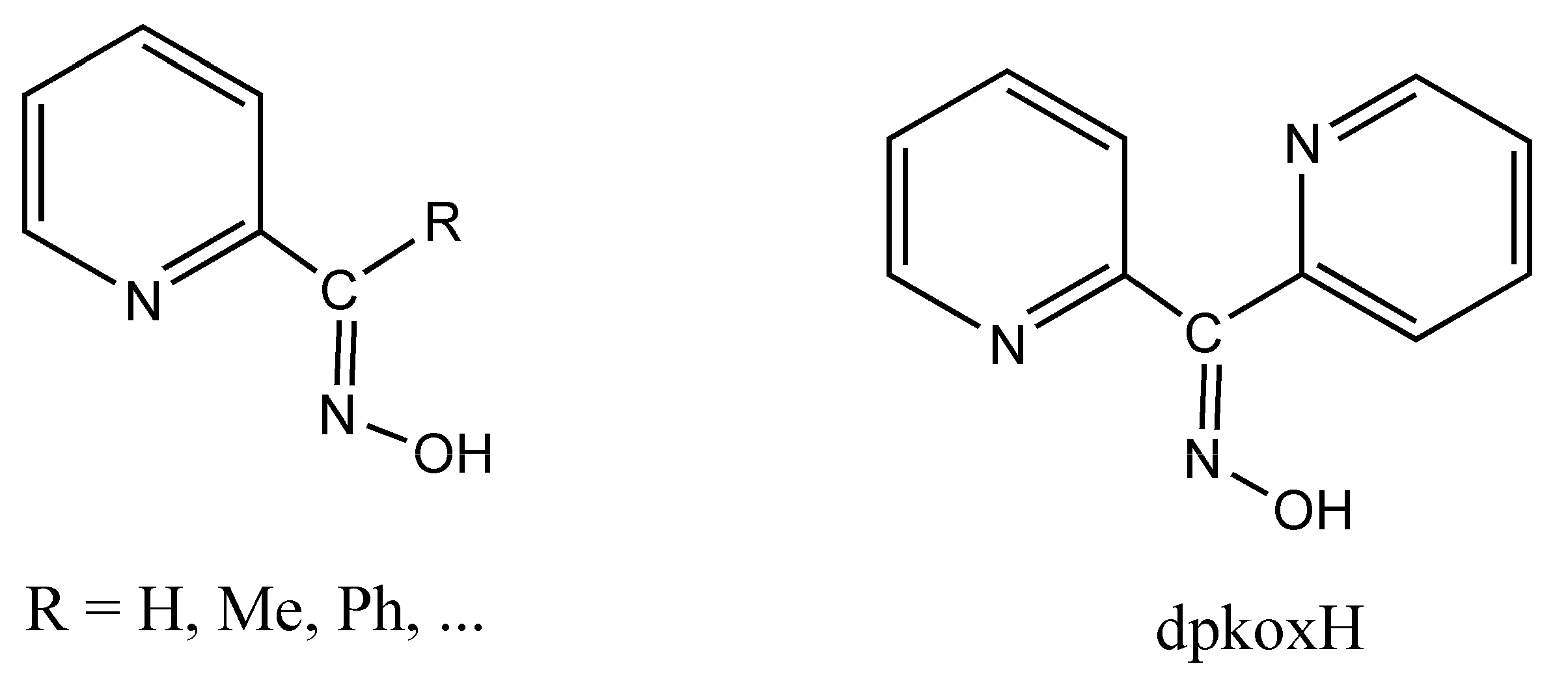
The oxime group (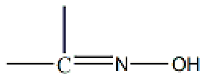 ), which is commonly produced by the reaction of a carbonyl group (from an aldehyde giving aldoximes and from a ketone resulting in ketoximes) with hydroxylamine, is a classical functional group in organic chemistry. It also plays a significant role in supramolecular chemistry and crystal engineering due to its capability to form several types of H bonds. The landmark of the use of oximes was the gravimetric determination of Ni(II) as the highly insoluble, red bis(dimethylglyoximato)nickel(II) solid [1]. Oxime and oximato metal complexes are central “players” in several aspects of coordination [2,3] and bioinorganic [4,5] chemistry, molecular magnetism [6,7], and homogeneous catalysis [8]. The reactivity of the coordinated oxime group is also of interest; this group has three sites (C, N, and O) for electrophilic or nucleophilic additions, most often promoted/assisted by metal ions [9,10,11]. Such reactions can proceed with rupture or preservation of the {CNO} moiety. In many cases, the oxime group is part of an organic molecule that contains one or more donor sites. Typical examples are the various 2-pyridyl oximes (Figure 1, left; R = a non-donor group). These ligands have been used among other purposes, including the following: (i) the linking (by coordination bonds) of smaller clusters to supramolecular entities [12]; (ii) the synthesis of single-chain magnets (SCMs) [13]; (iii) the isolation of 3d/4f-metal dinuclear and polynuclear complexes [14]; (iv) the “switching on” of single-molecule magnetism (SMM) properties [15]; and (v) the modeling of solvent extraction of Cd(II) from aqueous media using 2-pyridyl ketoxime extractants [16].
), which is commonly produced by the reaction of a carbonyl group (from an aldehyde giving aldoximes and from a ketone resulting in ketoximes) with hydroxylamine, is a classical functional group in organic chemistry. It also plays a significant role in supramolecular chemistry and crystal engineering due to its capability to form several types of H bonds. The landmark of the use of oximes was the gravimetric determination of Ni(II) as the highly insoluble, red bis(dimethylglyoximato)nickel(II) solid [1]. Oxime and oximato metal complexes are central “players” in several aspects of coordination [2,3] and bioinorganic [4,5] chemistry, molecular magnetism [6,7], and homogeneous catalysis [8]. The reactivity of the coordinated oxime group is also of interest; this group has three sites (C, N, and O) for electrophilic or nucleophilic additions, most often promoted/assisted by metal ions [9,10,11]. Such reactions can proceed with rupture or preservation of the {CNO} moiety. In many cases, the oxime group is part of an organic molecule that contains one or more donor sites. Typical examples are the various 2-pyridyl oximes (Figure 1, left; R = a non-donor group). These ligands have been used among other purposes, including the following: (i) the linking (by coordination bonds) of smaller clusters to supramolecular entities [12]; (ii) the synthesis of single-chain magnets (SCMs) [13]; (iii) the isolation of 3d/4f-metal dinuclear and polynuclear complexes [14]; (iv) the “switching on” of single-molecule magnetism (SMM) properties [15]; and (v) the modeling of solvent extraction of Cd(II) from aqueous media using 2-pyridyl ketoxime extractants [16].
), which is commonly produced by the reaction of a carbonyl group (from an aldehyde giving aldoximes and from a ketone resulting in ketoximes) with hydroxylamine, is a classical functional group in organic chemistry. It also plays a significant role in supramolecular chemistry and crystal engineering due to its capability to form several types of H bonds. The landmark of the use of oximes was the gravimetric determination of Ni(II) as the highly insoluble, red bis(dimethylglyoximato)nickel(II) solid [1]. Oxime and oximato metal complexes are central “players” in several aspects of coordination [2,3] and bioinorganic [4,5] chemistry, molecular magnetism [6,7], and homogeneous catalysis [8]. The reactivity of the coordinated oxime group is also of interest; this group has three sites (C, N, and O) for electrophilic or nucleophilic additions, most often promoted/assisted by metal ions [9,10,11]. Such reactions can proceed with rupture or preservation of the {CNO} moiety. In many cases, the oxime group is part of an organic molecule that contains one or more donor sites. Typical examples are the various 2-pyridyl oximes (Figure 1, left; R = a non-donor group). These ligands have been used among other purposes, including the following: (i) the linking (by coordination bonds) of smaller clusters to supramolecular entities [12]; (ii) the synthesis of single-chain magnets (SCMs) [13]; (iii) the isolation of 3d/4f-metal dinuclear and polynuclear complexes [14]; (iv) the “switching on” of single-molecule magnetism (SMM) properties [15]; and (v) the modeling of solvent extraction of Cd(II) from aqueous media using 2-pyridyl ketoxime extractants [16].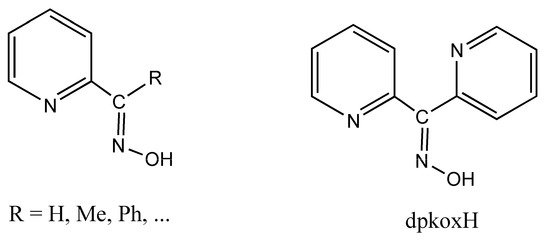
Figure 1.
(Left) The general structural formula of simple 2-pyridyl oximes (R is a non-donor group); (right) di-2-pyridyl ketone oxime (dpkoxH), the ligand used in this work.
Di-2-pyridyl ketone oxime (Figure 1, right, abbreviated as dpkoxH, where H denotes the potentially acidic hydrogen atom) occupies a special position among the 2-pyridyl oximes. Not only is the R group a donor site but also this group is a second 2-pyridyl unit. An attractive aspect of dpkoxH (and its anionic form, dpkox−) is its remarkable coordinative flexibility and versatility that have resulted in 3d- and 3d/4f-metal coordination clusters with interesting structural features and magnetic properties [17,18]. Another area to which the deprotonated ligand is relevant is the chemistry of metallacrowns (MCs), both regular and inverse [19,20]. Finally, an equally interesting aspect of the dpkoxH coordination chemistry is the activation of this molecule by 3d-metal ions, which appears to be useful in synthetic inorganic chemistry [21].
Cadmium(II)/pyridyl aldoxime complexes are known in the literature. This chemistry has been pioneered mainly by Forani’s group, and her work has led to complexes with remarkable structures and impressive properties, including luminescence and supramolecular isomerism [22,23,24,25,26].
One family of such Cd(II) compounds includes mixed carboxylate/pyridyl aldoxime complexes. The complexes {[Cd(1,3-bdc)(2-pyaoH)]}n and {[Cd(1,4-bdc)(4-pyaoH)2]∙DMF}n have laminar 2D structures, whereas {[Cd(fum)(2-pyaoH)2]}n, {[Cd(1,3-bdc)(4-pyaoH)(H2O)2]∙DMF∙H2O}n and {[Cd(1,4-bdc)(4-pyaoH)2(H2O)]∙DMF}n are 1D polymers; in these formulae, 2-pyaoH is pyridine-2-aldoxime, 4-pyaoH is pyridine-4-aldoxime, fum2− is the fumarate(-2) ligand, 1,3-bdc2− is the 1,3-benzenedicarboxylate(-2) ligand, and 1,4-bdc2− is the 1,4-benzenedicarboxylate(-2) ligand [22]. The complexes [Cd2(O2CMe)4(4-pyaoH)4]∙4H2O and [Cd(O2CMe)2(4-pyaoH)3]∙3H2O consist of discrete (dinuclear and mononuclear, respectively) molecules and display blue luminescence upon excitation with UV light [23]. The use of the sulfato co-ligand in Cd(II)/4-pyaoH chemistry has led to the 1D compound {[Cd(SO4)(4-pyaoH)2(H2O)2]}n, which displays ligand-based emission [24]; this complex is a supramolecular isomer with its Zn(II) counterpart. The complexes [Cd2(suc)(2-pyaoH)4(H2O)2](BF4)2, {[Cd(suc)(2-pyaoH)2]}n, {[Cd(mal)(4-pyaoH)(H2O)]}n, and {[Cd(adi)(4-pyaoH)2]∙DMF}n were isolated using the saturated aliphatic dicarboxylate(-2) ligands malonate (mal2−), succinate (suc2−), and adipate (adi2−). The polymeric succinate complex has a 1D array, whereas the other two coordination polymers have layered 2D structures [25]. Further use of the 2-pyaoH ligand in Cd(II) chemistry has resulted in the isolation of the mononuclear complex [Cd(HCO2)2(2-pyaoH)2], and the 1D coordination polymers {[Cd(1,4-bdc)(2-pyaoH)]∙1.5DMF}n and {[Cd(SO4)(2-pyaoH)(H2O)]}n; a rare dual light emission was observed in the sulfato compound which, according to TD/DFT calculations, originates from the nπ* (at 400 nm) and ππ* (at 650 nm) of the 2-pyaoH ligand [26]. The complex [Cd(O2CMe)2(2-pyaoH)2] [27] has a similar structure to that of [Cd(HClO2)2(2-pyaoH)2] [26], the two monodentate carboxylate ligands being in cis positions. Our group has also contributed to the Cd(II)/pyridyl aldoxime chemistry by isolating and characterizing the mononuclear complex [CdI2(2-pyaoH)2] and the 1D chain compounds {[CdI2(3-pyaoH)2]}n and {[CdI2(4-pyaoH)2]}n [16], where 3-pyaoH is pyridyl-3-aldoxime; the 2-pyaoH molecules behave as Npyridyl and Noxime-bidentate chelating ligands, whereas 3-pyaoH and 4-pyaoH act in a monodentate manner coordinating via their pyridyl nitrogen atom.
The Cd(II)/pyridyl ketoxime chemistry is much less studied, and almost all of the published work comes from our groups. The reaction between CdSO4∙8/3H2O and methyl 2-pyridyl ketone oxime (mepaoH, R = Me in Figure 1) in H2O gives {[Cd(SO4)(mepaoH)(H2O)]}n∙{[Cd(SO4)(mepaoH)(H2O)2]}n, whose unique structure consists of two different linear and ladder-type units [28]. The mononuclear complexes [Cd(phpaoH)3](NO3)2 [29], [CdCl2(phpaoH)2]∙H2O [30], and the 1D coordination polymer {[CdCl2(phpaoH)]}n [30], where phpaoH is phenyl 2-pyridyl ketone oxime (R = Ph in Figure 1), have also been prepared and structurally characterized. The mepaoH and phpaoH ligands chelate Cd(II) via their Npyridyl and Noxime atoms.
However, Cd(II)/dpkoxH complexes remain unknown in the literature. In this work, we are glad to report the first Cd(II) complexes with this ligand by describing the syntheses, structures, and spectroscopic characterization of the products derived from the reactions of cadmium(II) halides and dpkoxH; an attempt has also been made to study the behavior of the compounds in solution.
2. Results and Discussion
2.1. Synthetic Comments
For reasons outlined in the Introduction section, we sought the preparation of Cd(II)/dpkoxH complexes. The best results (from the crystallization viewpoint) were obtained using cadmium(II) halides. We started our efforts using neutral dpkoxH, i.e., by avoiding adding an external base in the reaction mixtures, which would lead to the deprotonation of the oxime group. Reactions with the addition of bases are in progress in our laboratories, and—if successful—they will be submitted in the future. A variety of CdX2/dpkoxH (X = Cl, Br, and I) reaction systems using different reagent molar ratios, solvent media, temperatures, and crystallization techniques were systematically employed before arriving at the optimized preparative conditions described in Section 3. Equations (1)–(3) illustrate the synthesis of the complexes.
The yields were 50–60%. The three complexes were prepared using 1:2 Cd(II)/dpkoxH reaction ratios, i.e., in the presence of an excess of the ligand. Using the stoichiometric 1:1 ratio, the same complexes were again isolated (microanalytical and IR evidence) in comparable yields but with lower crystallinity. In an attempt to prepare 1:2 “CdX2(dpkoxH)2” compounds, we performed reactions with a larger excess of the ligand, i.e., 1:3, but again, compounds 1∙2H2O, 2, and 3 were isolated, suggesting that these complexes are thermodynamically stable under the reaction conditions employed.
2.2. Description of Structures
The structures of 1∙2H2O, 2, and 3 were determined via single-crystal X-ray crystallography. Crystallographic data are listed in Table 1. Selected interatomic distances and angles are given in Table 2. Various structural plots are shown in Figure 2, Figure 3, Figure 4 and Figure 5 and Figures S1–S5. Further details can be found in the deposited CIF files.

Table 1.
Crystallographic data and refinement parameters for compounds 1∙2H2O, 2, and 3.
Table 2.
Selected bond lengths (Å) and angles (°) for the polymeric complexes 1∙2H2O, 2, and 3.
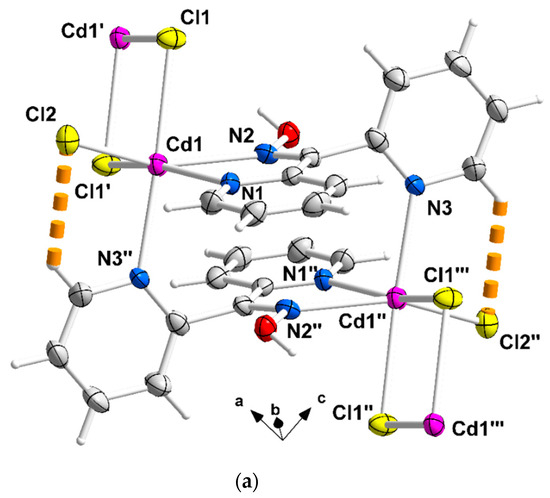
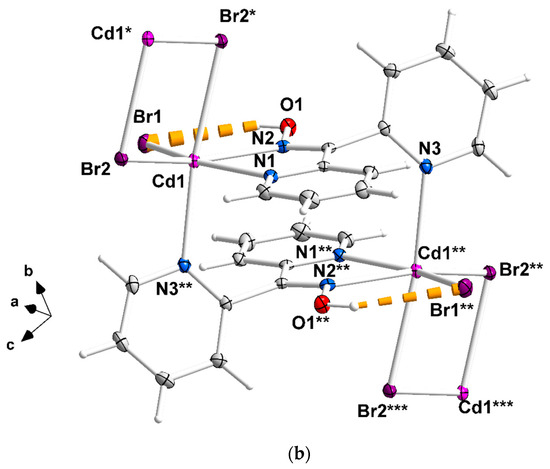
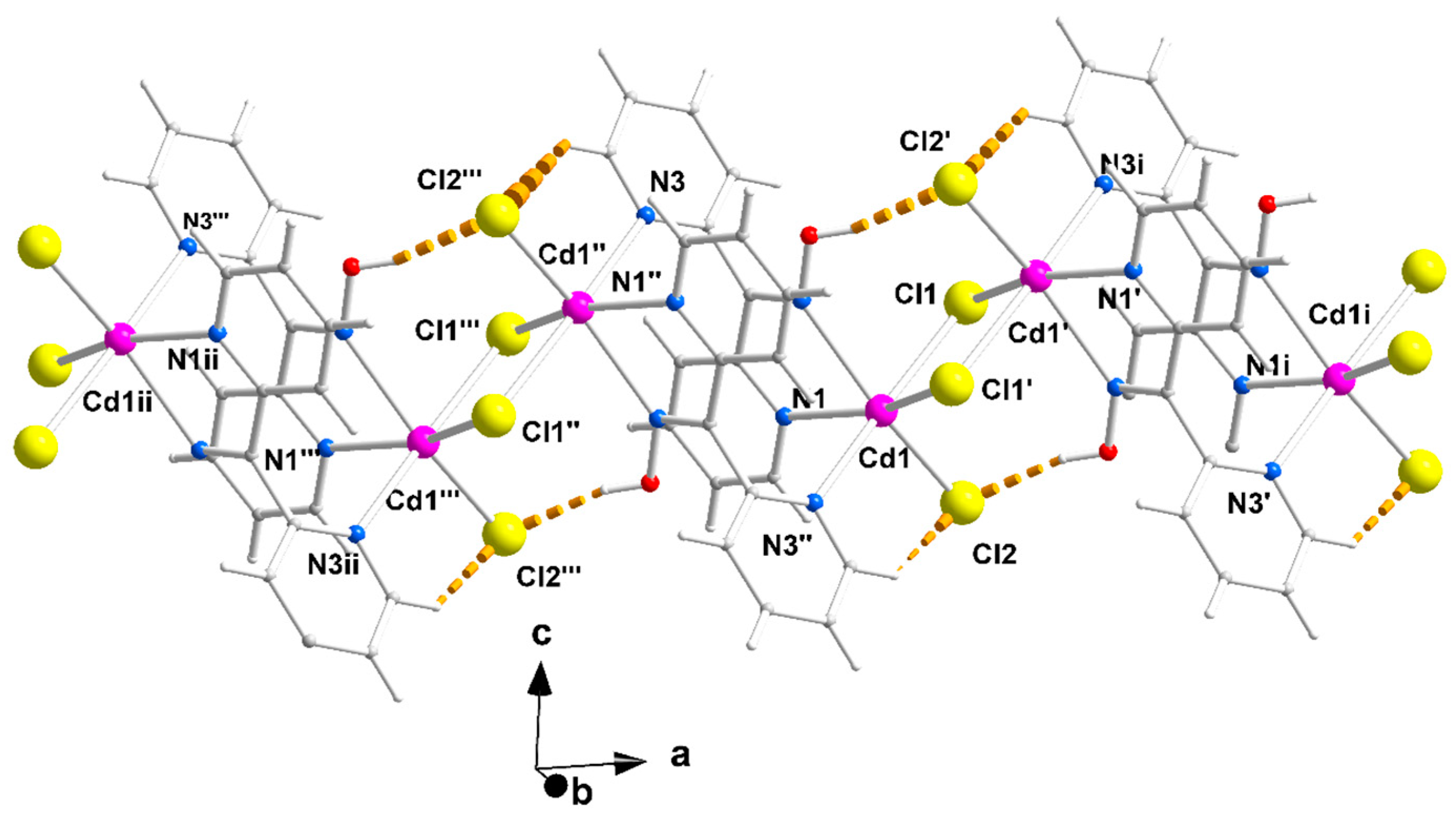
Figure 2.
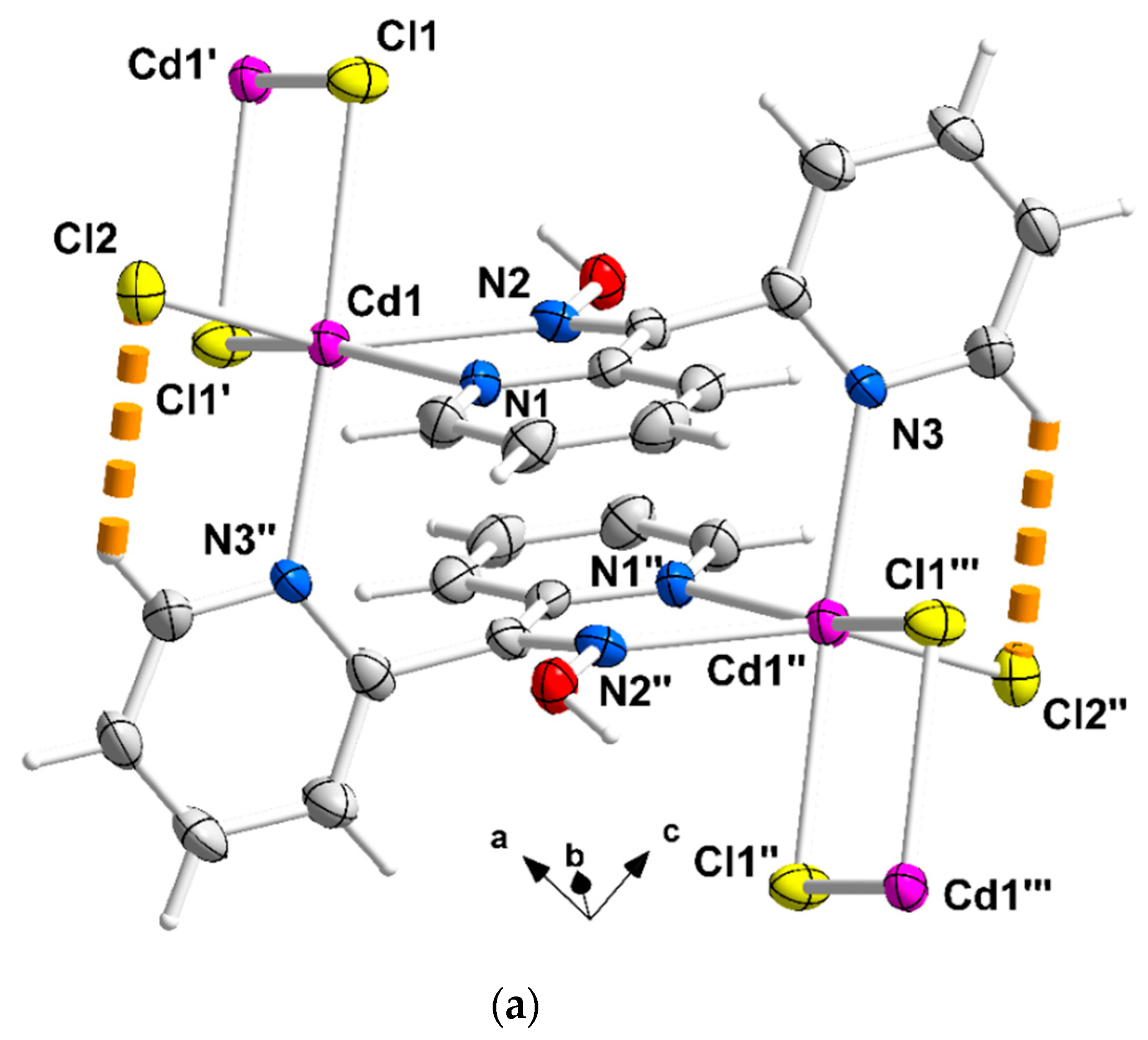
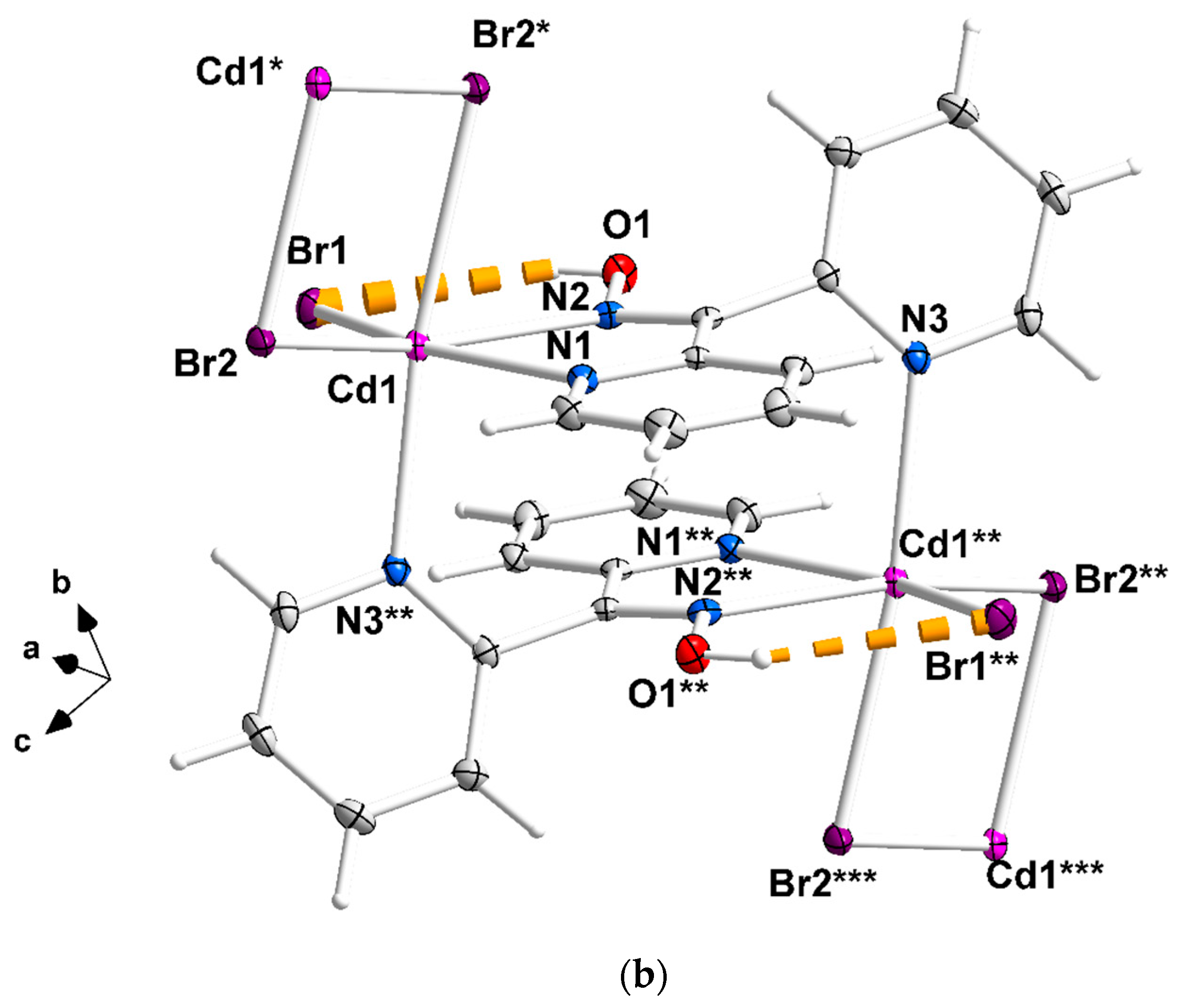
Complete coordination spheres of two neighboring CdII centers in the zigzag chains of complex 1∙2H2O (a) and 2 (b). Symmetry codes: (′) 2 − x, 2 − y, −z; (″) 1 − x, 2 − y, −z; (‴) −1 + x, y, z for complex 1∙2H2O, and (*) 1.5 − x, 1.5 − y, 1 − z; (**) 1 − x, 1 − y, 1 − z); (***) −0.5 + x, −0.5 + y, z for compound 2. The thick dashed orange lines represent C-H∙∙∙Clterminal (a) and Ooxime-H∙∙∙Brterminal (b) H bonds.
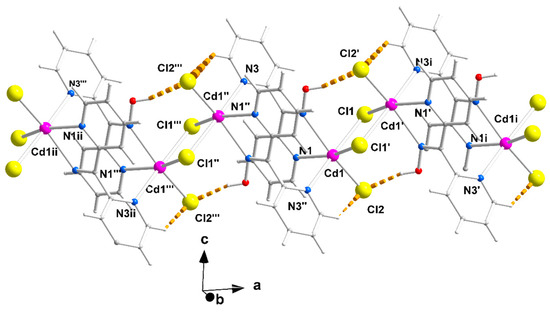
Figure 3.
A portion of one zigzag chain (developed along the α axis) of complex 1∙2H2O; the lattice H2O molecules are not drawn. The thick dashed orange lines represent intrachain C-H∙∙∙Clterminal and Ooxime-H∙∙∙Clterminal H bonds. The symmetry codes (′), (″), and (‴) are the same as those described in Figure 2a, and the additional codes are (i) 1 + x, y, z and (ii) −x, 2 − y, z.
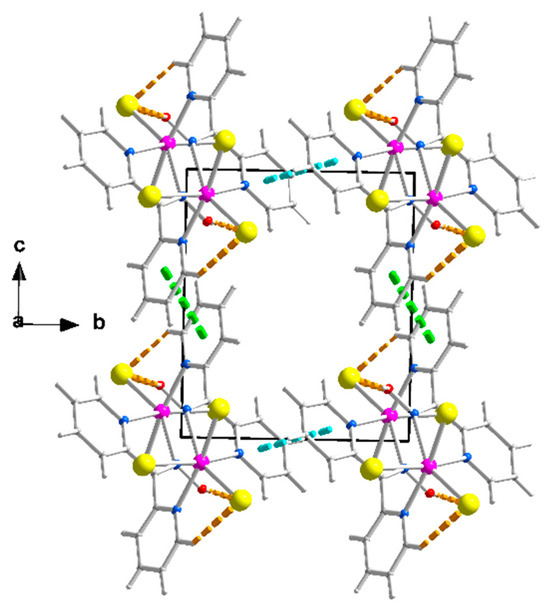
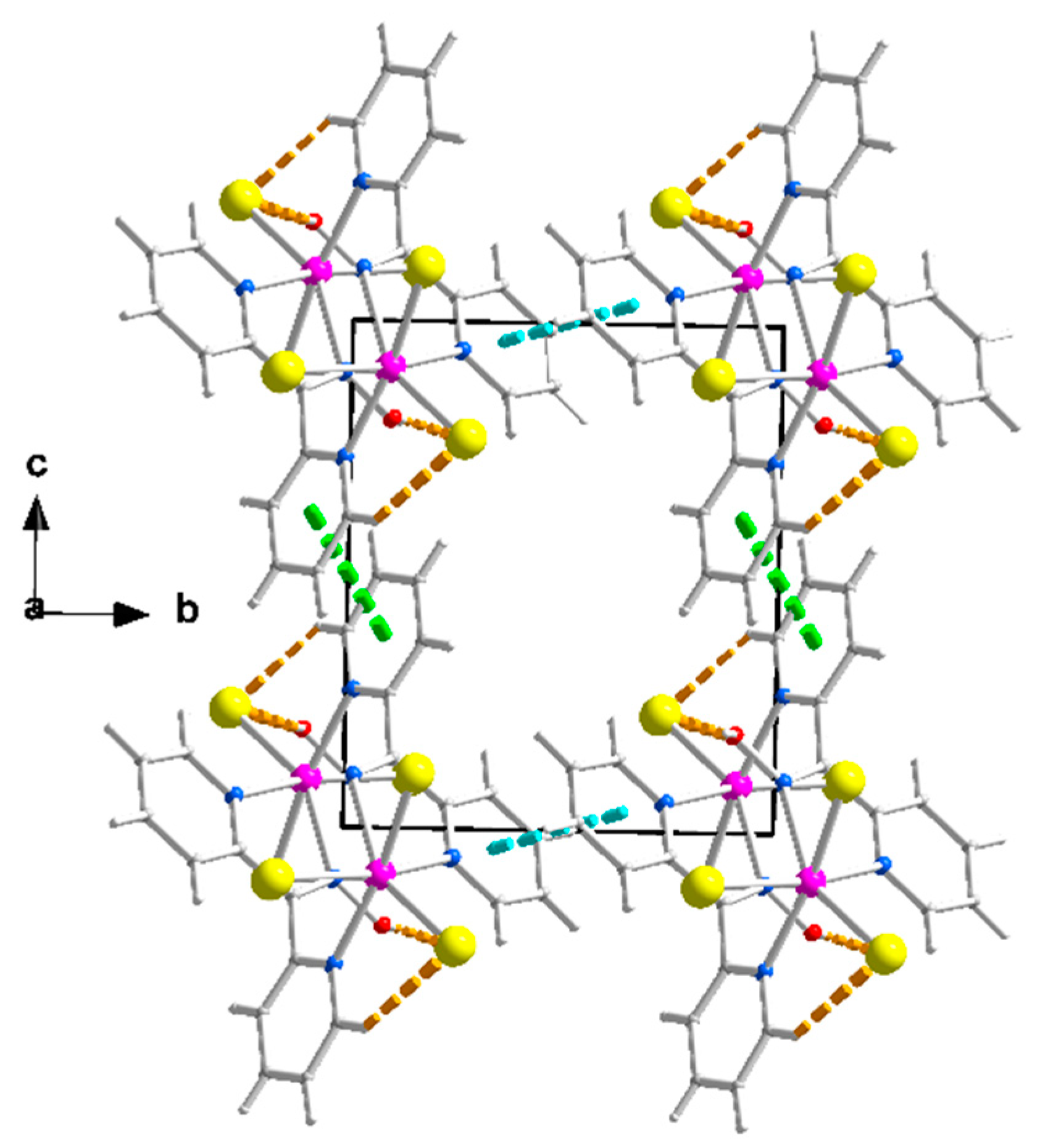
Figure 4.
Packing of chains in the crystal structure of complex 1∙2H2O. The π-π stacking interactions between the rings N1, C1, …, C5 and N1, C1, …, C5 (1 − x, 3 − y, −z), and between the rings N3, C7, …, C11 and N3, C7, …, C11 (1 − x, 2 − y, 1 − z) are indicated with thick cyan and light green lines, respectively. The thick dashed orange lines represent the intrachain H bonds; see Figure 3.
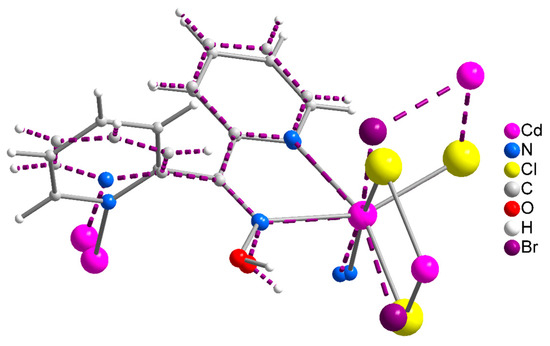
Figure 5.
A very small portion of the chains of complexes 1 and 2 in the overlay mode. The solid lines correspond to 1, and the dashed lines to 2. The color scheme of the atoms involved is shown on the right.
Compound 1∙2H2O crystallizes in the centrosymmetric space group Pī. Complexes 2 and 3 are isomorphous, both crystallizing in the centrosymmetric space group C2/c; thus, only the crystal structure of 2 will be described in detail. The structures consist of 1D zigzag chains. In compound 1∙2H2O, the chains extend along the α axis, while in complexes 2 and 3, along the [1, 1, 0] direction. The zigzag motif is proven by the angle which is formed by three neighboring metal ions (CdII∙∙∙CdII∙∙∙CdII), which is 115.7(1)° for 1∙2H2O, 133.3(1)° for 2 and 130.8(1)° for 3. Neighboring CdII atoms are alternately doubly bridged by halogeno and dpkoxH ligands which adopt the η1:η1:η1:μ (or 2.0111 using Harris notation [31]) coordination mode (Figure 6; see below). In each dimeric subunit, there is a crystallographically imposed inversion center. A terminal halogeno ligand completes 6-coordination at each metal ion. Thus, the coordination sphere of each CdII center is {CdII(η1 − X)(μ − X)2(Npyridyl)2(Noxime)}.
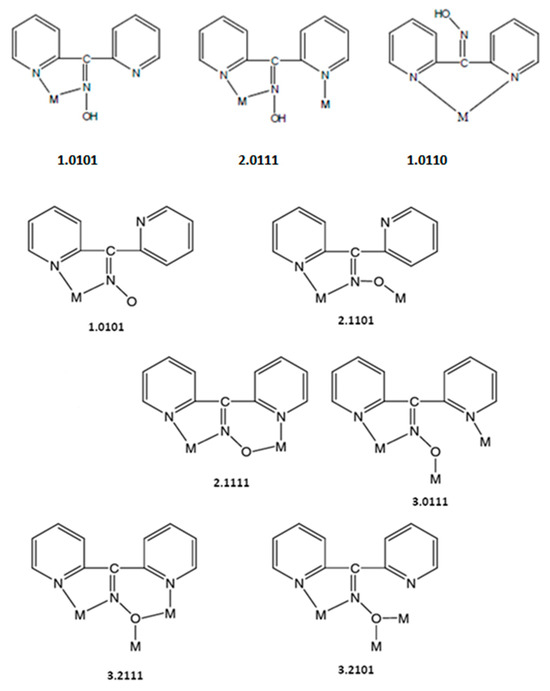
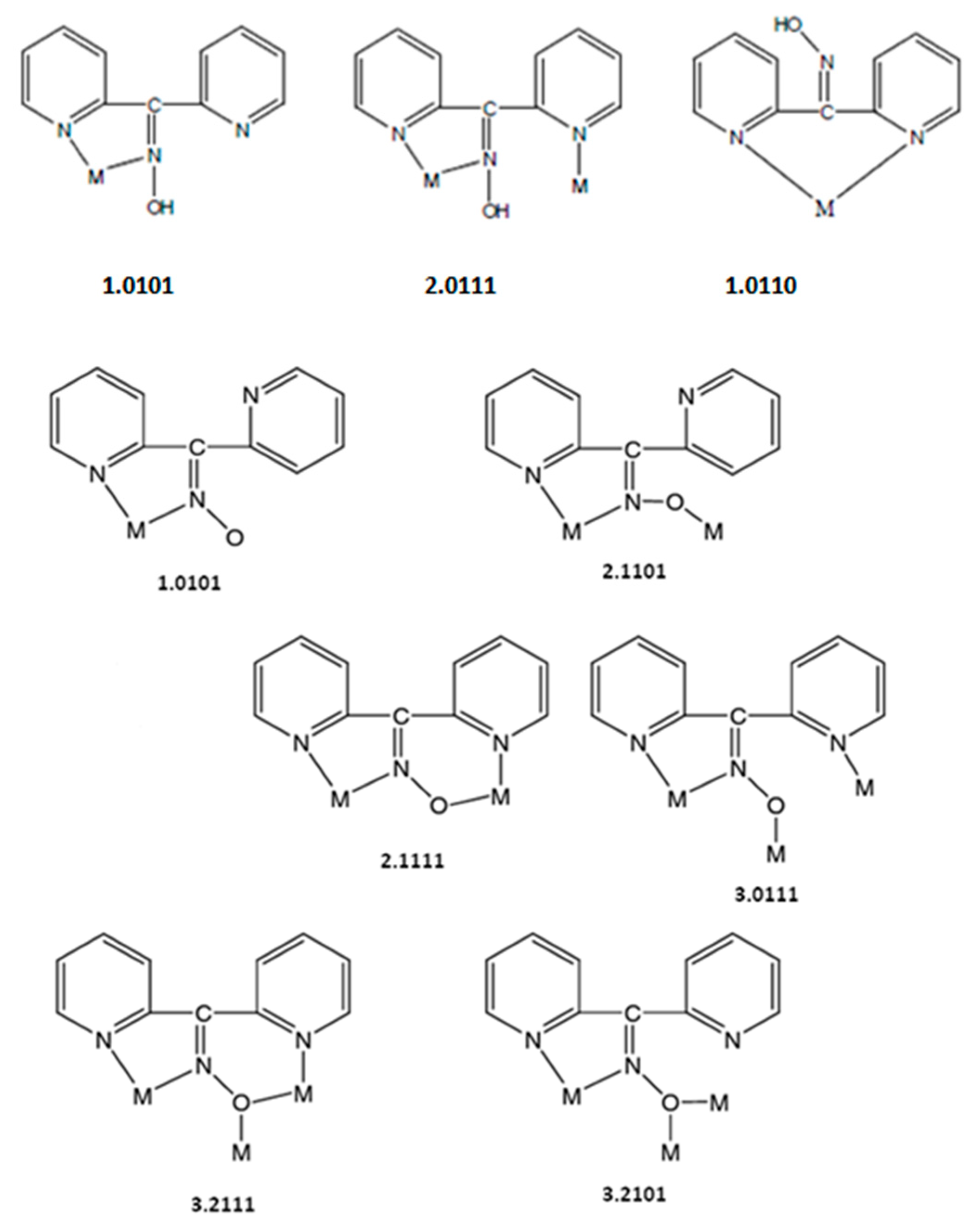
Figure 6.
The to-date crystallographically confirmed coordination modes of dpkoxH and dpkox−, and the Harris notation that describes these modes; the ligation mode observed in 1∙2H2O, 2, and 3 is shown in the middle of the upper row.
The halogeno bridges are asymmetric, see Table 2; the {Cd(μ − X)2Cd} subunit is strictly planar due to symmetry. The CdII∙∙∙CdII distances between the halogeno-bridged metal ions and the CdII-X-CdII angles are, respectively, 3.859(1) Å and 94.1(1)° for 1∙2H2O, 3.972(1) Å and 90.3(1)° for 2, and 4.281(1) Å and 88.7(1)° for 3. The CdII∙∙∙CdII distance becomes larger as the size of the halogeno group increases (I− > Br− > Cl−). On the contrary, the distances between the dpkoxH-bridged CdII centers are practically the same (~6.0 Å) in the three complexes due to the similar type of bridging ligation. For a given complex, the CdII-Xterminal bond length is shorter than the CdII-Xbridging bond lengths, as expected. For example, the Cd1-Br1 bond length in 2 is 2.607(1) Å, while the Cd1-Br2 and Cd1-Br2* [1.5 − x, 1.5 − y, 1 − z] lengths are 2.643(1) and 2.950(1) Å, respectively (Figure 2b, Table 2). The two CdII-Npyridyl bond lengths in each compound differ significantly; the bond that is part of the 5-membered chelating ring is stronger than the bond to the nitrogen of the pyridyl ring that does not participate in chelation. For example, the Cd1-N1 and Cd1-N3** [1 − x, 1 − y, 1 − z] bond lengths in 3 are 2.375(6) and 2.503(7) Å, respectively (Figure S1, Table 2). The CdII-Npyridyl and CdII-Noxime bond lengths are typical for 6-coordinate metal cadmium(II) centers in 2-pyridyl oxime complexes [16,22,23,24,25,26,27].
The coordination geometry of the metal ion in the three complexes is distorted octahedral. The trans-donor–atom pairs are Clterminal/Noxime and two Clbridging/Npyridyl in 1∙2H2O; on the contrary, the trans donor–atom pairs are Xterminal/Npyridyl, Xbridging/Noxime and Xbridging/Npyridyl (X = Br, I) in the isomorphous complexes 2 and 3. The trans angles are in the ranges 159.9(2)°–171.7(2)°, 155.2(1)–171.0(1)° and 155.6(2)–169.5(2)° in 1∙2H2O, 2 and 3, respectively. The distortion from the regular octahedral geometry arises primarily from the small bite angle of the 5-membered chelating “part” of the bridging dpkoxH ligand; the N1(pyridyl)-CdII-N2(oxime) values are considerably less than the ideal value of 90°, i.e., 67.9(2)° for 1∙2H2O and 2, and 67.1(2)° for 3.
There are two types of intrachain H-bonding interactions in the crystal structure of 1∙2H2O. The first type is shown in Figure 2a, while both types are illustrated in Figure 3. In the first type, the donor is the carbon atom (C11) next to the pyridyl nitrogen atom N3 and the acceptor the terminal chloro group (Cl2); its dimensions are C11∙∙∙Cl2″ 3.54(1) Å, H11∙∙∙Cl2″ 2.803(2) Å and C11-H11∙∙∙Cl2″ 135.3(5)° [(″) 1 − x, 2 − y, −z]. In the second type, the donor is the oxime oxygen atom and the acceptor again the terminal chloro group; its dimensions are O1∙∙∙Cl2′ 3.106(5) Å, H(O1)∙∙∙Cl2′ 2.315(2) Å and O1-H(O1)∙∙∙Cl2′ 157.1(4)° [(′) 2 − x, 2 − y, −z]. The second type of intrachain H bond is also present in the structures of the isomorphous complexes 2 (Figure 2b and Figure S2) and 3 (Figure S1). The dimensions are Ooxime∙∙∙Brterminal 3.408(1) Å, H∙∙∙Brterminal 2.699(1) Å, Ooxime-H∙∙∙Brterminal 143.1(1)° and Ooxime-H∙∙∙Iterminal 3.581(5) Å, H∙∙∙Iterminal 2.945(1) Å, and Ooxime-H∙∙∙Iterminal 136.6(4)°.
The packing of the chains in the structure of 1∙2H2O is illustrated in Figure 4. The π-π stacking interactions between similar pyridyl rings of neighboring chains along the crystallographic axes b and c result in the formation of voids around the middle (0, 0.5, 0.5) of the base of the cell where the lattice H2O molecules are hosted. The 3D architecture of the structure is built through the π-π overlap of centrosymmetrically related pyridyl rings N1, C1, …, C5/N1, C1, …, C5 (1 − x, 3 − y, −z) and N3, C7, …, C11/N3, C7, …, C11 (1 − x, 2 − y, 1 − z) along b and c, respectively. The planes of the first pair of rings are at a distance of 3.41(2) Å, while those of the second one are at a distance of 3.38(2) Å.
The supramolecular characteristics of 2 and 3 are similar due to their isomorphism but different compared to those of 1∙2H2O. Figure S3 illustrates the interactions between neighboring chains for compound 2. Neighboring chains are linked via pairs of C2-H2∙∙∙Cg1 interactions; C2 is an aromatic carbon of the N1-containing pyridyl ring, and Cg1 is the centroid of the N1, C1, …, C5 (0.5 + x, −0.5 + y, z) ring. Relevant dimensions are H2∙∙∙Cg1 2.731(1) Å, C2∙∙∙Cg1 3.592(2) Å and C2-H2∙∙∙Cg1 151.0(1) °. They also interact via pairs of π-π interactions between the N1, C1, …, C5 and N3, C7, …, C11 (1 − x, y, 0.5 − z) rings; the centroid–centroid distance is 4.392(1) Å and the angle that the planes form is 15.2(1)°. The former interactions among chains extending along the [1, 1, 0] direction result in the formation of layers parallel to the (001) plane (Figure S4), and the latter contributes to the 3D architecture of the structure. The layers are stacked along the c axis in a crossed-arrangement fashion relative to each other due to the 2-fold axis symmetry that relates the chains that belong to neighboring layers (Figure S5a). The chains in the top layer shown in Figure S5a are oriented along the [−1, 1, 0] direction. Layers of chains along the [1, 1, 0] or [−1, 1, 0] directions are stacked alternately along the c axis (Figure S5b). As mentioned above, the 3D architecture of 3 is built via the same types of interactions. Geometric characteristics are H2∙∙∙Cg1 2.874(1) Å, C2∙∙∙Cg1 3.75(1) Å and C2-H2∙∙∙Cg1 154.1(6)°; in the pair of π-π interactions between the N1, C1, …, C5 and N3, C7, …, C11 (1 − x, y, 0.5 − z) rings, the centroid–centroid distance is 4.683(1) Å, and the angle between the planes is 15.8(2)°.
Concluding the structural part of this work, we already described the different supramolecular structures of 1∙2H2O and 2, 3. However, careful inspection reveals that there is also a difference in the molecular conformation, despite the same formulae, the same coordination mode of dpkoxH, the similar coordination environment of the CdII centers, and the zigzag character of the chains; this difference is illustrated in the overlay diagram of Figure 5. The difference is also reflected (as reported earlier) in the CdII∙∙∙CdII∙∙∙CdII angles, which are smaller in 1∙2H2O (115.7°) than in 2 and 3 (133.3° for 2 and 130.8° for 3). We tentatively assign the differences in the molecular and supramolecular characteristics of the structures to the size of the halogeno ligands and the different nature of the pairs of the trans donor atoms between 1∙2H2O and 2 and 3 (see above); the lattice H2O molecules in the chloro complex might also play a role, probably being engaged in H-bonding interactions.
Compounds 1∙2H2O, 2, and 3 are the first structurally characterized cadmium(II) complexes with the neutral or/and anionic forms of di-2-pyridyl ketone oxime as ligands. The observed 2.0111 (or η1:η1:η1:μ) coordination mode is very rare in the coordination mode of neutral dpkoxH and has been confirmed previously in three complexes (Table 3); the previously reported complexes [32,33,34] are dinuclear and, thus, the present compounds are the first characterized polymeric complexes with any metal that contain dpkoxH with 2.0111 ligations. Since many metal complexes of dpkoxH or/and dpkox− are now known, we feel it is timely to collect the to-date crystallographically confirmed coordination modes of dpkoxH and dpkox−; these modes are illustrated in Figure 6.
Table 3.
The to-date structurally characterized metal complexes of neutral dpkoxH with the 2.0111 a coordination mode b.
We finalize this structural part by attempting to compare the present CdX2/dpkoxH complexes with their analogs of the other group 12 metal ions. The complexes [ZnCl2(dpkoxH)] (three polymorphs) [35,36] and [ZnBr2(dpkoxH)] [35] were characterized via single-crystal X-ray crystallography. The complexes consist of mononuclear (pseudo)tetrahedral molecules in which dpkoxH behaves as a Npyridyl, N’pyridyl-bidentate chelating ligand (1.0110 in Figure 6). This fact reflects the often-different coordination geometry between analogous Zn(II) and Cd(II) complexes. Comparison of 1∙2H2O, 2, and 3 with analogous Hg(II) complexes is not possible because HgX2/dpkoxH complexes are not known.
2.3. Spectroscopic Characterization in Brief
The complexes were characterized in the solid state using Raman and IR spectroscopies and in solution via NMR (1H, 13C, and 113Cd) techniques. To aid the solution studies, the molar conductivity values were also determined. Representative spectra are shown in Figure 7, Figure 8, Figure 9, Figure 10 and Figure 11 and Figures S6–S8. It should be mentioned at the outset that most bands are not expected to be pure vibrations, and thus, the assignments represent approximate descriptions of the vibrational modes.
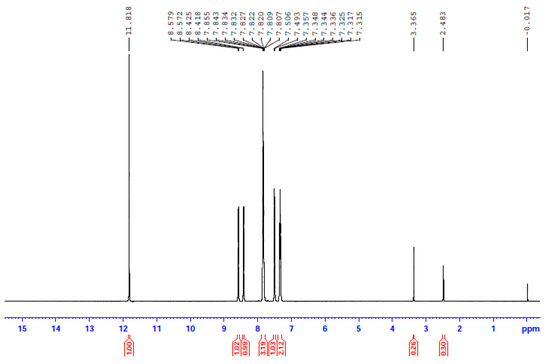
Figure 7.
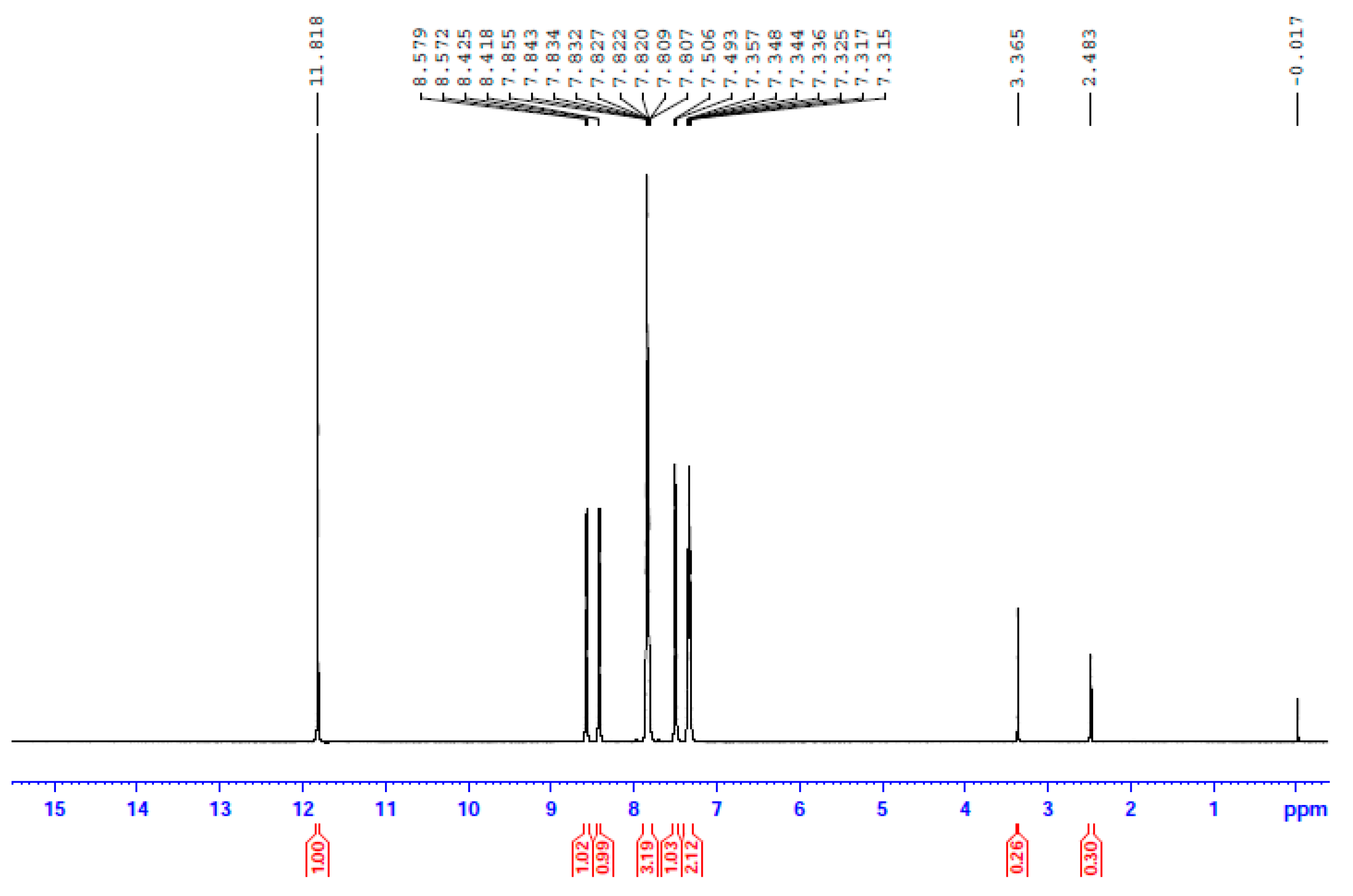
The 1H NMR spectrum of 3 in d6-DMSO. The signal at δ 2.48 ppm is due to the methyl groups of the non-deuterated amount of the solvent, and the signal at δ 3.37 ppm to the protons of the H2O content of the solvent.
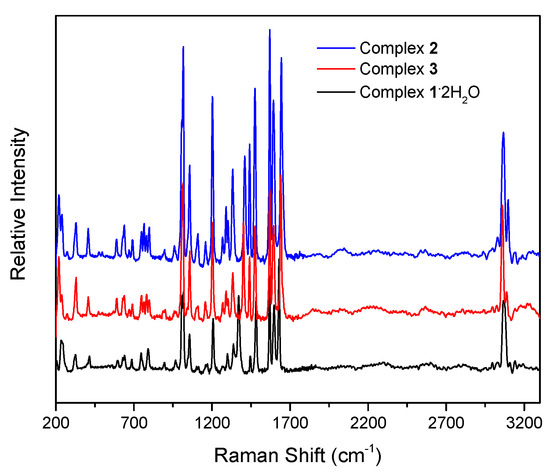
Figure 8.
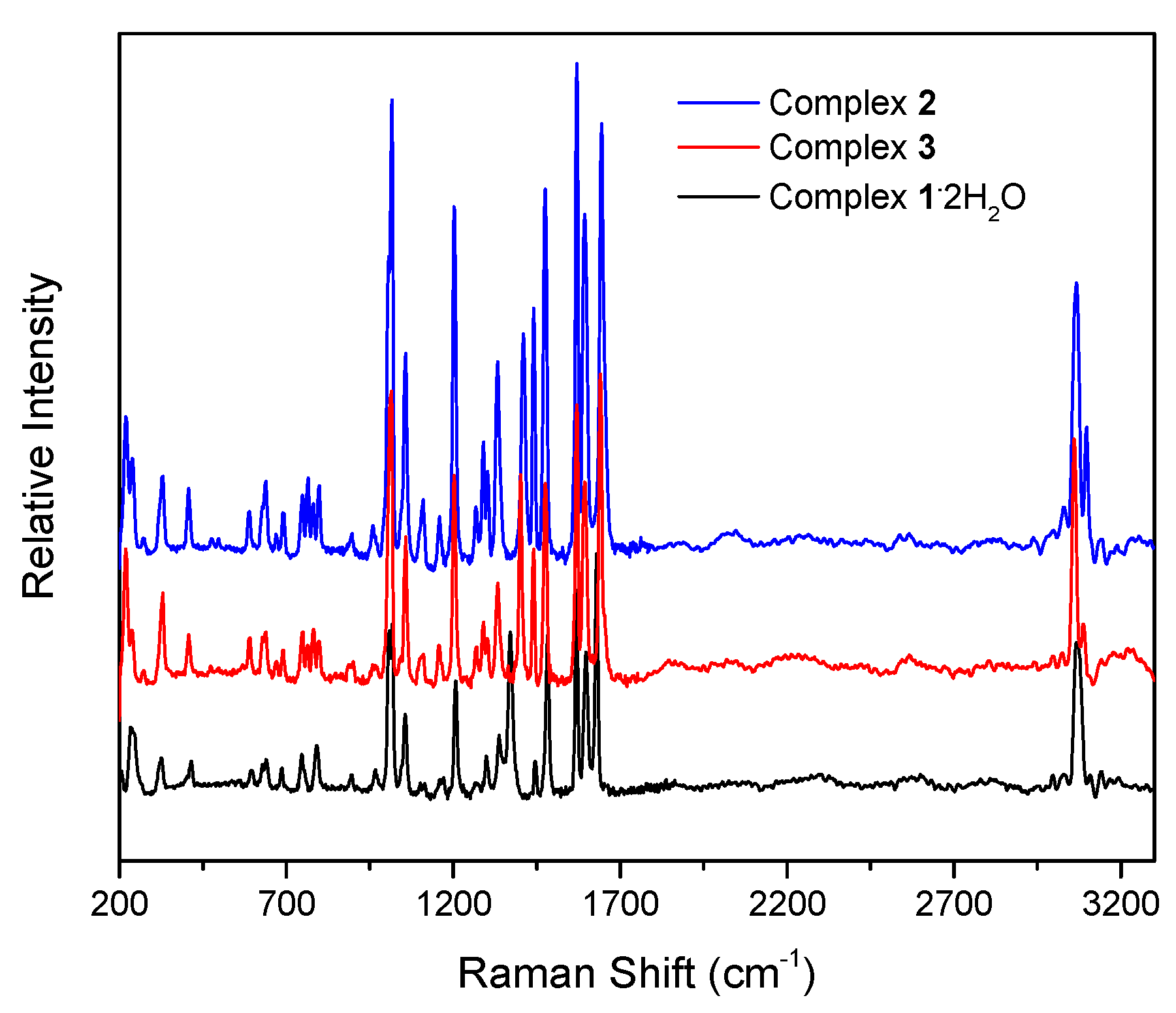
The Raman spectra of complexes 1∙2H2O (black), 2 (blue), and 3 (red).
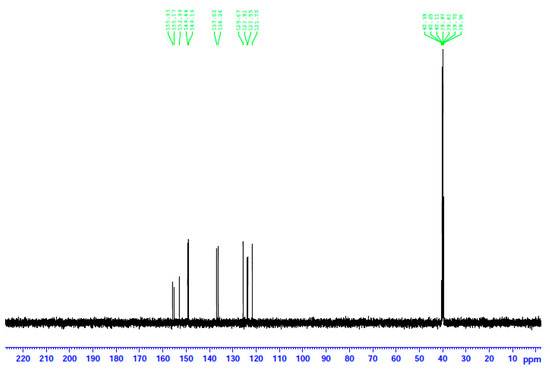
Figure 9.
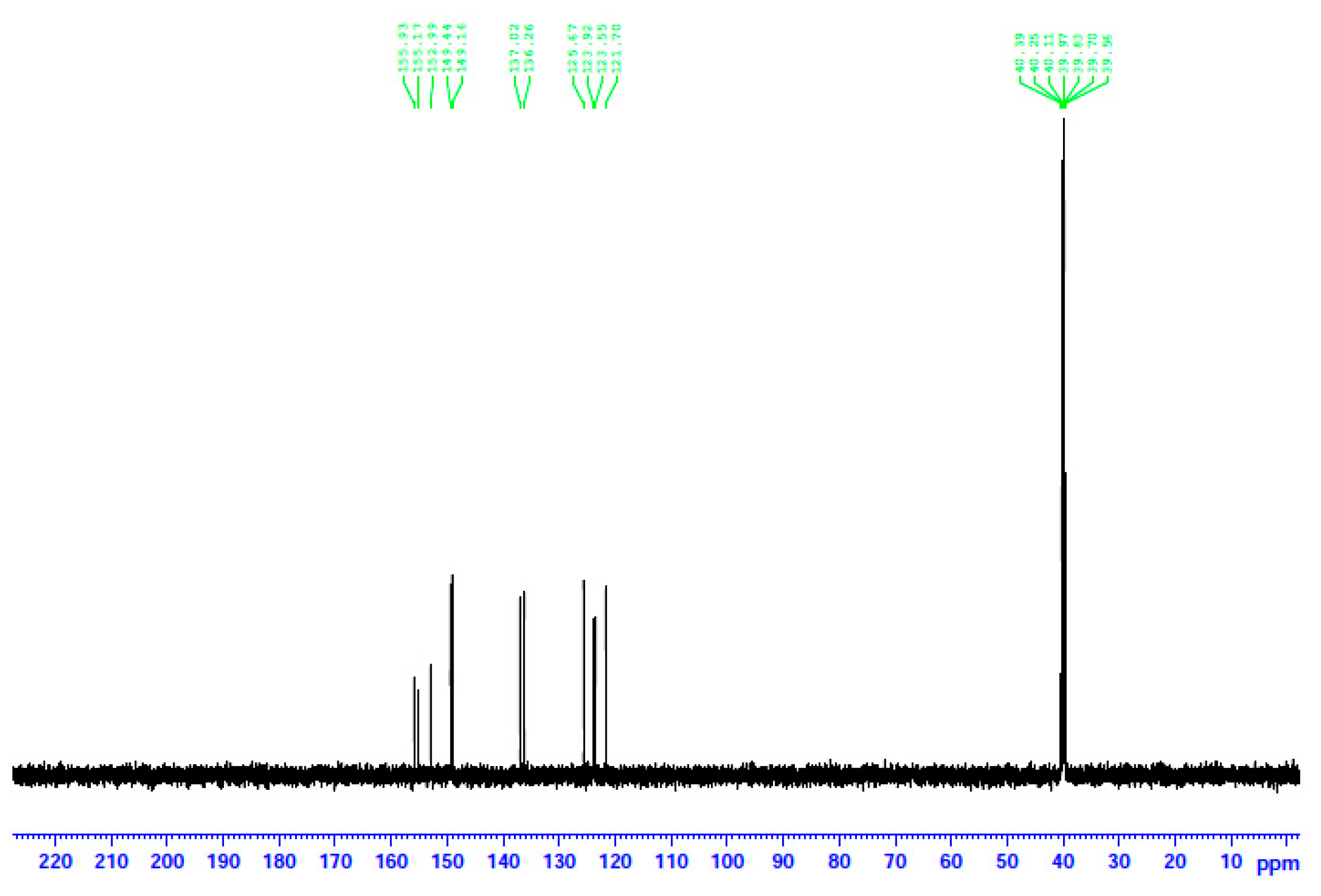
The 13C NMR spectrum of 2 in d6-DMSO. The signals at δ ~40 ppm are due to the methyl carbon atoms of the solvent.
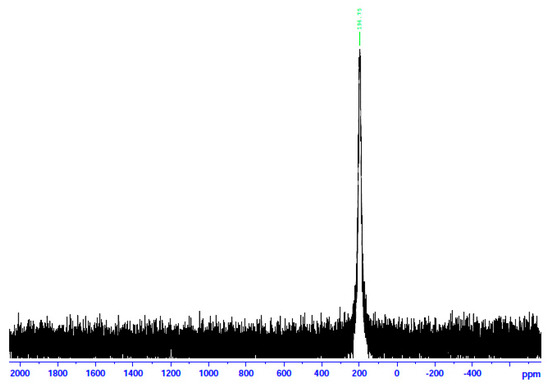
Figure 10.
The 113Cd NMR spectrum of 1∙2H2O in d6-DMSO.
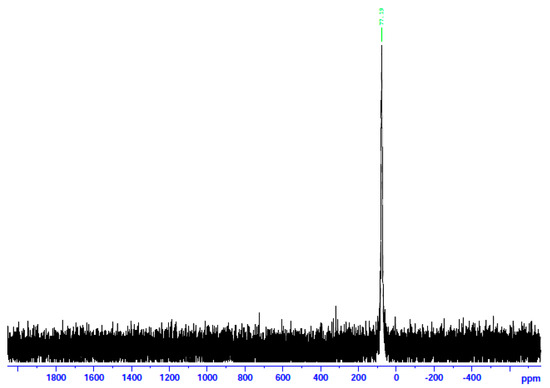
Figure 11.
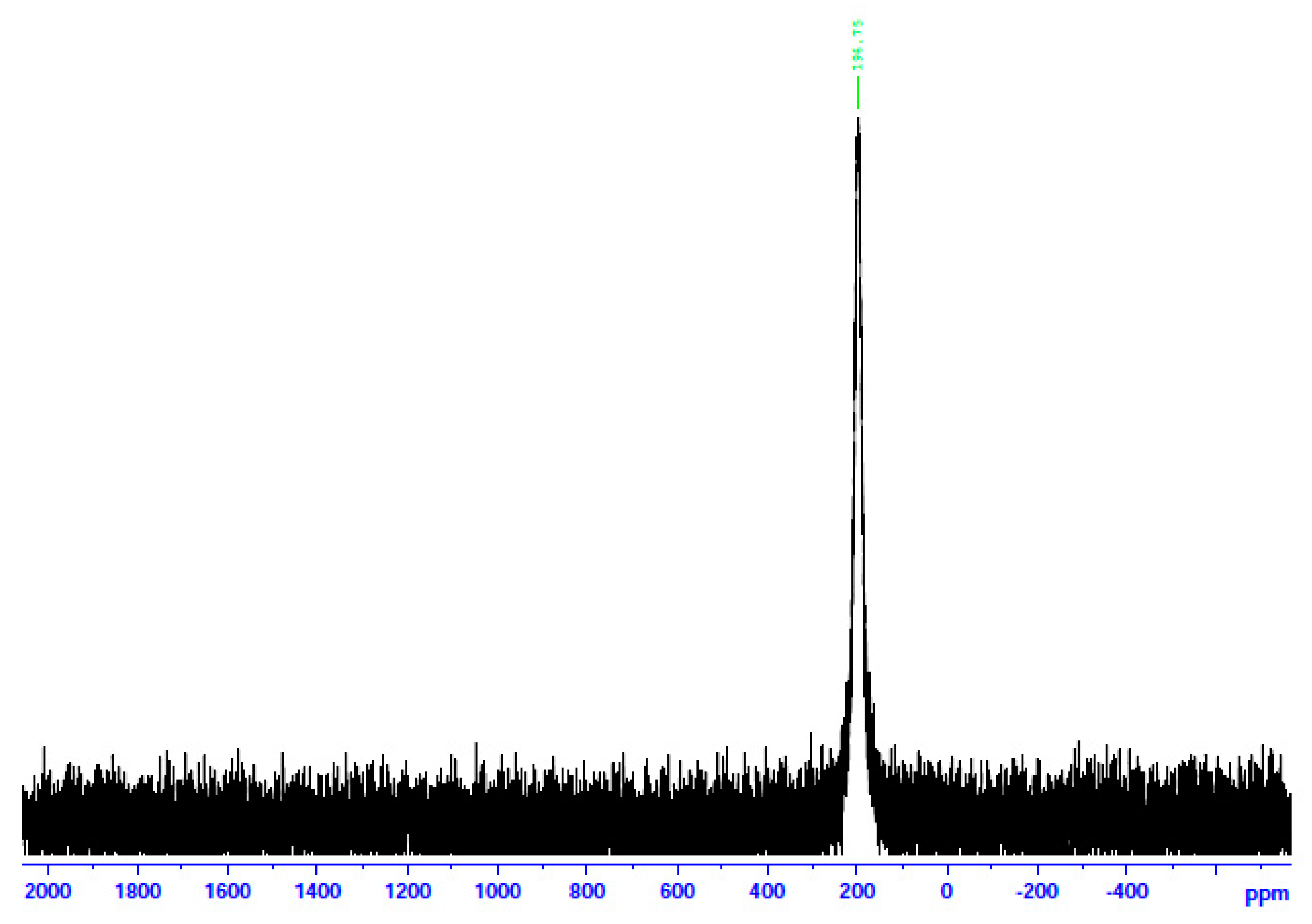
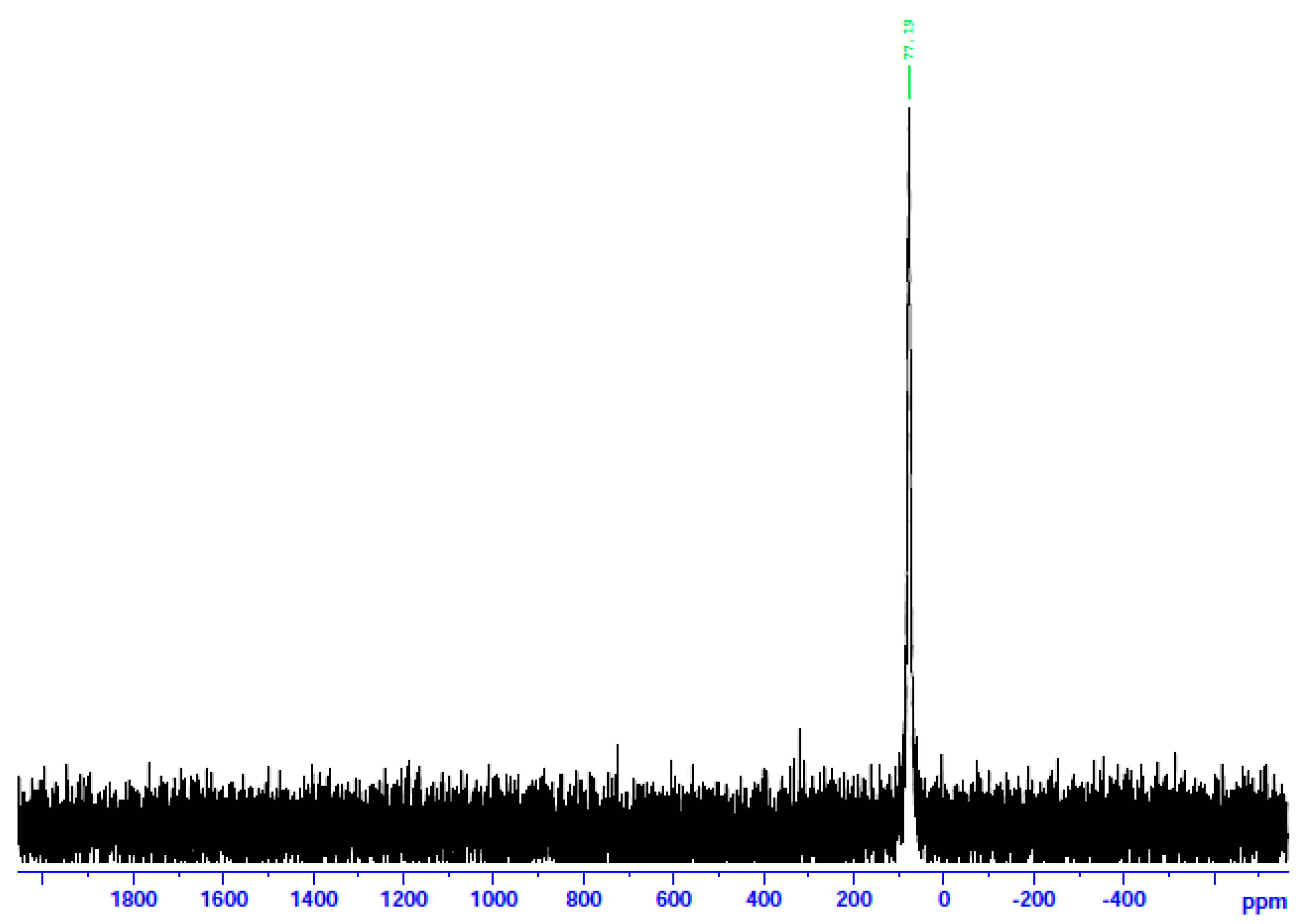
The 113Cd NMR spectrum of 3 in d6-DMSO.
The IR spectra of the complexes do not exhibit bands that are present in the free dpkoxH ligand, suggesting their purity. The presence of the neutral oxime group in 1∙2H2O, 2, and 3 by a broad band at ~3350 cm−1 assigned to ν(OH)oxime [16]; the broadness of this band is indicative of hydrogen bonding, which has also been observed by crystallography. In the spectrum of 1∙2H2O, the medium-intensity band at ~3430 cm−1 is attributed to the ν(OH) vibration of the lattice H2O [30]; its rather high wavenumber clearly reflects the non-coordinating nature of the water molecules. As expected, the O-H vibrations are hardly seen in the Raman spectra of the compounds. The strong IR band at 1636 cm−1 and the intense Raman peak at 1652 cm−1 in the spectra of free dpkoxH are assigned to the ν(C=N)oxime vibration [36]. This band/peak in the spectra of the complexes is located at lower wavenumbers, suggesting oxime-N coordination [16]. The shift is rather small in the Raman spectra (the peaks appear at 1628–1644 cm−1) but large in the IR spectra where the strong band appears at 1595–1590 cm−1, overlapping with an aromatic stretch of the 2-pyridyl groups [22]. The same trend is also evident in the ν(NO)oxime mode, which appears at ~1015 (IR) and at approximately the same wavenumber (Raman) in the spectra of the complexes [37], lower than the wavenumber of this mode in the free ligand. The medium-intensity peaks in the Raman spectra at 3067 (1∙2H2O), 3066 (2), and 3060 (3) cm−1 are assigned to the stretching vibration of the aromatic carbon–hydrogen bonds, ν(CH) [38]; the corresponding IR band is weak and appears at 3060 cm−1 [36]. Exact assignments of the other 2-pyridyl modes are not an easy task because the spectra are complicated, and thus, any discussion about coordination shifts might be risky. The Raman peaks in the 415–210 cm−1 region would be associated with the Cd-X (X = Cl, Br, and I), Cd-Noxime, and Cd-Npyridyl stretching vibrations [38,39]. No X−-dependent peak was noticed, and the ν(CdX)t and ν(CdX)b modes (t = terminal; b = bridging) are most probably located below 210 cm−1, which is the low-frequency limit of the experiments.
In an attempt to probe the solution behavior of 1∙2H2O, 2, and 3, the NMR spectra (1H, 13C, 113Cd) of the complexes in d6-DMSO were recorded at room temperature. The 1H and 13C NMR spectra of the three complexes are identical; however, the most remarkable feature is that the three spectra are also identical to the spectrum of free dpkoxH in the same solvent. This is evidence that the three complexes decompose in d6-DMSO, releasing dpkoxH, a fact attributable to the strong donor capacity of the solvent. The spectra show a singlet signal at δ 11.82 ppm assigned to the hydroxyl proton [16,30,36,40] and two doublets at δ 8.58 and 8.43 ppm attributed to the two non-equivalent H atoms of the aromatic carbons adjacent to the ring-N atoms [30,36]; the integration ratio of the three signals is 1:1:1, as expected. The six remaining aromatic protons appear as two multiple signals at δ 7.84 and 7.37 ppm and one doublet at δ 7.50 ppm with an integration ratio of 3:2:1, respectively.
The decomposition of the complexes in DMSO is corroborated by the 13C NMR spectra. The spectra of the three complexes and the free dpkoxH compound are identical. This means that the coordinated dpkoxH is released in solution, i.e., the solution Cd(II) species do not contain the oxime ligand. Thus, the 13C NMR spectra of the decomposition products are identical to those of the “free”, i.e., uncoordinated, dpkoxH compound. The spectra exhibit the 11 signals expected for the carbon atoms of the organic molecule in the δ 121.7–155.9 ppm region [36,41,42,43,44]. The signal at δ 155.2 ppm is due to the carbon atom of the oxime group [36]; exact assignments of the pyridyl 13C resonances would be risky.
In an attempt to probe the solution Cd(II) species in more detail, we recorded the 113Cd NMR spectra of the complexes in d6-DMSO, combined with molar conductivity values (25 °C, 10−3 M) in DMSO. 113Cd NMR spectroscopy is a diagnostic tool for Cd(II) species in solution. The reasons are the nuclear spin I of ½ and its relatively good receptivity (R = 7.59 relative to 13C (R = 1)) [45]. It should be mentioned at this point that there is a danger of oversimplification of the chemical shift data. This is because chemical exchange often occurs between different complexation or binding sites (including ligand and solvent) for Cd(II), and the observed line is a time average of the different chemical shifts from these different sites [45,46]. The ΛΜ values of 1∙2H2O and 2 are 71 and 66 S cm2 mol−1, respectively, suggesting 1:2 electrolytes [47]. This is strong evidence that chlorides and bromides do not participate in coordination with Cd(II) in solution. On the contrary, the ΛΜ value of 3 is 5 S cm2 mol−1, indicating a negligible ionization in solution, i.e., a non-electrolyte behavior [47]. This fact suggests that the iodides are bonded to Cd(II) in solution. In analogy, the 13Cd NMR spectra of 1∙2H2O and 2 in d6-DMSO are identical and different from the corresponding spectrum of 3. The spectra of the two former complexes consist of a single resonance at δ 196.7 ppm, while the spectrum of the iodo complex displays again one signal at a completely different δ value (77.2 ppm). The appearance of one signal indicates that there is one Cd(II) species in solution for every complex. Moreover, this spectral behavior shows that the species of 1∙2H2O and 2 are identical and different from the species arising from 3. The δ values indicate Cd(II) species in a predominantly O-environment [48]. The δ values of 77.2 ppm for complex 3 indicate the coordination of iodo ligands to CdII [45] since values of +43 and +101 ppm have been recorded in Cd(II) iodo complexes in solution. Taken together, the above molar conductivity and spectral data suggest that the decomposition of the complex most probably occurs via the processes represented by Equations (4)–(6), where DMSO is O-bonded.
The different solution behavior can be attributed to the nature of the halogeno ligand. The CdII-Cl and CdII-Br coordination bonds have a higher polar contribution than the CdII-I bonds, which should be more covalent. Thus, the chloro and bromo 1D polymers release the halogeno ligand from the coordination sphere of CdII easier than 3, which keeps the iodo groups bonded with the metal ion more tightly. The different coordination behavior of Cl−, Br−, and I− ligands in Cd(II)/DMSO complexes has also been observed and discussed in solid-state structures [49].
Efforts to further study the solution behavior of the complexes via ESI-MS were unsuccessful. The compounds are insoluble in the common ESI-MS solvents (MeCN, MeOH, H2O) but also in DMSO:MeOH (1:9 v/v) mixtures.
3. Experimental Section
3.1. Materials and Instrumentation
All manipulations were performed under aerobic conditions. Deionized water was received from the in-house facility. Solvents and reagents were purchased from Sigma-Aldrich (Tanfrichen, Germany) and Alfa Aesar (Karlsruhe, Germany) and used as received. The purity of dpkoxH was checked by 1H NMR spectroscopy. Safety note: Cd(II) compounds are toxic, and small quantities should be used; the use of gloves is recommended.
Microanalyses (C, H, and N) were performed by the Instrumental Analysis Center of the University of Patras. Conductivity measurements in DMSO were carried out at room temperature (24–26 °C) with a Metrohm-Herisau E-527 bridge and a cell of standard design; the concentration of the solutions was ~10−3 M. FT-IR spectra (4000–400 cm−1) were recorded using a Perkin-Elmer 16PC spectrometer (Waltham, MA, USA); the samples were in the form of KBr pellets. For the Raman measurements, the T64000 Horiba Jobin Yvon micro-Raman setup was used. The excitation wavelength was 514.5 nm emitted from a DPSS laser (Cobolt Fandango TMISO laser, Norfolk, UK). The laser power on the sample was 2.5 mW. The backscattered radiation was collected from a single configuration of the monochromator after passing through an appropriate edge filter (LP02-633RU-25, laser2000 UK Ltd., Huntingdon, Cambridgeshire, UK). The calibration of the instrument was achieved via the standard Raman peak position of Si at 520.5 cm−1. The spectral resolution was 5 cm−1. 1H and 13C NMR spectra in d6-DMSO were recorded on a Bruker Avance DPX spectrometer (Bruker AVANCE, Billerica, MA, USA) at resonance frequencies of 400.13 MHz (1H) and 100.62 MHz (13C); Me4Si was used as an internal standard. 113Cd NMR spectra in d6-DMSO were recorded on a Bruker Ascend (600 MHz) spectrometer; the reference was an aqueous Cd(ClO4)2∙6H2O 0.1 M solution (δ = 0 ppm).
3.2. Preparation of the Complexes
{[CdCl2(dpkoxH)]∙2H2O}n (1∙2H2O): A solution of CdCl2∙2H2O (0.022 g, 0.10 mmol) in MeOH (8 mL) was added to a solution of dpkoxH (0.039 g, 0.20 mmol) in the same solvent (2 mL). The resulting colorless solution was stirred for 10 min and stored in a closed vial at 25 °C. X-ray quality, colorless crystals of the product were precipitated within 48 h. The needle-like crystals were collected via filtration, washed with ice-cold EtOH (1 mL), and Et2O (2 × 1 mL), and dried in air. The yield was 54% (based on the metal ion available). Anal. Calcd. (%) for C11H13CdCl2N3O3: C, 31.56; H, 3.14; N, 10.04. Found (%): C, 31.41; H, 3.63; N, 9.89. IR (KBr, cm−1): 3430m, 3315sb, 3062w, 1620w, 1594m, 1560m, 1534w, 1474m, 1436m, 1408w, 1374m, 1332m, 1292w, 1202w, 1166w, 1064s, 1044s, 1018s, 1006sh, 976w, 788m, 752m, 688w, 676w, 628m, 598w, 570w, 474w, 406w. Raman (cm−1): 3067m, 1628s, 1597m, 1569s, 1483s, 1445w, 1371s, 1337w, 1299w, 1206m, 1056m, 1009s, 790w, 745w, 687w, 628w, 414w, 325w, 233m. 1H NMR (d6-DMSO, δ/ppm): 11.81 (s, 1H), 8.61 (d, 1H, J = 2.9 Hz), 8.47 (d, 1H, J = 2.9 Hz), 7.82 (mt, 3H), 7.53 (d, 1H, J = 5.2 Hz), 7.40 (mt, 2H). 13C NMR (d6-DMSO, δ/ppm): 122.3, 123.9, 124.2, 126.0, 136.7, 137.7, 149.3, 149.6, 152.5, 154.7, 155.2. 113Cd NMR (d6-DMSO, δ/ppm): 196.8. ΛΜ (DMSO, 10−3 M, 25 °C) = 71 S cm2 mol−1.
{[CdBr2(dpkoxH)]}n (2): A solution of CdBr2∙4H2O (0.034 g, 0.10 mmol) in MeCN (25 mL) was added to a solution of dpkoxH (0.040 g, 0.20 mmol) in the same solvent (10 mL). The resulting colorless solution was stirred for 20 min and allowed to slowly evaporate at room temperature. X-ray quality, colorless crystals of the product were obtained within 3 d. The plate-like crystals were collected by filtration, washed with ice-cold EtOH (2 × 1 mL) and Et2O (4 × 2 mL), and dried in air. The yield was 53% (based on the cadmium available). Anal. Calcd. (%) for C11H9CdBr2N3O3: C, 28.02; H, 1.93; N, 8.92. Found (%): C, 27.90; H, 2.15; N, 9.14. IR (KBr, cm−1): 3342mb, 3280sh, 3058w, 1618w, 1590s, 1566m, 1474m, 1438m, 1408w, 1362w, 1330s, 1292w, 1198m, 1158w, 1094w, 1062s, 1042s, 1016s, 1006sh, 966m, 794m, 752m, 742m, 686m, 672m, 628m, 594w, 540wb, 466w, 408m. Raman (cm−1): 3097m, 3066s, 3027w, 1644s, 1593s, 1571s, 1476s, 1441m, 1410m, 1333m, 1303w, 1290w, 1268w, 1202s, 1159w, 1110w, 1058m, 1017s, 1006sh, 799w, 785w, 748w, 689w, 639w, 589w, 407w, 329w, 238w, 219m. 1H NMR (d6-DMSO, δ/ppm): 11.81 (s, 1H), 8.58 (d, 1H, J = 2.8 Hz), 8.43 (d, 1H, J = 2.8 Hz), 7.84 (mt, 3H), 7.50 (d, 1H, J = 5.2 Hz), 7.37 (mt, 2H). 13C NMR (d6-DMSO, δ/ppm): 122.4, 123.8, 124.0, 125.9, 136.8, 137.8, 149.2, 149.5, 152.5, 154.8, 155.3. 113Cd NMR (d6-DMSO, δ/ppm): 196.3. ΛΜ (DMSO, 10−3 M, 25 °C) = 66 S cm2 mol−1.
{[CdI2(dpkoxH)]}n (3): A solution of CdI2 (0.036 g, 0.10 mmol) in MeOH (3 mL) was added to a solution of dpkoxH (0.039 g, 0.20 mmol) in the same solvent (7 mL). The resulting colorless solution was stored in a closed vial for 30 d. During this time, the color of the solution slowly became pale yellow, and after one month (total of 60 d), X-ray quality, colorless plate-like crystals of the product were precipitated. The crystals were collected via filtration, washed with ice-cold EtOH (2 × 1 mL) and Et2O (3 × 2 mL), and dried in air. The yield was 51% (based on the cadmium available). Anal. Calcd. (%) for C11H9CdI2N3O: C, 23.37; H, 1.61; N, 7.43. Found (%): C, 23.87; H, 1.69; N, 7.09. IR (KBr, cm−1): 3355mb, 3065w, 1614w, 1590m, 1560m, 1474m, 1438s, 1398m, 1330w, 1286w, 1198w, 1062sh, 1040s, 1014m, 960w, 899w, 880w, 796m, 776m, 758m, 690m, 668s, 636sh, 626m, 586w, 550m, 496m, 470w, 408m. Raman (cm−1): 3087w, 3060s, 1642s, 1593s, 1571s, 1476s, 1441m, 1402s, 1333m, 1304w, 1290w, 1202s, 1058m, 1015s, 781w, 750w, 693w, 592w, 407w, 330w, 238w, 219m. 1H NMR (d6-DMSO, δ/ppm): 11.82 (s, 1H), 8.58 (d, 1H, J = 2.8 Hz), 8.43 (d, 1H, J = 2.8 Hz), 7.83 (mt, 3H), 7.50 (d, 1H, J = 5.2 Hz), 7.37 (mt, 2H). 13C NMR (d6-DMSO, δ/ppm): 121.9, 123.7, 124.0, 125.8, 136.4, 137.3, 149.2, 149.5, 152.4, 154.9, 155.2. 113Cd NMR (d6-DMSO, δ/ppm): 77.2. ΛΜ (DMSO, 10−3 M, 25 °C) = 5 S cm2 mol−1.
3.3. Single-Crystal X-ray Crystallography
Colorless crystals of 1∙2H2O and 3 were taken directly from the mother liquor and immediately cooled to −103(2) and −93(2) °C, respectively. Diffraction data for these compounds were collected on a Rigaku R-AXIS Image Plate diffractometer (Rigaku Americas Corporation, The Woodlands, TX, USA) using graphite-monochromated Cu Κα radiation. Data collection (ω-scans) and processing (cell refinement, data reduction, and empirical/numerical absorption correction) were performed using the CrystalClear program package [50]. The structures were solved by direct methods using SHELXS, ver. 2013/1 [51] and refined by full-matrix least-squares techniques on F2 with SHELXL, ver. 2014/6 [52]. The H atoms were located by difference maps and refined isotropically. All non-H atoms were refined anisotropically. For the chloro compound, the SQUEEZE procedure [53] was applied, and an estimated number of two lattice H2O molecules per formula unit was derived based on the accessible solvent voids volume or electron counts within the voids volume. The complex has been abbreviated as 1∙2H2O throughout this work. Diffraction data for complex 2 were collected with a Bruker APEX II Quasar diffractometer (Bruker Analytical X-Ray Systems, Madison, WI, USA), equipped with a graphite monochromator on the path of Mo Kα radiation. Crystals, taken directly from the reaction solution, were coated with CargilleTM NHV immersion oil and mounted on a fiber loop, followed by data collection at −153 °C. The program SAINT was used to integrate the data, which were thereafter corrected using SADABS [54]. The structure was solved using SHELXT [55] and refined by the full-matrix least-squares technique on F2 using SHELXL-2018 [52]. All H atoms were located by difference maps and refined isotropically; all non-H atoms were refined with anisotropic displacement parameters. Plots of the structures were drawn using the Diamond 3 program package [56].
Crystallographic data were deposited with the Cambridge Crystallographic Data Centers, Nos 2311945 (1∙2H2O), 2311946 (2), and 2311947 (3). Copies of the data can be obtained free of charge upon application to CCDC, 12 Union Road, Cambridge, CB2 1EZ, UK: Tel.: +(44)-1223-762910; Fax: +(44)-1223-336033; E-mail: deposit@ccdc.cam.ac.uk.
4. Conclusions in Brief and Perspectives
In this work, we reported the employment of dpkoxH in reactions with Cd(II) halides which has resulted in the isolation of three 1D polymeric complexes. Thus, the first Cd(II)/dpkoxH complexes have been prepared and characterized. The most important features of this work are: (a) The reactions lead to 1D zigzag polymers with a 1:1 metal-to-ligand ratio; no monomeric 1:2 complexes could be obtained. (b) The three complexes have interesting molecular and supramolecular structures, with the neutral ligand exhibiting a rare coordination mode. (c) The nature of the halogeno ligand slightly affects the molecular structure and significantly the supramolecular characteristic, with the bromo and iodo complexes being isomorphous. Isostructurality among the non-fluorine halogens is a well-documented phenomenon in organic chemistry [57]. However, there are some limits to this kind of halogen equivalence in supramolecular organic chemistry. In molecular and supramolecular inorganic chemistry, however, non-equivalence is common [58]. This has been attributed to size, electronegativity, and polarization effects. Contrary to the general trend according to which the chloro and bromo ligands play the same role in the crystal structures, whereas the iodo ligand is distinct [58], we have observed a different trend in the present work; complexes 2 and 3 are isomorphous, whereas 1∙2H2O is different. The increase in the size of the halogeno ligand results in the distortion of the CdII coordination sphere by increasing the Xbridging-CdII-Xbridging angle (85.9° for 1∙2H2O, 86.7° for 2, 91.3° for 3); at the same time, the two Xbridging-CdII-Xterminal angles are significantly smaller in the chloro complex (103.2° and 87.3° in 1∙2H2O, compared to 112.1°, 95.4° for 2, and 110.0°, and 93.2° for 3). These variations, in turn, affect other coordination bond angles and eventually the nature of the trans-donor pairs, which are similar in 2 and 3 and different from that in 1∙2H2O (see structural descriptions in Section 2.2). The differences in the molecular characteristics affect the supramolecular features; the presence of lattice H2O in the chloro complex might also play a role. (d) The complexes do not retain their solid-state structures in DMSO solution, and again the nature of the halogeno ligand affects the species identity; contrary to the solid state, the chloro and bromo complexes have an identical behavior in solution compared to that of the iodo compound; and (e) Comparison of the coordination chemistry of neutral dpkoxH toward CdX2 and ZnX2 (X = Cl, Br, …) reveals differences. Although the stoichiometry in both cases is 1:1, the Zn(II) complexes are monomeric tetrahedral [35,36], whereas the Cd(II) ones are polymeric octahedral, reflecting the well-known differences in the chemistry of these group 12 metal ions.
Comparison of the present Cd(II)/dpkoxH complexes with Cd(II) complexes of other 2-pyridyl aldoximes and ketoximes is not direct; the reason is that dpkoxH has another site capable of coordination. In all the previously characterized complexes, the neutral 2-pyridyl oxime molecule behaves as a Npyridyl and Noxime -bidentate chelating ligand. The mononuclear [16,26,27,29,30] and dinuclear [25] complexes have a 1:2 or 1:3 metal-to-ligand stoichiometry. As mentioned above, mononuclear or dinuclear Cd(II)/dpkoxH halide complexes could not be isolated. Most of the polymeric Cd(II)/2-pyridyl oxime complexes have a 1:1 metal-to-ligand stoichiometry [22,26,28,30], with notable exceptions being the 1D coordination polymers {[Cd(fum)(2-pyaoH)2]}n [22] and {[Cd(suc)(2-pyaoH)2]}n [25]. In all polymeric Cd(II)/2-pyridyl oxime compounds, bridging of the neighboring CdII atoms is achieved via the ancillary dicarboxylate [22,25,26], sulfate [26,28] or halogeno [30] ligands. In the Cd(II)/dpkoxH complexes of the present work (1∙2H2O, 2, 3), the linking of neighboring CdII atoms is attained via alternate bis(halogeno) and bis(dpkoxH) bridges; this behavior is the result of the second 2-pyridyl nitrogen which allows the 2.0111 coordination mode (Figure 6).
A possible scenario for the formation of the polymeric Cd(II) complexes is the following: First, a monomeric “CdX2(dpkoxH)” complex (with terminal halogeno ligands, and a Npyridyl, Noxime-bidentate chelating dpkoxH molecule) or a dimeric “Cd2X4(dpkoxH)2” species (either with exclusively terminal halogeno groups and two N, N′, and N″-bridging dpkoxH molecules, or with both terminal and bridging halogeno groups and two Npyridyl and Noxime-bidentate chelating dpkoxH molecules) are formed in solution. The CdII atoms are coordinatively unsaturated in these species (coordination numbers 4 or 5) and then polymerization occurs during the precipitation of the products.
With the knowledge and experience obtained in this work, our future research efforts are directed, among others, to (1) the enrichment of the Cd(II) position in the “Periodic Table” of dpkoxH by using other Cd(II) sources, such as nitrate, perchlorate, sulfate, and carboxylates, and by adding an external base (e.g., Et3N, LiOH, and Bu4nNOH) in the reaction systems in order to obtain complexes (clusters and coordination polymers) containing the deprotonated dpkox− ligand; the expected coordination of the deprotonated oximate oxygen atom gives enormous possibilities for different and interesting chemistry. (2) The preparation of HgX2/dpkoxH (X = Cl, Br, I) complexes with the goal of discovering similarities or/and differences with the corresponding Cd(II) compounds; and (3) The study of the reactions between CdX2 (X = Br, I) and phenyl 2-pyridyl ketoxime (phpaoH; R = Ph in Figure 1) and comparison of the products with the complexes of the present work; the ligands dpkoxH and phpaoH have identical steric properties and rather similar electronic properties, but they differ in the number of donor atoms (phpaoH lacks the second 2-pyridyl N atom). Our previous work [30] on the CdCl2/phpaoH reaction system revealed the existence of both monomeric [CdCl2(phpaoH)2] and polymeric {[CdCl2(phpaoH)]}n (with a different structure, as expected, compared with the structures of 1∙2H2O) compounds and this also gives hopes for interesting CdX2/phpaoH (X = Br, I) chemistry. Some of our efforts along the three above-mentioned topics are already well advanced, and the results will be submitted soon.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/molecules29020509/s1: Figure S1: Complete coordination spheres of two neighboring CdII centers in one zigzag chain of complex 3; Figure S2: A portion of one zigzag chain in the crystal structure of 2; Figure S3: Interactions between neighboring chains in the structure of compound 2; Figure S4: Layers of chains in the crystal structures of 2 and 3; Figure S5: (a) Neighboring layers of 2 and 3; (b) alternate stacking interactions in 2 and 3; Figure S6: The IR spectrum (KBr, cm−1) of the 1D polymer 2; Figure S7: The 1H NMR spectrum of 1∙2H2O in d6-DMSO; Figure S8: The 1H NMR spectrum of 2 in d6-DMSO in the aromatic region.
Author Contributions
C.S., P.G. and V.R. contributed to the synthesis, crystallization, and conventional characterization of the complexes. Z.G.L. performed the Raman experiments and interpreted the spectral results. P.D., C.P.R. and V.P. collected single-crystal crystallographic data, solved the structures, and performed their refinements; the latter also investigated the supramolecular characteristics and wrote the relevant part of the paper. C.T.C. recorded and studied the NMR spectra. K.F.K., C.T.C. and S.P.P. coordinated the research and wrote the paper based on the reports of their collaborators. S.P.P. coordinated the cooperation between the teams and submitted the manuscript. All the authors exchanged opinions concerning the progress of the experiments and commented on the various drafts of the paper. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are contained within the article and Supplementary Materials.
Acknowledgments
The authors thank the head of the Laboratory of Applied Molecular Spectroscopy Research, George A. Voyiatzis (ICE-HT/FORTH), for the access to the Raman setup and for the purchase of quality dpkoxH.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Tschugaeff, L. Ueber ein neues, empfindliches reagens auf nickel. Ber. Dtsch. Chem. Ges. 1885, 18, S.2728–S.2734. [Google Scholar] [CrossRef]
- Smith, A.G.; Tasker, P.A.; White, D.J. The structures of phenolic oximes and their complexes. Coord. Chem. Rev. 2003, 241, 61–85. [Google Scholar] [CrossRef]
- Tasker, P.A.; Tong, C.C.; Westra, A.N. Co-extraction of cations and anions in base metal recovery. Coord. Chem. Rev. 2007, 251, 1868–1877. [Google Scholar] [CrossRef]
- Gerasimchuk, N.; Maher, T.; Durham, P.; Domasevitch, K.V.; Wilking, J.; Mokhir, A. Tin(IV) cyanoximates: Synthesis, characterization, and cytotoxicity. Inorg. Chem. 2007, 46, 7268–7274. [Google Scholar] [CrossRef] [PubMed]
- Sahyoun, T.; Arrault, A.; Schneider, R. Amidoximes and oximes. Synthesis, structure, and their key role as NO donors. Molecules 2019, 24, 2470. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.-J.; Zhang, Z.Z.; Lin, S.-B. A review of manganese-based molecular magnets and supramolecular architectures from phenolic oximes. Coord. Chem. Rev. 2015, 289–290, 289–314. [Google Scholar] [CrossRef]
- Chaudhuri, P. Homo- and hetero-polymetallic exchange coupled metal-oximates. Coord. Chem. Rev. 2003, 243, 143–190. [Google Scholar] [CrossRef]
- Kopylovich, M.N.; Kukushkin, V.Y.; Haukka, M.; Fraústo da Silva, J.J.R.; Pombeiro, A.J.L. Zinc(II)/ketoxime system as a simple and efficient catalyst for hydrolysis of organonitriles. Inorg. Chem. 2002, 41, 4798–4804. [Google Scholar] [CrossRef]
- Garnovskii, D.A.; Kukushkin, V.Y. Metal-mediated reactions of oximes. Russ. Chem. Rev. 2006, 75, 111–124. [Google Scholar] [CrossRef]
- Bolotin, D.S.; Bokach, N.A.; Demakova, M.Y.; Kukushkin, V.Y. Metal-involving synthesis and reactions of oximes. Chem. Rev. 2017, 117, 13039–13122. [Google Scholar] [CrossRef]
- Lada, Z.G.; Soto Beobide, A.; Savvidou, A.; Raptopoulou, C.P.; Psycharis, V.; Voyiatzis, G.A.; Turnbull, M.M.; Perlepes, S.P. A unique copper(II)-assisted transformation of acetylacetone dioxime in acetone that leads to one-dimensional, quinoxaline-bridged coordination polymers. Dalton Trans. 2017, 46, 260–274. [Google Scholar] [CrossRef] [PubMed]
- Mowson, A.M.; Nguyen, T.N.; Abboud, K.A.; Christou, G. Dimeric and tetrameric supramolecular aggregates of single-molecule magnets via carboxylate substitution. Inorg. Chem. 2013, 52, 12320–12322. [Google Scholar] [CrossRef]
- Clérac, R.; Miyasaka, H.; Yamashita, M.; Coulon, C. Evidence for single-chain magnet behavior in a MnIIINiII chain designed with high spin magnetic units: A route to high temperature metastable magnets. J. Am. Chem. Soc. 2002, 124, 12837–12844. [Google Scholar] [CrossRef] [PubMed]
- Lada, Z.G.; Polyzou, C.D.; Nika, V.; Stamatatos, T.C.; Konidaris, K.F.; Perlepes, S.P. Adventures in the coordination chemistry of 2-pyridyl oximes: On the way to 3d/4f-metal coordination clusters. Inorg. Chim. Acta 2022, 539, 120954. [Google Scholar] [CrossRef]
- Stamatatos, T.C.; Foguet-Albiol, D.; Lee, S.C.; Stoumpos, C.C.; Raptopoulou, C.P.; Terzis, A.; Wernsdorfer, W.; Hill, S.O.; Perlepes, S.P.; Christou, G. “Switching on” the properties of single-molecule magnetism in triangular manganese(III) complexes. J. Am. Chem. Soc. 2007, 129, 9484–9499. [Google Scholar] [CrossRef] [PubMed]
- Routzomani, A.; Lada, Z.G.; Angelidou, V.; Raptopoulou, C.P.; Psycharis, V.; Konidaris, K.F.; Chasapis, C.T.; Perlepes, S.P. Confirming the Molecular Basis of the Solvent Extraction of Cadmium(II) Using 2-Pyridyl Oximes through a Synthetic Inorganic Chemistry Approach and a Proposal for More Efficient Extractants. Molecules 2022, 27, 1619. [Google Scholar] [CrossRef] [PubMed]
- Mori, F.; Ishida, T.; Nogami, T. Structure and magnetic properties of 3d-4f heterometallic complexes containing di-2-pyridyl ketoximate. An approach to single-molecule magnets. Polyhedron 2005, 24, 2588–2592. [Google Scholar] [CrossRef]
- Mori, F.; Ishida, T.; Nogami, T.; Choi, K.Y.; Nojiri, H. Oximate-bridged trinuclear Dy-Cu-Dy complex behaving as a single-molecule magnet and its mechanistic investigation. J. Am. Chem. Soc. 2006, 128, 1440–1441. [Google Scholar] [CrossRef]
- Stemmler, A.J.; Kampf, J.W.; Pecoraro, V.L. Synthesis and Crystal Structure of the First Inverse 12-Metallacrown-4. Inorg. Chem. 1995, 34, 2771–2772. [Google Scholar] [CrossRef]
- Psomas, G.; Stemmler, A.J.; Dendrinou-Samara, C.; Bodwin, J.J.; Schneider, M.; Alexiou, M.; Kampf, J.W.; Kessissoglou, D.P.; Pecoraro, V.L. Preparation of Site-Differentiated Mixed Ligand and Mixed Ligand/Mixed Metal Metallacrowns. Inorg. Chem. 2001, 40, 1562–1570. [Google Scholar] [CrossRef]
- Kumagai, H.; Endo, M.; Kondo, M.; Kawata, S.; Kitagawa, S. Reactions of di-2-pyridyl ketone oxime in the presence of vanadium(III): Crystal structures of the coordination products. Coord. Chem. Rev. 2003, 237, 197–203. [Google Scholar] [CrossRef]
- Croitor, L.; Coropceanu, E.B.; Duca, G.; Siminel, A.V.; Fonari, M.S. Nine Mn(II), Zn(II), and Cd(II) mixed-ligand coordination networks with rigid dicarboxylate and pyridine-n-aldoxime ligands: Impact of the second ligand in the structures’ dimensionality and solvent capacity. Polyhedron 2017, 129, 9–21. [Google Scholar] [CrossRef]
- Coropceanu, E.B.; Croitor, L.; Siminel, A.V.; Fonari, M.S. Preparation, structural characterization and luminescence studies of mono- and binuclear Zn(II) and Cd(II) acetates with pyridine-4-aldoxime and pyridine-4-amidoxime ligands. Polyhedron 2014, 75, 73–80. [Google Scholar] [CrossRef]
- Croitor, L.; Coropceanu, E.B.; Masunov, A.A.; Rivea-Jacquez, H.J.; Siminel, A.V.; Fonari, M.S. Mechanism of Nonlinear Optical Enhancement and Supramolecular Isomerism in 1D Polymeric Zn(II) and Cd(II) Sulfates with Pyridine-4-aldoxime Ligands. J. Phys. Chem. C 2014, 118, 9217–9227. [Google Scholar] [CrossRef]
- Croitor, L.; Coropceanu, E.B.; Masunov, A.E.; Rivera-Jacquez, H.J.; Siminel, A.V.; Zelentsov, V.I.; Datsko, T.Y.; Fonari, M.S. Polymeric Luminescent Zn(II) and Cd(II) Dicarboxylates Decorated by Oxime Ligands: Tuning the Dimensionality and Adsorption Capacity. Cryst. Growth Des. 2014, 14, 3935–3948. [Google Scholar] [CrossRef]
- Croitor, L.; Coropceanu, E.B.; Siminel, A.V.; Masunov, A.E.; Fonari, M.S. From discrete molecules to one-dimensional coordination polymers containing Mn(II), Zn(II) or Cd(II) pyridine-2-aldoxime building unit. Polyhedron 2013, 60, 140–150. [Google Scholar] [CrossRef]
- Shirvan, S.A.; Dezfuli, S.H. Bis(acetato,κO)bis(2-pyridinealdoxime-κ2N, N′)cadmium. Acta Crystallogr. Sect. E 2012, 68, m1080–m1081. [Google Scholar] [CrossRef]
- Papatriantafyllopoulou, C.; Kostakis, G.E.; Raptopoulou, C.P.; Terzis, A.; Perlepes, S.P.; Plakatouras, J.C. Investigation of the MSO4∙XH2O (M = Zn, x = 7; M = Cd, x = 8/3)/methyl 2-pyridyl ketone oxime reaction system: A novel Cd(II) coordination polymer versus mononuclear and dinuclear Zn(II) complexes. Inorg. Chim. Acta 2009, 362, 2361–2370. [Google Scholar] [CrossRef]
- Yan, J.; Liu, G.-X. Tris(phenyl 2-pyridyl ketone oxime- κ2Ν, Ν′)cadmium(II) dinitrate. Acta Crystallogr. Sect. E 2009, 65, m461. [Google Scholar] [CrossRef]
- Mazarakioti, E.C.; Soto Beobide, A.; Angelidou, V.; Efthymiou, C.G.; Terzis, A.; Psycharis, V.; Voyiatzis, G.A.; Perlepes, S.P. Modeling the Solvent Extraction of Cadmium(II) from Aqueous Chloride Solutions by 2-pyridyl Ketoximes: A Coordination Chemistry Approach. Molecules 2019, 24, 2219. [Google Scholar] [CrossRef]
- Coxall, R.A.; Harris, S.G.; Henderson, D.K.; Parsons, S.; Tasker, P.A.; Winpenny, R.E.P. Inter-ligand reactions: In situ formation of new polydentate ligands. J. Chem. Soc. Dalton Trans. 2000, 2349–2356. [Google Scholar] [CrossRef]
- Sommerer, S.O.; Westcott, B.L.; Jircitano, A.J.; Abboud, K.A. The synthesis and structure of two novel metal-di-2-pyridyl ketone oxime dimers. Inorg. Chim. Acta 1995, 238, 149–153. [Google Scholar] [CrossRef]
- Milios, C.J.; Kyritsis, P.; Raptopoulou, C.P.; Terzis, A.; Vicente, R.; Escuer, A.; Perlepes, S.P. Di-2-pyridyl ketone oxime [(py)2CNOH] in manganese carboxylate chemistry: Mononuclear, dinuclear and tetranuclear complexes, and partial transformation of (py)2CNOH to the gem-diolate(2-) derivative of di-2-pyridyl ketone leading to the formation of NO3−. Dalton Trans. 2005, 501–511. [Google Scholar] [CrossRef]
- Goher, M.A.S.; Mautner, F.A. Dimeric and polymeric copper(I) complexes. Synthesis and characterization of copper(I) complexes of di-2-pyridyl ketone oxime (DPKox) and crystal structures of [Cu(DPKox)Cl]2∙2H2O and [Cu(DPKox)(NCS)]n. Polyhedron 1999, 18, 3425–3431. [Google Scholar] [CrossRef]
- Westcott, B.L.; Crundwell, G.; Remesic, M.; Knopf, K.; Chandler, K.; McMaster, J.; Davies, E.S. Crystal structure and magnetic properties of di-copper and di-zinc complexes with di-2-pyridyl ketone oxime. Inorg. Chem. Commun. 2016, 74, 79–81. [Google Scholar] [CrossRef]
- Gökce, H.; Alpaslan, G.; Alasalvar, C. Crystal structure, spectroscopic characterization, DFT computations and molecular docking study of a synthesized Zn(II) complex. J. Coord. Chem. 2019, 72, 1075–1096. [Google Scholar] [CrossRef]
- Holynska, M. Formation of Ni(II) oxime-bridged basket- like complexes and their structural aspects. Curr. Inorg. Chem. 2015, 5, 64–70. [Google Scholar] [CrossRef]
- Dollish, F.R.; Fateley, W.G.; Bentley, F.F. Characteristic Raman Frequencies of Organic Compounds; Wiley: New York, NY, USA, 1974; pp. 134–137. [Google Scholar]
- Adams, D.M. Metal-Ligand and Related Vibrations: A Critical Survey of the Infrared and Raman Spectra of Metallic and Organometallic Compounds; Edward Arnold Publishers: London, UK, 1967; pp. 45, 79. [Google Scholar]
- Jackman, L.M.; Sternhell, S. Applications of Nuclear Magnetic Resonance in Organic Chemistry, 2nd ed.; Pergamon Press: Oxford, UK, 1969; pp. 216, 220. [Google Scholar]
- Thomas, S.; Brühl, I.; Heilmann, D.; Kleinpeter, E. 13C NMR chemical shift calculations for some substituted pyridines: A comparative consideration. J. Chem. Inf. Comput. Sci. 1997, 37, 726–730. [Google Scholar] [CrossRef]
- Oszczapowicz, J. Substituent Effects in the 13C-NMR spectra of six-membered nitrogen heteroatomic compounds. Int. J. Mol. Sci. 2005, 6, 11–17. [Google Scholar] [CrossRef]
- Silverstein, R.M.; Bassler, G.C.; Morrill, T.C. Spectrometric Identification of Organic Compounds, 4th ed.; Wiley: New York, NY, USA, 1981; pp. 134, 135, 266, 267. [Google Scholar]
- Batterham, T.J. NMR Spectra of Simple Heterocycles; Wiley: New York, NY, USA, 1973; pp. 8–67. [Google Scholar]
- Kolehmainen, E. NMR Spectroscopy, Heteronuclei, Y-Cd. In Encyclopedia of Spectroscopy and Spectrometry, 3rd ed.; Lindon, J.C., Koppenaal, D.W., Tranter, G.E., Eds.; Elsevier: New York, NY, USA, 2017; pp. 366–374. [Google Scholar]
- Elis, P.D. Cadmium-113 Magnetic Resonance Spectroscopy. Science 1983, 221, 1141–1146. [Google Scholar] [CrossRef]
- Geary, W.J. The use of conductivity measurements in organic solvents for the characterization of coordination compounds. Coord. Chem. Rev. 1971, 7, 81–122. [Google Scholar] [CrossRef]
- Frost, J.M.; Kobera, L.; Pialat, A.; Zhang, Y.; Southern, S.A.; Gabidullin, B.; Bryce, D.L.; Murugesu, M. From discrete molecule, to polymer, to MOF: Mapping the coordination chemistry of CdII using 113Cd solid-state NMR. Chem. Commun. 2016, 52, 10680–10683. [Google Scholar] [CrossRef] [PubMed]
- Nieuwenhuyzen, M.; Wilkins, C.J. Crystal Structures and Significance of Complexes Formed Between Cadmium Bromide and Dimethyl Sulfoxide. J. Chem. Soc. Dalton Trans. 1993, 2673–2681. [Google Scholar] [CrossRef]
- CrystalClear; Rigaku: The Woodlands, TX, USA; MSC Inc.: The Woodlands, TX, USA, 2005.
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELX. Acta Crystallogr. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Spek, A.L. PLATON SQUEEZE: A tool for the calculation of the disordered solvent contribution to the calculated structure factors. Acta Crystallogr. Sect. C 2015, 71, 9–18. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SADABS, Version 2.03; Bruker Analytical X-ray Systems: Madison, WI, USA, 2000. [Google Scholar]
- Sheldrick, G.M. SHELXT-Intergrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Diamond, Crystal and Molecular Structure Visualization, Version 3.1; Crystal Impact: Bonn, Germany, 2018.
- Dey, A.; Jetti, R.K.R.; Boese, R.; Desiraju, G.R. Supramolecular equivalence of halogen, ethynyl and hydroxyl groups. A comparison of the crystal structures of some 4-substituted anilines. Cryst. Eng. Comm. 2003, 5, 248–252. [Google Scholar] [CrossRef]
- Hsu, Y.-F.; Hsu, W.; Wu, C.-J.; Cheng, P.-C.; Yeh, C.-W.; Chang, W.-J.; Chen, J.-D.; Wang, J.-C. Roles of halide anions in the structural diversity of Zn(II) complexes containing the flexible N, N’’-di(4-pyridyl)adipoamide ligand. Cryst. Eng. Comm. 2010, 12, 702–710. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).