
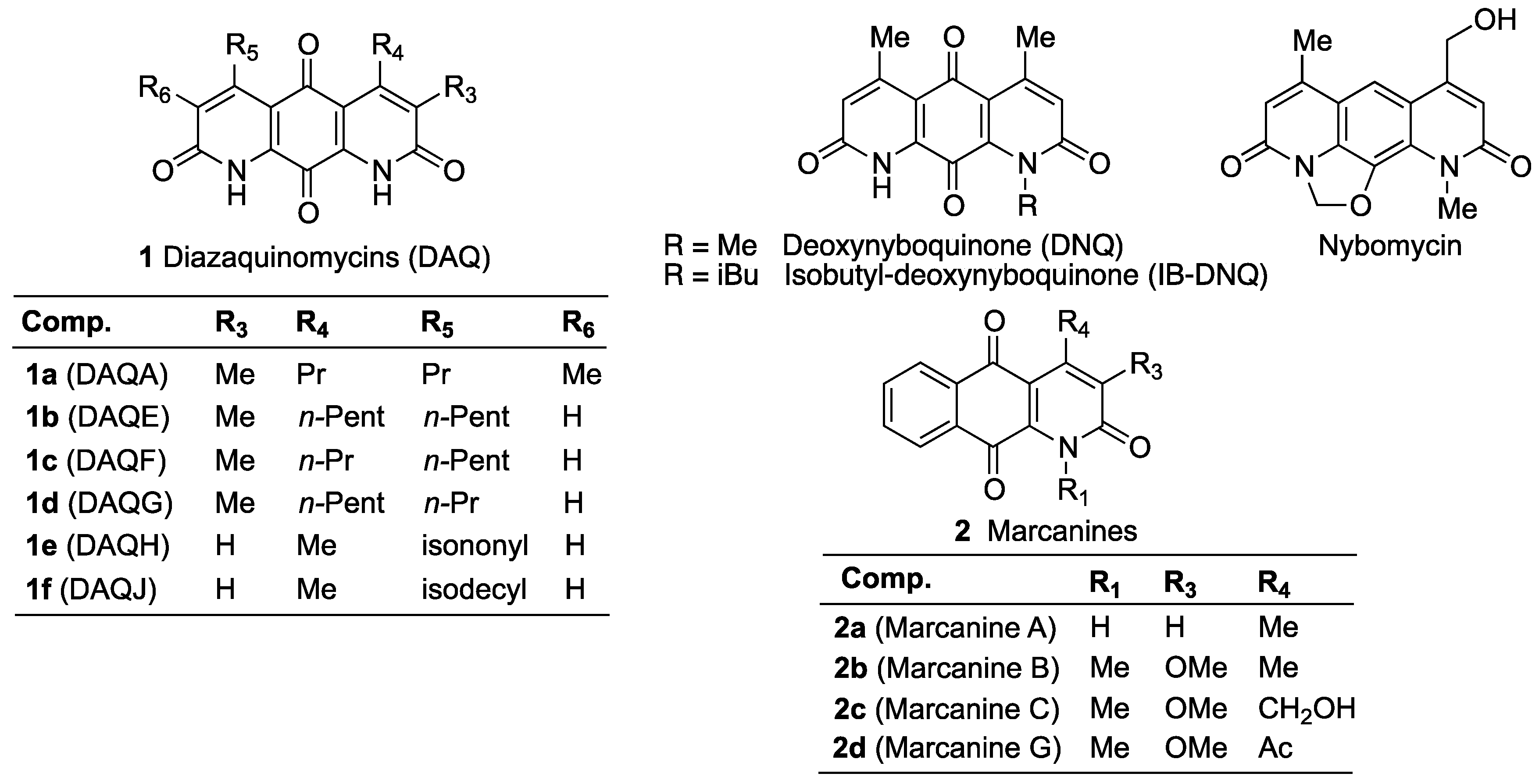
Structure-Antitumor Activity Relationships of Aza- and Diaza-Anthracene-2,9,10-Triones and Their Partially Saturated Derivatives



Abstract

1. Introduction
2. Results and Discussion
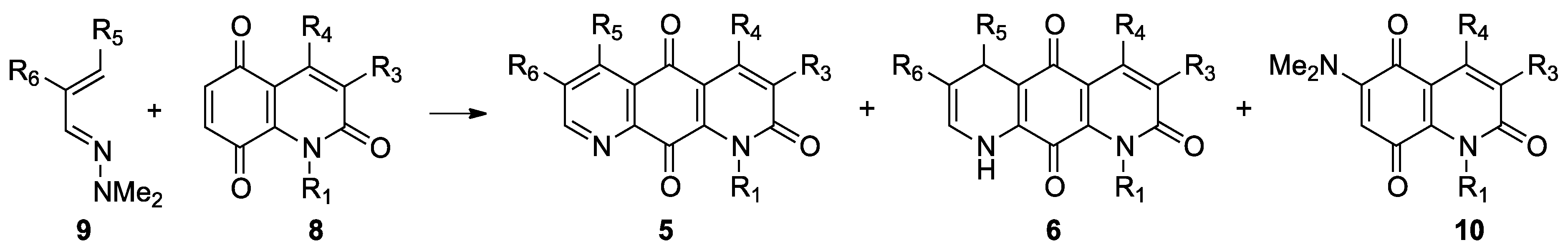
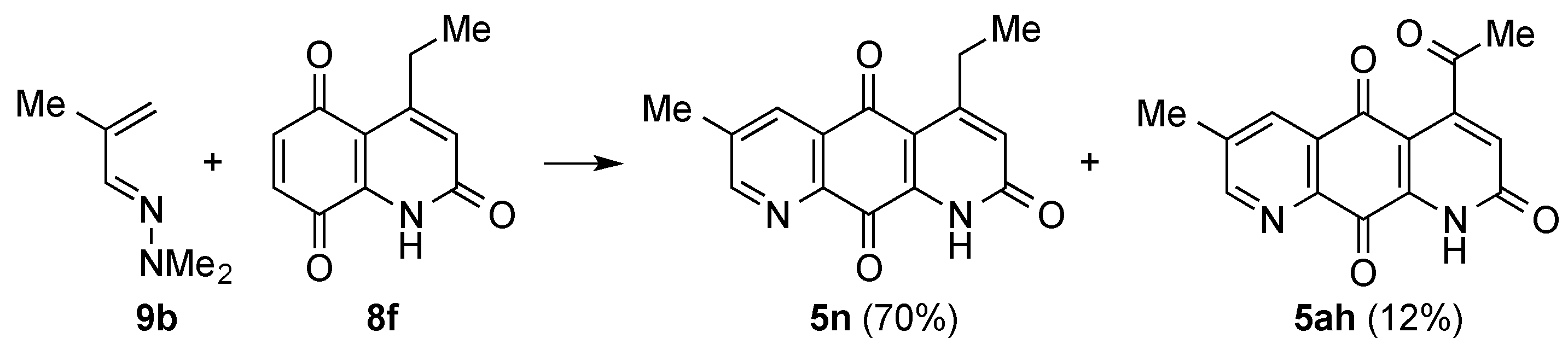
2.1. Preparation of 1,8-Diazaanthracene-2,9,10-trione Derivatives
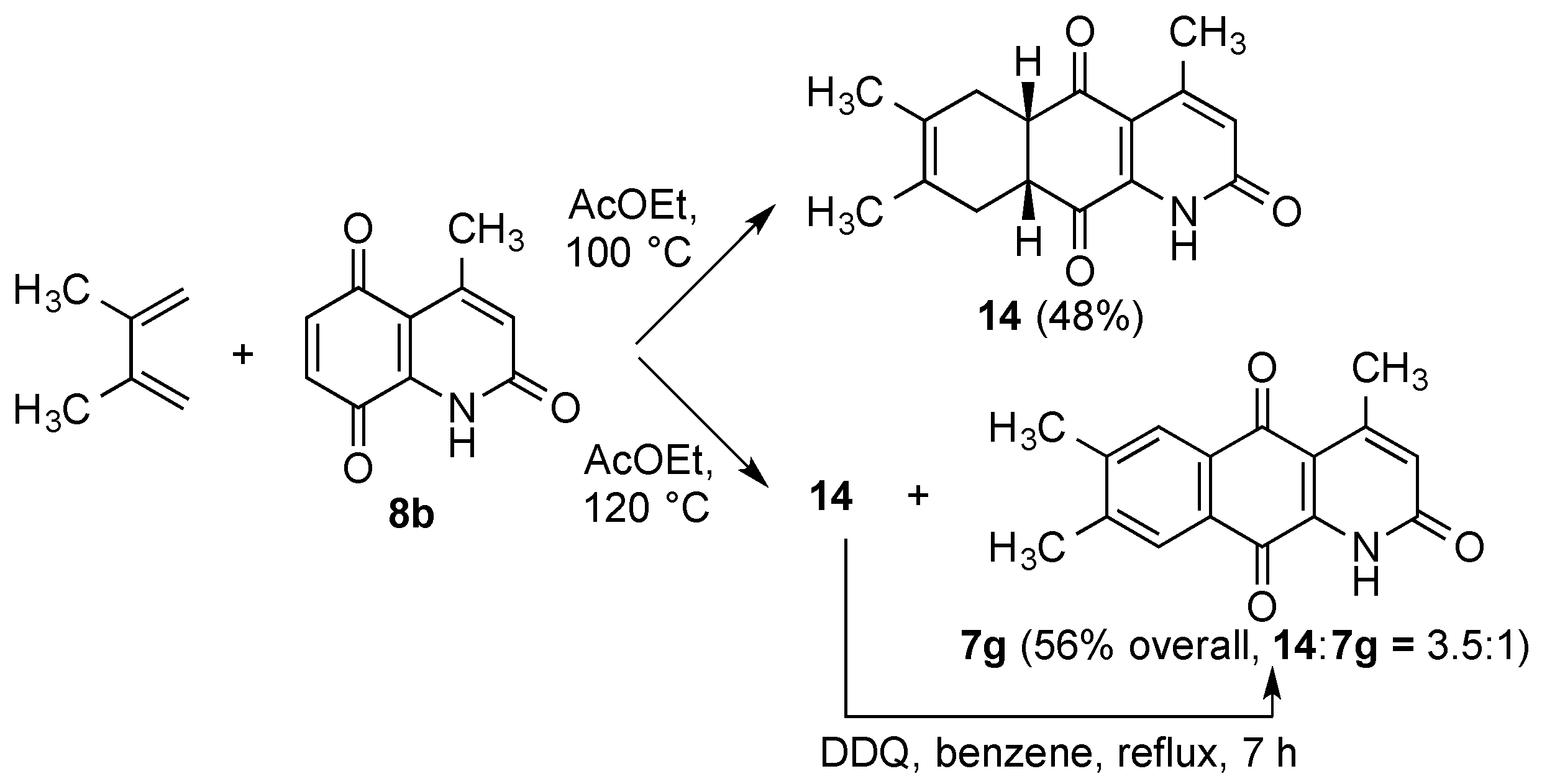
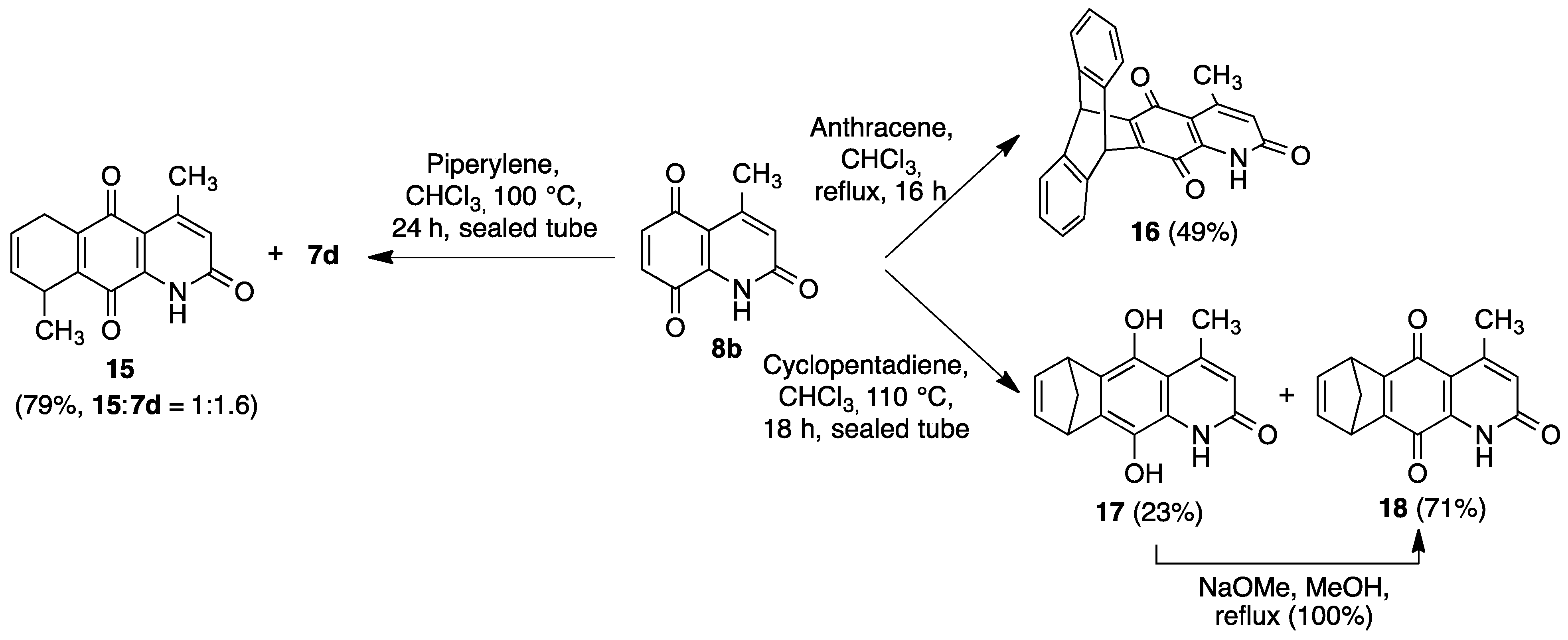
2.2. Preparation of 1-Azaanthracene-2,9,10-trione Derivatives 7
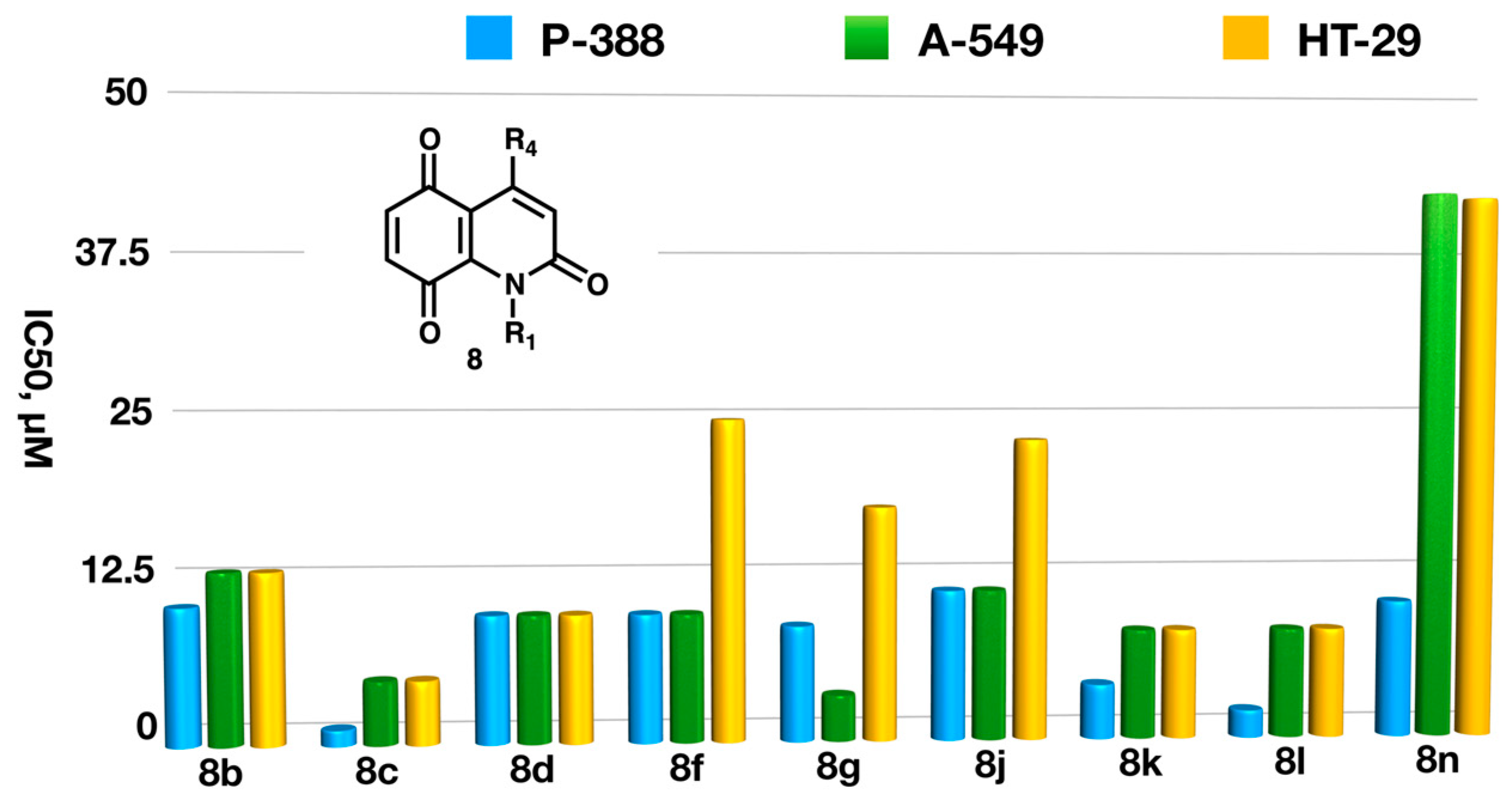
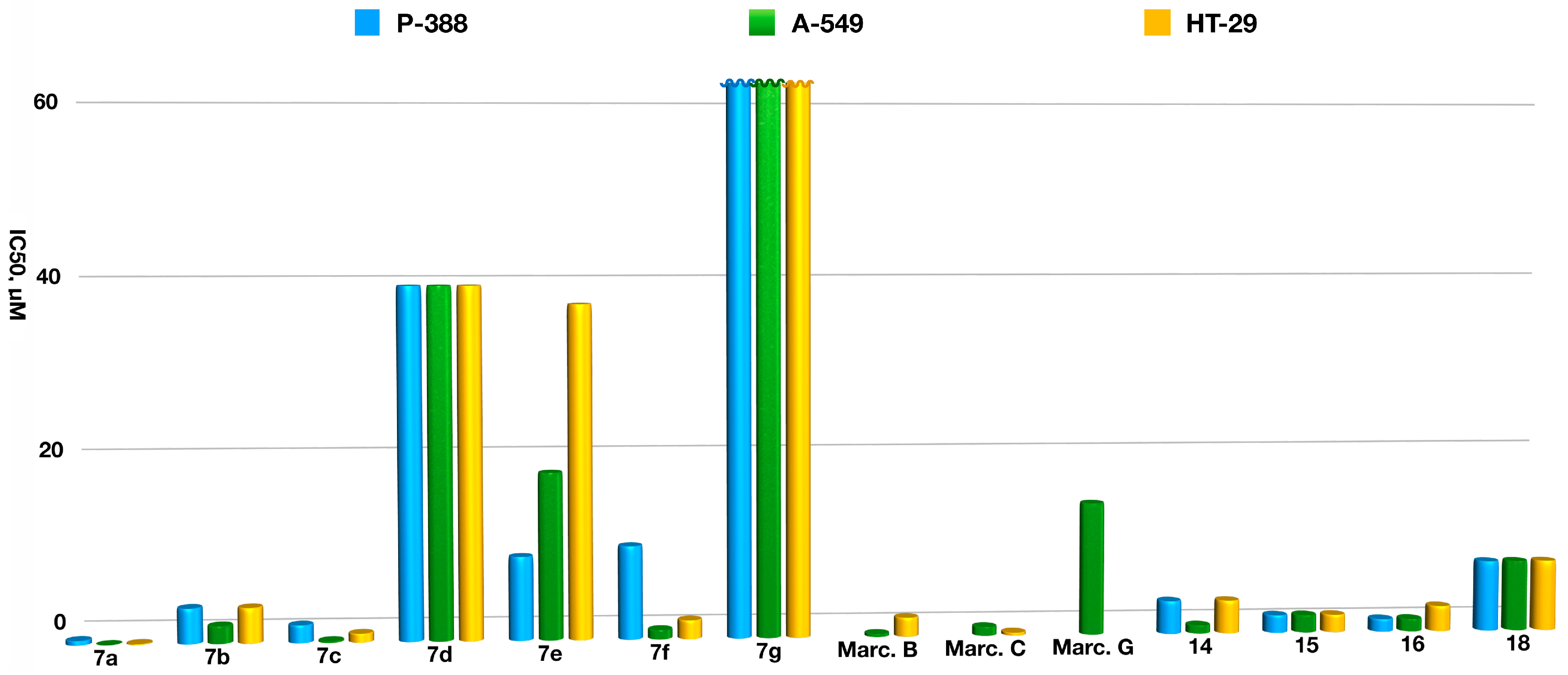
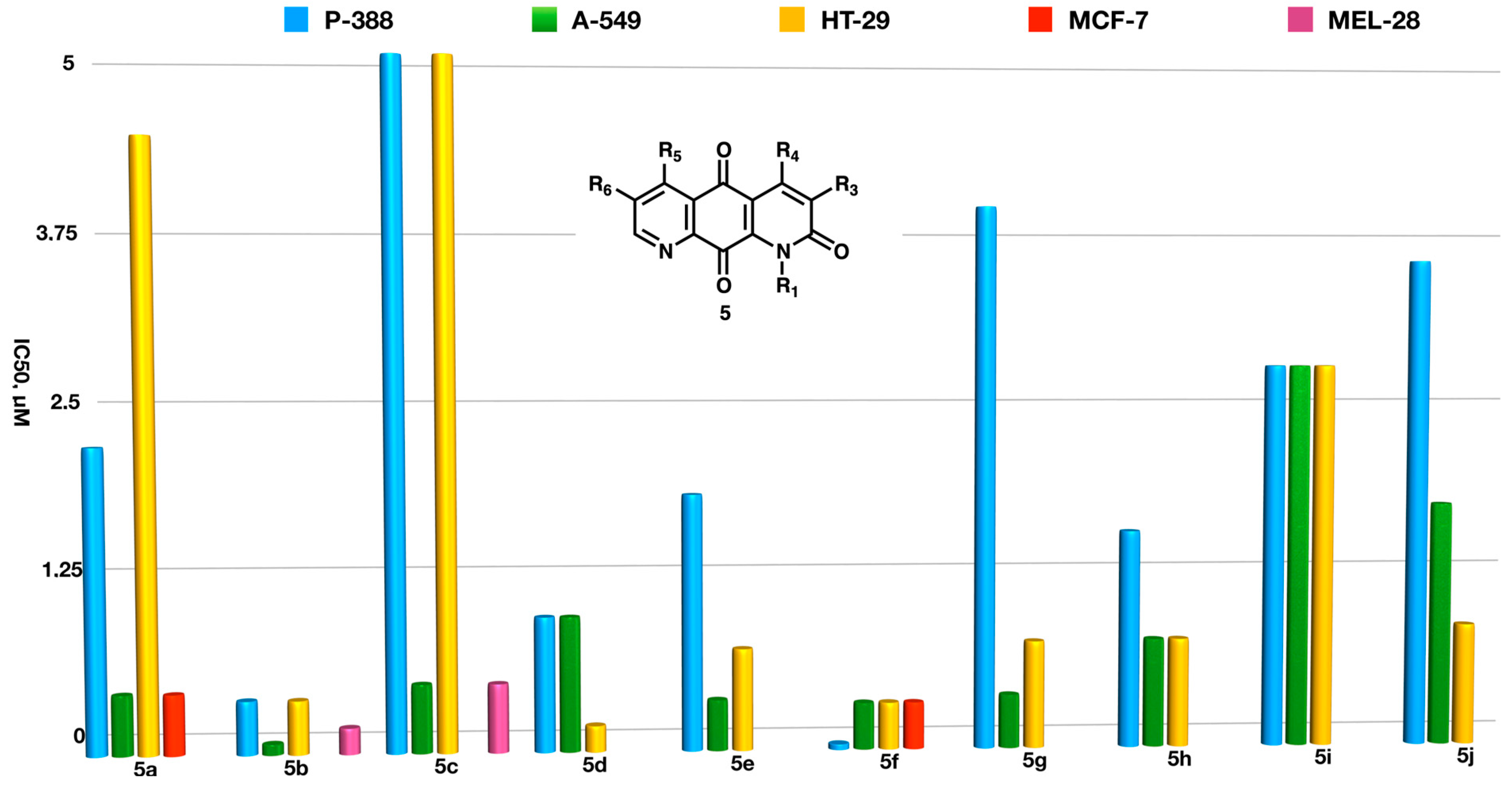
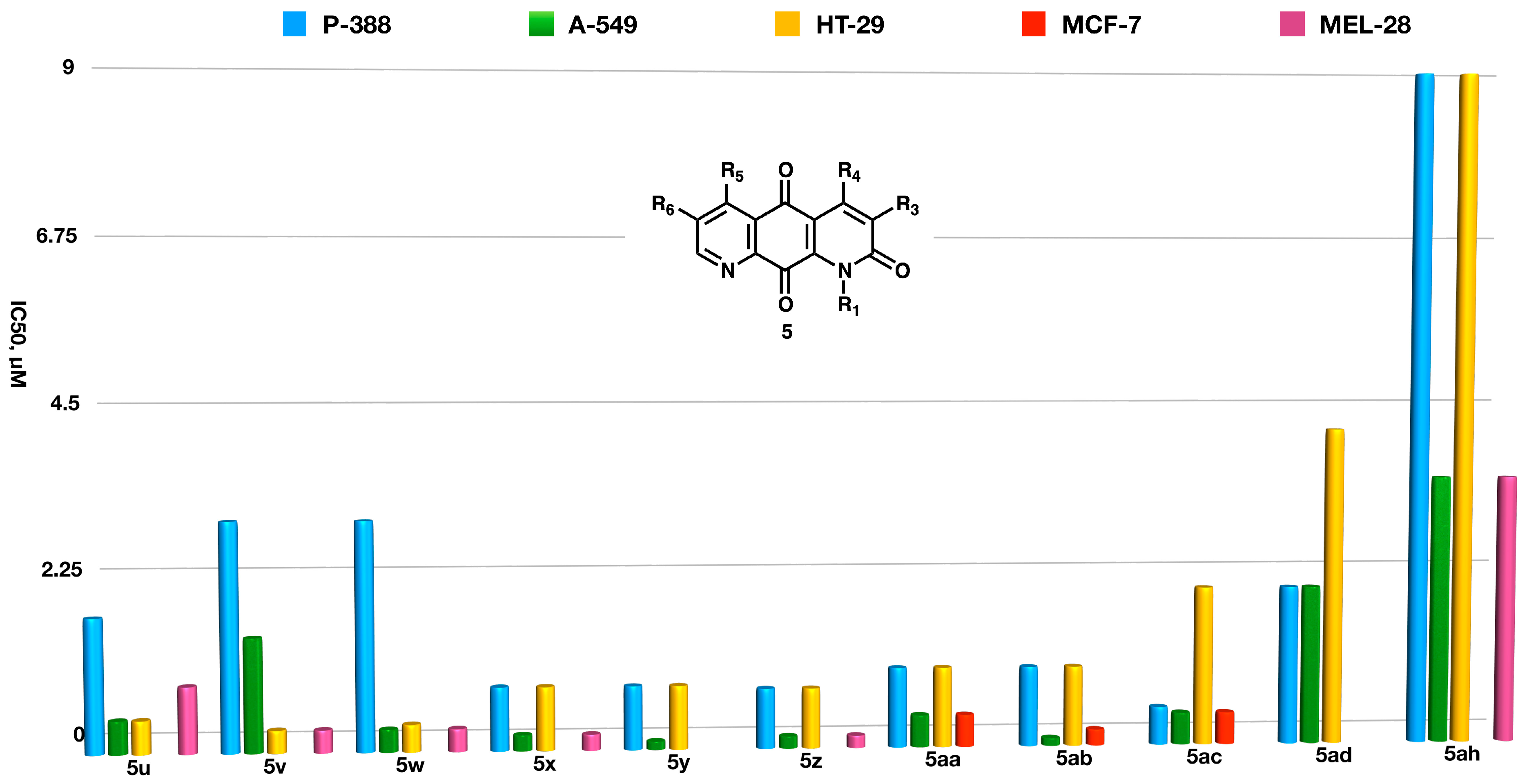
2.3. Biological Studies
- (a)
- Aromatic substituents at N-1 lower both activity and selectivity against solid tumors, as shown by the comparison of the data for compounds 5d and 5p.
- (b)
- Aromatic substituents at C-3 do not improve the activity on solid tumor cell lines with respect to alkyl groups when directly attached to the anthraquinone ring (5q vs. 5l and 5w vs. 5r)
- (c)
- On the other hand, when an aromatic ring is attached to C3 via a methylidene spacer, not only is considerable antitumor activity observed, but also a notable selectivity towards certain solid tumors. Thus, compound 5m is ten times more active in lung carcinoma and melanoma than in colon carcinoma and ca. 50 times more active than on the P-388 lymphoma cells. This difference also exists but is not so marked in the derivatives, which only present alkyl substituents at C3 such as 5l.
- (d)
- The presence of simultaneous substituents at C3, C4, and C5, leading to a steric interaction between the C10=O groups with R4 and R4, increases the activity against P-388 compared to the same compounds when they only have substituents at C4 and C5 or C3 and C4 (5u vs. 5t).
- (e)
- An increase in the length of the R5 chain seems to be accompanied by a slight decrease in activity in lung cancer cells (compounds 5g, 5j, and 5k). Aromatic substituents, especially when bearing an electron-releasing group, also lead to decreased activity in these cells (compounds 5h and 5i).
- (f)
- The presence of alkyl substituents at R6 is generally favorable for activity on the human colorectal adenocarcinoma cell line, as shown by the comparison of the data for 5c and 5d–f.
- (g)
- Electron-withdrawing groups decrease the activity when they are in the C3 and C4 positions with respect to their alkyl counterparts, as shown by the comparison between the compounds (5y, 5z, and 5aa vs. 5ad and 5ah vs. 5d).
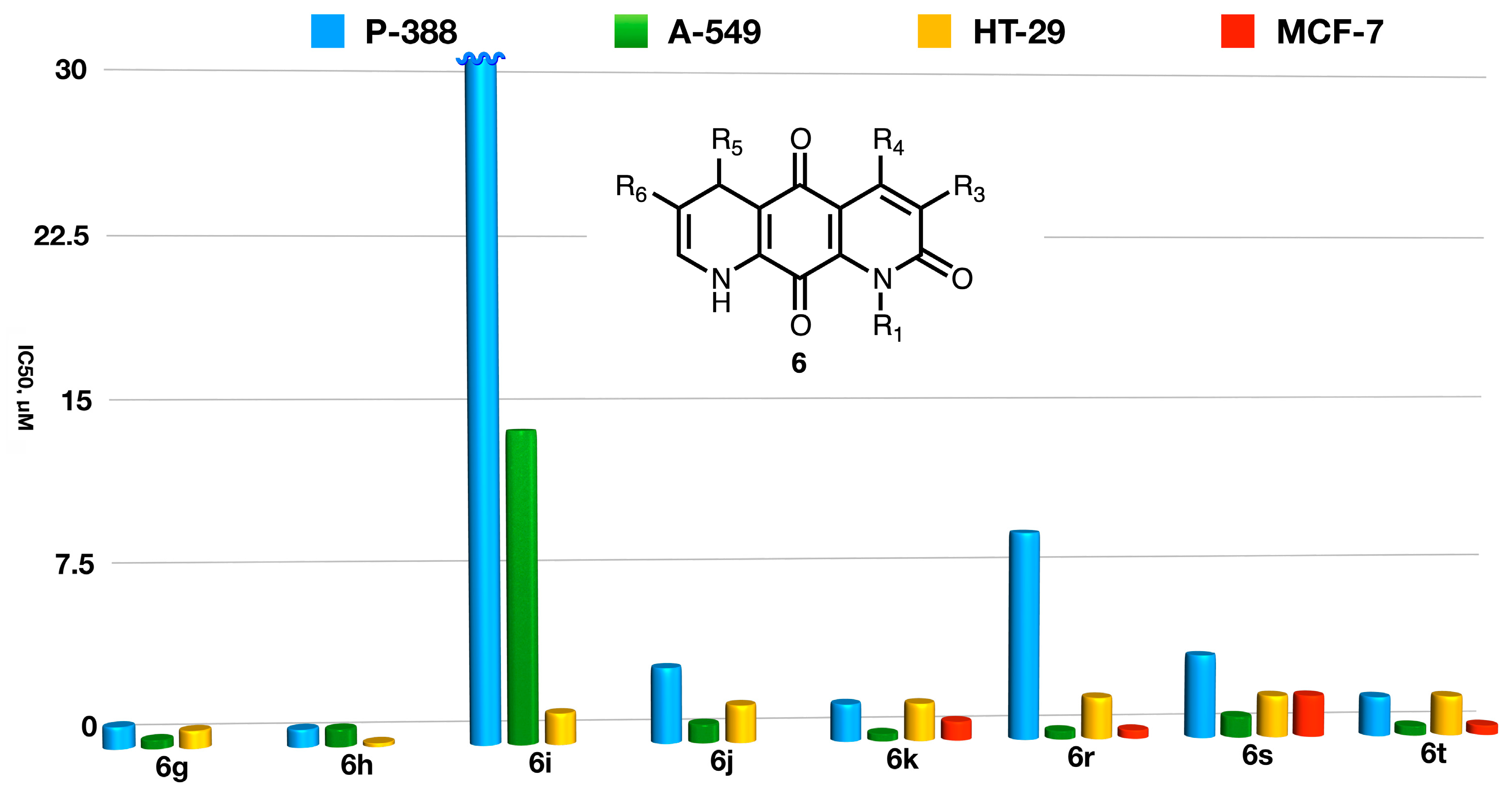
- (a)
- Generally speaking, the aromatic compounds 5 and their 5,8-dihydro counterparts 6 show similar activities, but some exceptions to this rule were observed, as summarized below.
- (b)
- The presence of aromatic derivatives at C-5 increases the activity and selectivity against colon cancer cells in the dihydro derivatives such as 6h and 6i compared to their aromatized counterparts 5h and 5i. For N-aryl substitution, dihydro derivatives seem less selective for solid tumors (5v vs. 6v).
- (c)
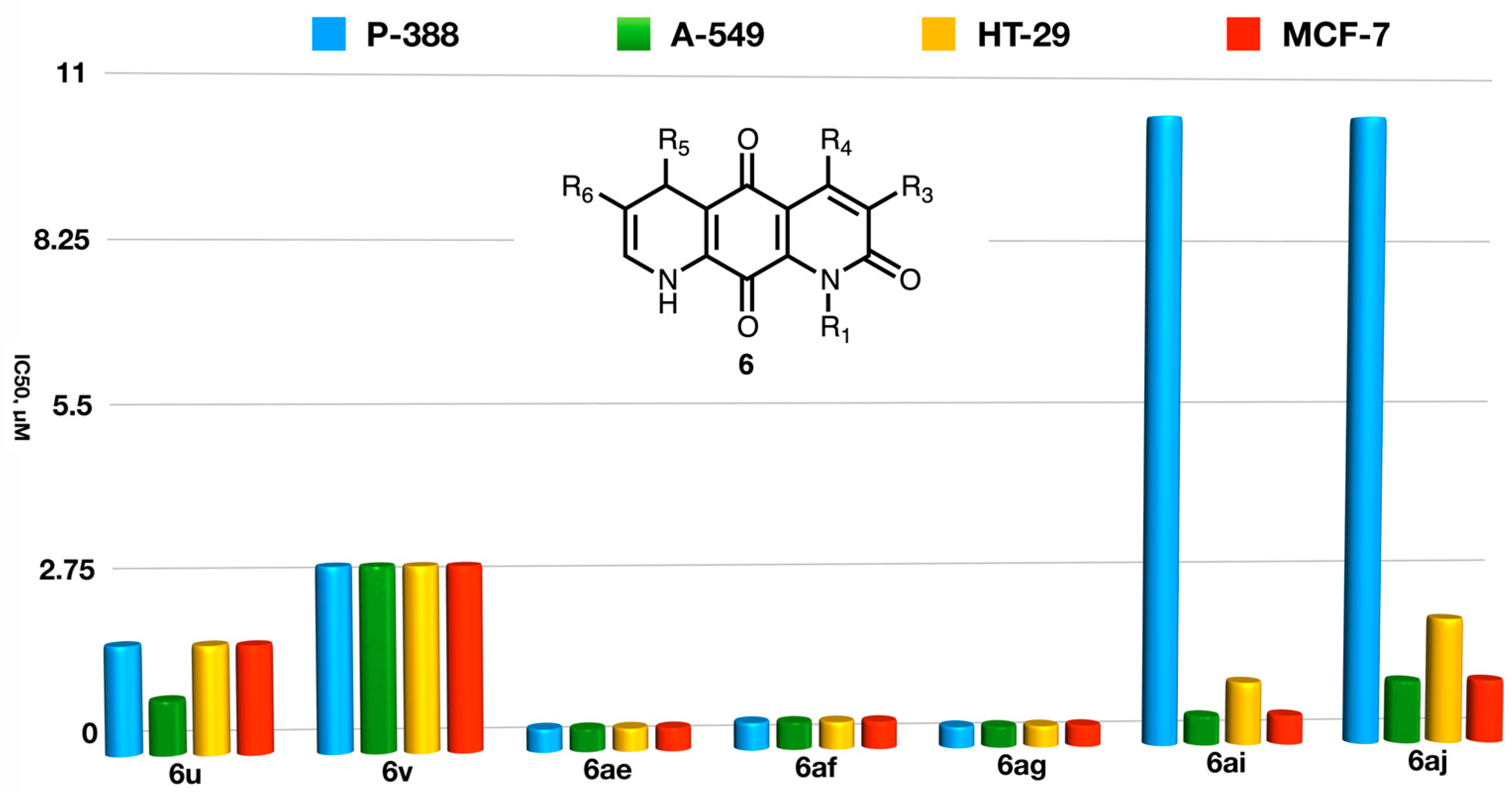
- On the other hand, for the case of alkyl substitution the selectivity trend is the opposite one, with the aromatic systems 5 showing a higher selectivity for solid tumors than the corresponding compounds 6 (5s vs. 6s, 5t vs. 6t, 5u vs. 6u). Even though a comparison cannot be established in these cases, the lack of selectivity found in compounds 6ae, 6af, and 6ag supports this conclusion.
- (d)
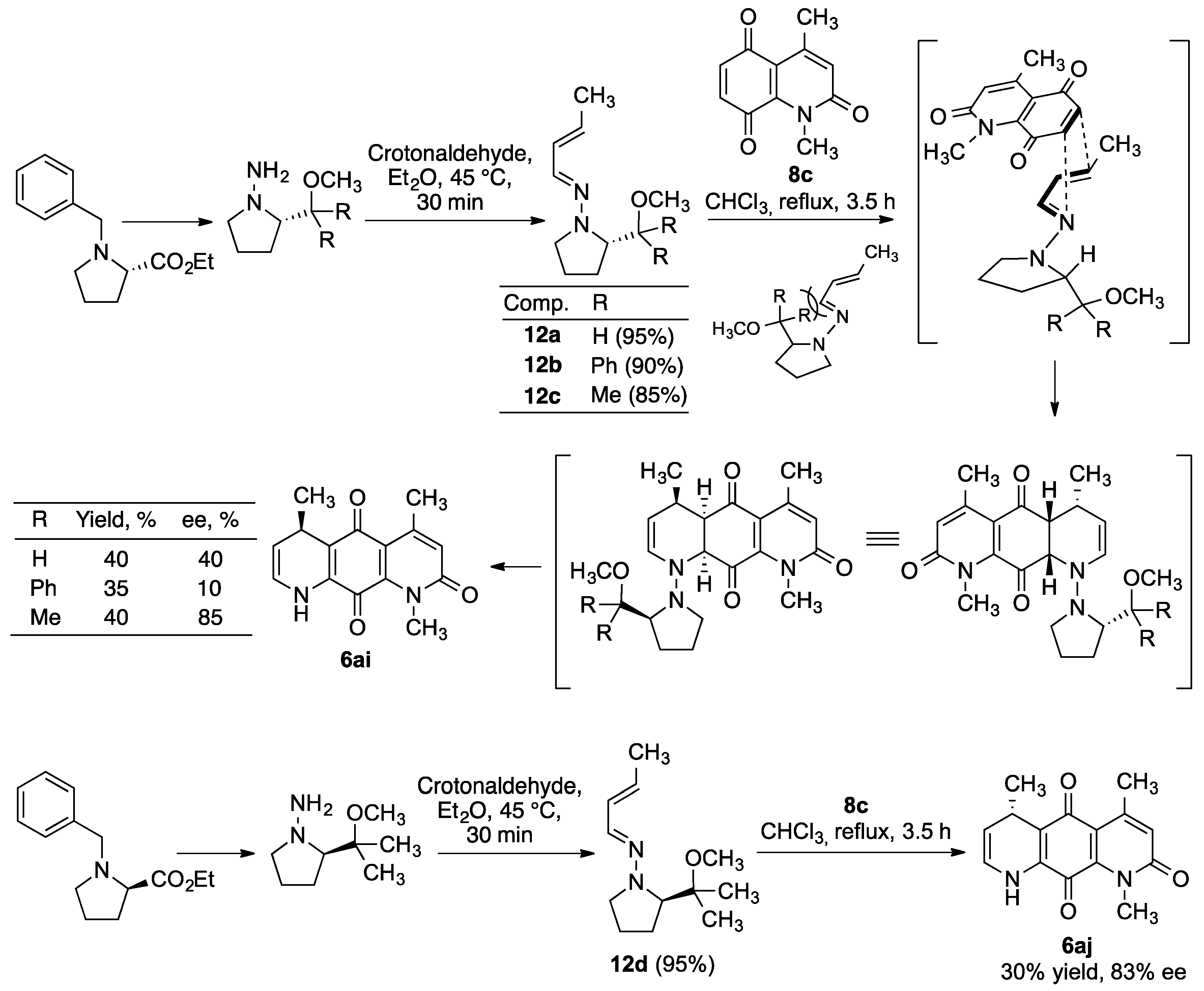
- A comparison of the activities of the enantio-enriched compounds 6ai and 6aj leads to the conclusion that the configuration of the stereogenic center at C-5 does not greatly influence the anti-tumor activity.
3. Materials and Methods
3.1. General Information
3.2. Preparation of Enantiomerically Pure 1-Azadienes—General Procedure
3.2.1. (S)-N-(-But-2-en-1-ylidene)-2-(methoxymethyl)pyrrolidin-1-amine (12a)
3.2.2. (S)-N-(But-2-en-1-ylidene)-2-(methoxydiphenylmethyl)pyrrolidin-1-amine (12b)
3.2.3. (S)-N-(2-(2-Methoxypropan-2-yl)pyrrolidin-1-yl)but-2-en-1-imine (12c)
3.2.4. (R)-N-(2-(2-Methoxypropan-2-yl)pyrrolidin-1-yl)but-2-en-1-imine (12d)
3.3. Hetero Diels–Alder Reactions between Dimethylhydrazones 9 and Quinones 8—General Procedure
3.3.1. 6-Butyl-4-methyl-1H-1,8-diazaanthracene-2,9,10-trione (5f)
3.3.2. 5,8-Dihydro-4,6-dimethyl-5-propyl-1H-1,8-diazaanthracene-2,9,10-trione (6k)
3.3.3. 3,4,6-Trimethyl-1H-1,8-diazaanthracene-2,9,10-trione (5l)
3.3.4. 3-Benzyl-4,6-dimethyl-1H-1,8-diazaanthracene-2,9,10-trione (5m)
3.3.5. 4-Ethyl-6-methyl-1H-1,8-diazaanthracene-2,9,10-trione (5n)
3.3.6. 4-Acetyl-6-methyl-1H-1,8-diazaanthracene-2,9,10-trione (5ah)
3.3.7. 6-Methyl-4-(2-phenylethyl)-1H-1,8-diazaanthracene-2,9,10-trione (5o)
3.3.8. 6-Methyl-1-(4-tolyl)-1,8-diazaanthracene-2,9,10-trione (5p)
3.3.9. 7-Methyl-1-(4-tolyl)-1,5-diazaanthracene-2,9,10-trione (11)
3.3.10. 6-Methyl-3-(p-tolyl)-1,8-diazaanthracene-2,9,10-trione (5q)
3.3.11. 5,8-Dihydro-3,4,5-trimethyl-1H-1,8-diazaanthracene-2,9,10-trione (6r)
3.3.12. 5,8-Dihydro-4-ethyl-5-methyl-1H-1,8-diazaanthracene-2,9,10-trione (6s)
3.3.13. 5,8-Dihydro-5-methyl-4-propyl-1H-1,8-diazaanthracene-2,9,10-trione (6t)
3.3.14. 5,8-Dihydro-5-methyl-1-(4-tolyl)-1,8-diazaanthracene-2,9,10-trione (6v)
3.3.15. 5-Methyl-3-(4-tolyl)-1H-1,8-diazaanthracene-2,9,10-trione (5w)
3.3.16. 5,8-Dihydro-5-ethyl-4-methyl-1,8-diazaanthracene-2,9,10-trione (6ae)
3.3.17. 5,8-Dihydro-4-methyl-5-propyl-1,8-diazaanthracene-2,9,10-trione (6af)
3.3.18. 5,8-Dihydro-5-butyl-4-methyl-1H-1,8-diazaanthracene-2,9,10-trione (6ag)
3.4. Diels–Alder Reaction with Chiral 1-Dialkylamino-1-Azadienes—General Procedure
3.4.1. (5R)-1,4,5-Trimethyl-5,8 Dihydro-1H-1,8-diazaanthracene-2,9,10-trione (6ai)
3.4.2. (5S)-1,4,5-Trimethyl-5,8 Dihydro-1H-1,8-diazaanthracene-2,9,10-trione (6aj)
3.5. Dehydrogenation of Dihydro Derivatives 6 to 1,8-Diazaanthracene-2,9,10-triones 5—General Procedures
3.5.1. 5-(p-(Dimethylamino)phenyl)-4-methyl-1H-1,8-diazaanthracene-2,9,10-trione (5i)
3.5.2. 4,6-Dimethyl-5-propyl-1H-1,8-diazaanthracene-2,9,10-trione (5k)
3.5.3. 3,4,5-Trimethyl-1H-1,8-diazaanthracene-2,9,10-trione (5r)
3.5.4. 4-Ethyl-5-methyl-1H-1,8-diazaanthracene-2,9,10-trione (5s)
3.5.5. 5-Methyl-4-propyl-1H-1,8-diazaanthracene-2,9,10-trione (5t)
3.5.6. 5-Methyl-1-(4-tolyl)-1,8-diazaanthracene-2,9,10-trione (5v)
3.6. Diels–Alder Reaction of Quinone 8b and 2,3-Dimethyl-1,3-butadiene
3.7. Diels–Alder Reaction of Quinone 8b and Anthracene—Synthesis of 4-Methyl-6,11-[1,2]Benzenonaphtho[2,3-g]Quinoline-2,5,12(1H,6H,11H)-trione 16
3.8. Diels–Alder Reaction of Quinone 8b and Cyclopentadiene
3.8.1. 9,10-Dihydroxy-5,8-methano-4-methyl-5,8-dihydro-1H-1-azaanthracen-2-one (17)
3.8.2. 5,8-Methano-4-methyl-5,8-dihydro-1H-1-azaanthracen-2,9,10-trione (18)
3.9. Bioassays
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dulo, B.; Phan, K.; Githaiga, J.; Raes, K.; De Meeste, S. Natural quinone dyes: A review on structure, extraction techniques, analysis and application potential. Waste Biomass Valor. 2021, 12, 6339–6374. [Google Scholar] [CrossRef]
- Malik, M.S.; Alsantali, R.I.; Jassas, R.S.; Alsimaree, A.A.; Syed, R.; Alsharif, M.A.; Kalpana, K.; Morad, M.; Althagafia, I.I.; Ahmed, S.A. Journey of anthraquinones as anticancer agents—A systematic review of recent literature. RSC Adv. 2021, 11, 35806. [Google Scholar] [CrossRef] [PubMed]
- Martins-Teixeira, M.B.; Carvalho, I. Antitumor anthracyclines: Progress and perspectives. Chem. Med. Chem. 2020, 15, 933–948. [Google Scholar] [CrossRef]
- Moiseeva, A.A. Anthracycline derivatives and their anticancer activity. INEOS Open 2019, 2, 9–18. [Google Scholar] [CrossRef]
- Shandukya, M.; Sharma, S.; Prasad Das, P.; Charak, S. Molecular-level understanding of the anticancer action mechanism of anthracyclines. In Advances in Precision Medicine Oncology; Arnouk, H., Hassan, B.A.R., Eds.; Intech Open: London, UK, 2020; Chapter 9. [Google Scholar] [CrossRef]
- Hasen, M.R.; Hurley, L.H. Pluramycins. Old drugs having modern friends in structural biology. Acc. Chem. Res. 1996, 29, 249–258. [Google Scholar] [CrossRef]
- Kitamura, K.; Ando, Y.; Matsumoto, T.; Suzuki, K. Synthesis of the pluramycins 1: Two designed anthrones as enabling platforms for flexible bis-C-glycosylation. Angew. Chem. Int. Ed. 2014, 53, 1258–1261. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, K.; Maezawa, Y.; Ando, Y.; Kusumi, T.; Matsumoto, T.; Suzuki, K. Synthesis of the pluramycins 2: Total synthesis and structure assignment of saptomycin B. Angew. Chem. Int. Ed. 2014, 53, 1262–1265. [Google Scholar] [CrossRef]
- Gredicak, M.; Jerik, I. Enediyne compounds—New promises in anticancer therapy. Acta Pharm. 2007, 57, 133–150. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Smith, A.L. The enediyne antibiotics. J. Med. Chem. 1996, 39, 2103–2117. [Google Scholar] [CrossRef]
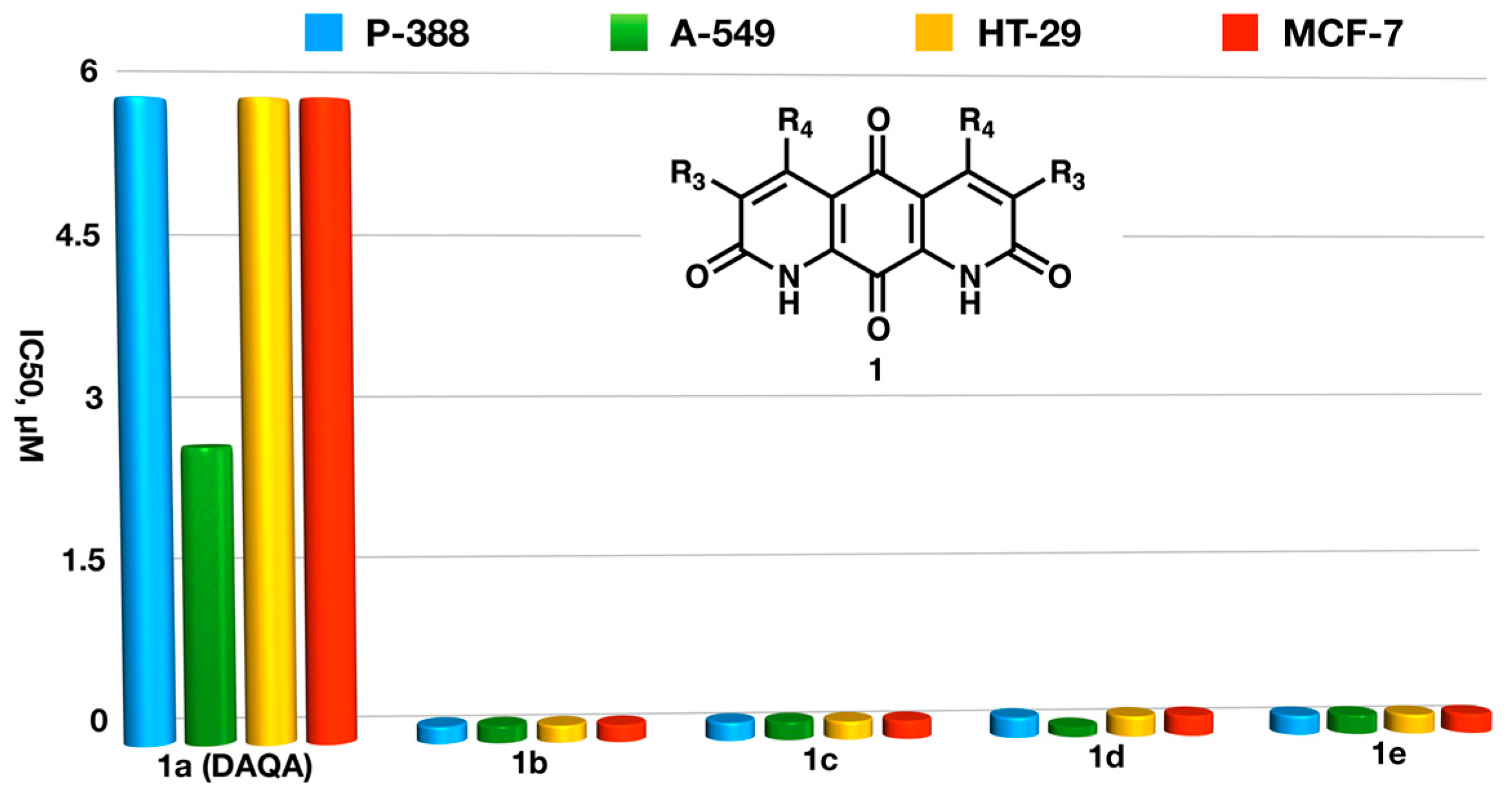
- Krapcho, A.P.; Maresch, M.J.; Hacker, M.P.; Menta, E.; Oliva, A.; Giuliani, F.C.; Spinelli, S. Aza and diaza bioisosteric anthracene-9,10-diones as antitumor agents. Acta Biochem. Polon. 1995, 42, 427–432. [Google Scholar] [CrossRef]
- Krapcho, A.P.; Maresch, M.J.; Hacker, M.P.; Hazelhurst, L.; Menta, E.; Oliva, A.; Spinelli, S.; Beggiolin, G.; Giuliani, F.C.; Pezzoni, G.; et al. Anthracene-9,10-diones and aza bioisosteres as antitumor agents. Curr. Med. Chem. 1995, 2, 803–824. [Google Scholar] [CrossRef]
- Bair, J.S.; Palchaudhuri, R.; Hergenrother, P.J. Chemistry and biology of deoxynyboquinone, a potent inducer of cancer cell death. J. Am. Chem. Soc. 2010, 132, 5469–5478. [Google Scholar] [CrossRef] [PubMed]
- Parkinson, E.I.; Hergenrother, P.J. Deoxy-nyboquinones as NQO1-activated cancer therapeutics. Acc. Chem. Res. 2015, 48, 2715–2733. [Google Scholar] [CrossRef] [PubMed]
- Forbis, R.M.; Rinehart, R.L. Nybomycin VII. Preparative routes to nybomycin and deoxynybomycin. J. Am. Chem. Soc. 1973, 95, 500–5013. [Google Scholar] [CrossRef]
- Lee, H.; Anderson, W.K. Total synthesis of of 4-acetyloxymethyl-1,6,9-trimethyl-1,-9-diazaanthracene-2,5,8,10-tetraone, a nybomycin acetate analogue. Tetrahedron Lett. 1990, 31, 4405–4408. [Google Scholar] [CrossRef]
- Ōmura, S.; Iwai, Y.; Hinotozawa, K.; Tanaka, H.; Takahashi, Y.; Nakagawa, A. OM-704 A, a new antibiotic active against gram-positive bacteria produced by Streptomyces sp. J. Antibiot. 1982, 35, 1425–1429. [Google Scholar] [CrossRef] [PubMed]
- Ōmura, S.; Nakagawa, A.; Aoyama, H.; Hinotozawa, K.; Sano, H. The structures of diazaquinomycins A and B, new antibiotic metabolites. Tetrahedron Lett. 1983, 24, 3643–3646. [Google Scholar] [CrossRef]
- Ōmura, S.; Murata, M.; Kimura, K.; Matsukura, S.; Nishihara, T.; Tanaka, H. Screening for new antifolates of microbial origin and a new antifolate AM-8402. J. Antibiot. 1985, 38, 1016–1024. [Google Scholar] [CrossRef]
- Murata, M.; MiYasaka, T.; Tanaka, H.; Omura, S. Diazaquinomycin A, a new antifolate antibiotic, inhibits thymidylate synthase. J. Antibiot. 1985, 38, 1025–1033. [Google Scholar] [CrossRef]
- Mullowney, M.W.; ÓhAinmhire, E.; Shikh, A.; Wei, X.; Tanouye, U.; Santarsiero, B.D.; Burdette, J.; Murphy, B.T. Diazaquinomycins E-G, novel diaza-anthracene analogs from a marine-derived Streptomyces sp. Mar. Drugs 2014, 12, 3574–3586. [Google Scholar] [CrossRef]
- Mullowney, M.W.; Hwang, C.H.; Newsome, A.G.; Wei, X.; Tanouye, U.; Wan, B.; Carlson, S.; Barranis, N.J.; hAinmhire, E.O.; Chen, W.-L.; et al. Diaza-anthracene antibiotics from freshwater-derived actinomycete with selective antibacterial activity toward Mycobacterim tuberculosis. ACS Infect. Dis. 2015, 1, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Braesel, J.; Lee, J.-H.; Arnould, B.; Murphy, B.T.; Eustáquio, A.S. Diazaquinomycin biosynthetic gene clusters from marine and freshwater actinomycetes. J. Nat. Prod. 2019, 82, 937–946. [Google Scholar] [CrossRef] [PubMed]
- Soonthornchareonnon, N.; Suwanborirux, K.; Bavovada, R.; Patarapanich, C.; Cassady, J.M. New cytotoxic 1-azaanthraquinones and 3-aminonapthoquinone from the stem bark of Goniothalamus marcanii. J. Nat. Prod. 1999, 62, 1390–1394. [Google Scholar] [CrossRef]
- Thanuphol, P.; Asami, Y.; Shiomi, K.; Wongnoppavich, A.; Tuchinda, P.; Soonthornchareonnon, N. Marcanine G, a new cytotoxic 1-azaanthraquinone from the stem bark of Goniothalamus marcanii Craib. Nat. Prod. Res. 2018, 32, 1682–1689. [Google Scholar] [CrossRef]
- Roumy, V.; Fabre, N.; Souard, F.; Massou, S.; Bourdy, G.; Maurel, S.; Valentin, A.; Moulis, C. Isolation and antimalarial activity of alkaloids from Pseudoxandra cuspidate. Planta Med. 2006, 72, 894–898. [Google Scholar] [CrossRef]
- Ichino, C.; Soonthornchareonnon, N.; Chuakul, W.; Kiyohara, H.; Ishiyama, A.; Sekiguchi, H.; Namatame, M.; Otoguro, K.; Ōmura, S.; Yamada, H. Screening of Thai medicinal plant extracts and their active constituents for in vitro antimalarial activity. Phytother. Res. 2006, 20, 307–309. [Google Scholar] [CrossRef]
- Jacobs, N.; Lang, S.; Panisch, R.; Wittstock, G.; Groth, U.; Nasiria, H.R. Investigation on the electrochemistry and cytotoxicity of the natural product marcanine A and its synthetic derivates. RSC Adv. 2015, 5, 58561–58565. [Google Scholar] [CrossRef]
- Thanuphol, T.; Noulsri, E.; Suwanborirux, K.; Soonthornchreannon, N.; Pattanapanyasal, K. Cytotoxic activity of marcanine C and some other aminoquinone derivates against MCF-7 cells. Mahidol Univ. J. Pharm. Sci. 2019, 43, 203–210. [Google Scholar] [CrossRef]
- Tsuzuki, K.; Yokozuka, T.; Murata, M.; Tanaka, H.; Ōmura, S. Synthesis and biological activity of analogues of diazaquinomycin A, a new thymidylate synthase inhibitor. J. Antibiot. 1989, 42, 727–737. [Google Scholar] [CrossRef]
- Gesto, C.; De la Cuesta, E.; Avendaño, C.; Emling, F. Synthesis and biological activity of new 1,8-diaza-2,9,10-anthracenetrione derivatives. J. Pharm. Sci. 1992, 8, 815–816. [Google Scholar] [CrossRef]
- Ramos, M.T.; Díaz-Guerra, L.M.; García-Copín, S.; Avendaño, C.; García-Grávalos, D.; De Quesada, T. Synthesis and Antitumor activity of fluorinated 1-aza and 1,8-diazaantraquinones. Il Farmaco 1996, 51, 375–379. [Google Scholar] [CrossRef] [PubMed]
- Pérez, J.M.; Avendaño, C.; Menéndez, J.C. 1-Acylamino-1-azadienes as an alternative to 1-dimethylamino-1-azadienes in the preparation of 1,8-diazaanthracene-2,9,10-triones. Tetrahedron 1995, 51, 6573–6586. [Google Scholar] [CrossRef]
- Pérez, J.M.; López-Alvarado, P.; Alonso, M.A.; Avendaño, C.; Menéndez, J.C. Silica gel-supported hetero Diels-Alder reactions of quinolinetriones. Tetrahedron Lett. 1996, 37, 6955–6958. [Google Scholar] [CrossRef]
- Pascual-Alfonso, E.; Avendaño, C.; Menéndez, J.C. Improvements in the hetero Diels-Alder reactions of 1-dimethylamino-1-azadienes in the presence of an electrophilic scavenger resin. Synlett 2000, 2000, 205–208. [Google Scholar] [CrossRef]
- Blanco, M.M.; Alonso, M.A.; Avendaño, C.; Menéndez, J.C. New findings in hetero Diels-Alder reactions of quinolinetriones. Tetrahedron 1996, 52, 5933–5944. [Google Scholar] [CrossRef]
- Villacampa, M.; Pérez, J.M.; Avendaño, C.; Menéndez, J.C. Ultrasound assisted Diels-Alder reactions of 1-azadienes with “normal” electronic demand. Tetrahedron 1994, 50, 10047–10054. [Google Scholar] [CrossRef]
- Waldner, A. [4+2]-Cycloadditionen von α,β-ungesättigten Hydrazonen. Teil 2. Pyridin-2,3-dicarboximide aus 1,4-Dihydropyridin-Derivaten. Helv. Chim. Acta 1988, 71, 493–497. [Google Scholar] [CrossRef]
- Echavarren, A. Lewis acid catalyzed reactions of α,β-Unsaturated N,N-dimethylhydrazones with 1,4-benzoquinone. Formation of indoles by a novel oxidative rearrangement. J. Org. Chem. 1990, 55, 4255–4260. [Google Scholar] [CrossRef]
- Satsumabayashi, S.; Nakajo, K.; Soneda, R.; Motoki, S. Preparation of Some α,β-Unsaturated Aldehydes. Bull. Chem. Soc. Jap. 1970, 43, 1586. [Google Scholar] [CrossRef]
- López-Alvarado, P.; Avendaño, C.; Menéndez, J.C. A General synthesis of quinoline-2,5,8 (1H)-trione via acylation of 2,5-dimethoxyaniline with S-tert-butyl thioacetates by application of the Knorr Cyclization. Synthesis 1998, 1998, 186–194. [Google Scholar] [CrossRef]
- Gesto, C.; De la Cuesta, E.; Avendaño, C. A Comparative study of synthetic approaches to 1-methyl-2,5,8(1H)-quinolinetrione and 4-methyl-2,5,8(1H)-quinolinetrione. Synthesis 1991, 1991, 727–730. [Google Scholar] [CrossRef]
- Pettit, G.R.; Fleming, W.C.; Paull, K.D. Synthesis of the 6- and 7-hydroxy-5,8-dioxocarbostyrils. J. Org. Chem. 1968, 33, 1089–1092. [Google Scholar] [CrossRef] [PubMed]
- Pérez, J.M.; López-Alvarado, P.; Pascual-Alfonso, E.; Avendaño, C.; Menéndez, J.C. Concise preparation of 1,8-diazaanthracene-2,7,9,10-tetraones. Two alternative syntheses of the natural antifolate diazaquinomycin A. Tetrahedron 2000, 56, 4575–4583. [Google Scholar] [CrossRef]
- Alonso, M.A.; Blanco, M.M.; Avendaño, C.; Menéndez, J.C. Synthesis of 2,5,8(1H)-quinolinetrione derivatives through Vilsmeier-Haack formylation of 2,5-dimethoxyanilides. Heterocycles 1993, 36, 2315–2325. [Google Scholar] [CrossRef]
- Gesto, C.; De la Cuesta, E.; Avendaño, C. Synthesis of diaza-anthraquinones by hetero Diels-Alder cycloaddition reactions. Tetrahedron 1989, 45, 4477–4484. [Google Scholar] [CrossRef]
- Enders, D.; Kipphardt, H.; Gerdes, P.; Breña-Valle, L.J.; Bhunsan, V. Large scale preparation of versatile chiral auxiliaries derived from (S)-proline. Bull. Soc. Chim. Belg. 1988, 97, 691. [Google Scholar] [CrossRef]
- Beaudgnies, R.; Ghosez, L. Asymmetric Diels-Alder reactions with chiral 1-azadienes. Tetrahedron Asymmetry 1994, 5, 557–560. [Google Scholar] [CrossRef]
- Pérez, J.M.; Vidal, L.; Grande, M.T.; Menéndez, J.C.; Avendaño, C. Regioselectivity of the Diels-Alder reaction of 2,5,8 (1H)-quinolinetriones. Tetrahedron 1994, 50, 7923–7932. [Google Scholar] [CrossRef]
- Kelly, T.R.; Field, J.A.; Li, Q. Synthesis of diazaquinomycin A and B: The first double Knorr cyclization. Tetrahedron Lett. 1988, 29, 3545–3546. [Google Scholar] [CrossRef]
- Prior, A.M.; Sun, D. Total synthesis of diazaquinomycins H and J using double Knorr cyclization in the presence of triisopropylsilane. RSC Adv. 2019, 9, 1759–1771. [Google Scholar] [CrossRef]
- Kovacic, P.; Osuna, J.A. Mechanisms of anti-cancer agents: Emphasis on oxidative stress and electron transfer. Curr. Pharm. Des. 2000, 6, 277–309. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Karakhanova, S.; Werner, J.; Bazhin, A.V. Reactive oxygen species in cancer biology and anticancer therapy. Curr. Med. Chem. 2013, 20, 3677–3692. [Google Scholar] [CrossRef]
- Denmark, S.E.; Edwards, J.P.; Weber, T.; Piotrowski, D.W. Organocerium additions to proline-derived hydrazones: Synthesis of enantiomerically enriched amines. Tetrahedron Asymmetry 2010, 21, 1278–1302. [Google Scholar] [CrossRef]
- Beebe, X.; Schore, N.E.; Kurth, M.J. Polymer-supported synthesis of cyclic ethers: Electrophilic cyclization of isoxazolines. J. Org. Chem. 1995, 60, 4196. [Google Scholar] [CrossRef]
- Bergeron, R.J.; Gavanaugh, P.F., Jr.; Kline, S.J.; Hughes, R.G., Jr.; Elliott, G.T.; Porter, C.W. Antineoplastic and antiherpetic activity of spermidine catecholamide iron chelators. Biochem. Bioph. Res. Comm. 1984, 121, 848–854. [Google Scholar] [CrossRef]
- Schroeder, A.C.; Hughes, R.G.; Bloch, A. Synthesis and biological effects of acyclic pyrimidine nucleoside analogs. J. Med. Chem. 1981, 24, 1078–1083. [Google Scholar] [CrossRef]





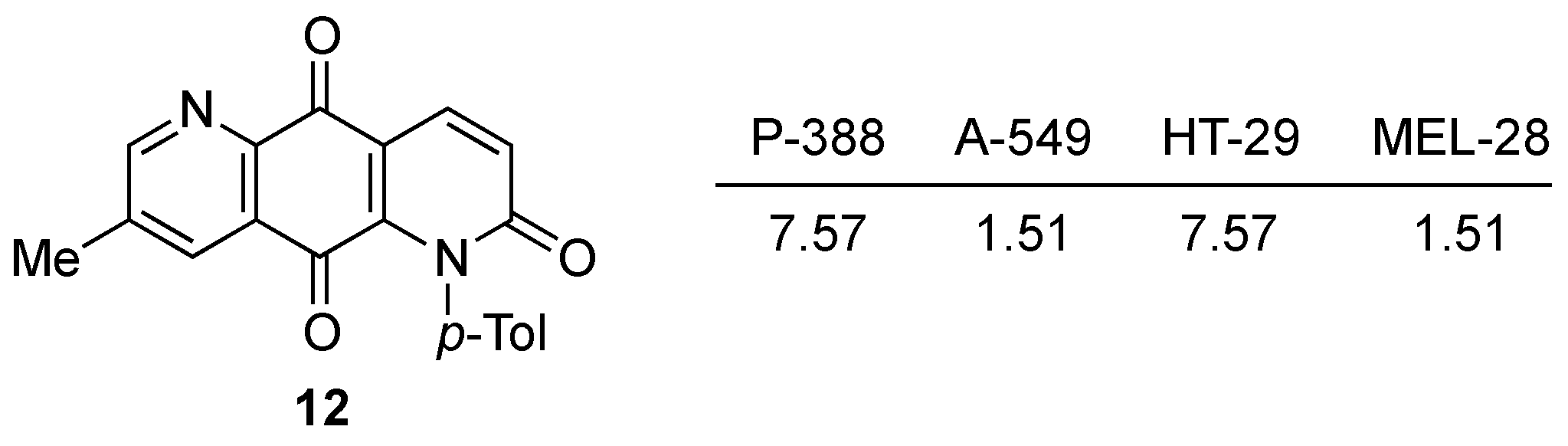


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
| Comp. | R5 | R6 | Comp. | R5 | R6 |
| a | H | H | f | Ph | H |
| b | H | Me | g | 4-Me2NC6H4 | H |
| c | H | Et | h | Et | Me |
| d | H | nBu | i | nPr | Me |
| e | Me | H | j | nBu | Me |
 | |||||||
| Comp. | R1 | R3 | R4 | Comp. | R1 | R3 | R4 |
| a | H | H | H | h | p-Tol | H | H |
| b | H | H | Me | i | H | p-Tol | H |
| c | Me | H | Me | j | H | H | nPr |
| d | H | Me | Me | k | H | Me | H |
| e | H | Bn | Me | l | H | Et | H |
| f | H | H | Et | m | H | Ph | H |
| g | H | H | (CH2)2Ph | n | H | CO2Et | H |
| Comp. | R1 | R3 | R4 | R5 | R6 | Method a | Yield, % | ||
|---|---|---|---|---|---|---|---|---|---|
| 5 | 6 | 10 | |||||||
| a | H | H | H | H | H | A | 81 | 0 | 8 b |
| b | H | H | H | H | Et | A | 70 | 0 | 0 b |
| c | H | H | Me | H | H | A | 70 | 0 | 14 b |
| d | H | H | Me | H | Me | A (B) | 48 (88) | 0 | 20 b (0) c |
| e | H | H | Me | H | Et | A | 42 | 0 | 33 b |
| f | H | H | Me | H | nBu | A (C) | 30 (68) | 0 | 50 (8) f |
| g | H | H | Me | Me | H | A (C) | 0 | 51 (53) | 35 b (15) f |
| h | H | H | Me | Ph | H | A | 0 | 34 | 65 b |
| i | H | H | Me | 4-Me2NC6H4 | H | A | 0 | 11 | 61 e |
| j | H | H | Me | Et | Me | A | 0 | 47 | 45 b |
| k | H | H | Me | nPr | Me | A | 0 | 32 | 40 |
| l | H | Me | Me | H | Me | A | 76 | 0 | 11 |
| m | H | Bn | Me | H | Me | B | 68 | 0 | 0 |
| n | H | H | Et | H | Me | A | 70 | 0 | 9 |
| o | H | H | (CH2)2Ph | H | Me | A (B) | 49 (82) | 0 (0) | 15 (0) c |
| p | p-Tol | H | H | H | Me | D | 52 | 0 | 9 |
| q | H | p-Tol | H | H | Me | B | 94 | 0 | 0 |
| r | H | Me | Me | Me | H | A (B) | 0 | 32 (70) | 42 (6) c |
| s | H | H | Et | Me | H | A (B) | 0 | 29 (84) | 59 (0) c |
| t | H | H | nPr | Me | H | A(B) | 0 | 32 (92) | 50 (0) c |
| u | H | Me | nPr | nPr | Me | B | 24 | 46 | 0 g |
| v | p-Tol | H | H | Me | H | B | 0 | 80 | 0 |
| w | H | p-Tol | H | Me | H | B | 64 | 0 | 0 |
| x | H | Me | H | H | Me | A | 50 | 0 | 0 d |
| y | H | Et | H | H | Me | A | 45 | 0 | 0 d |
| z | H | Ph | H | H | Me | A | 50 | 0 | 0 d |
| aa | H | Me | H | Me | H | D | 46 | 6 | 0 d |
| ab | H | Et | H | Me | H | A (B) | 18 (44) | 27 (0) | 7 (0) d |
| ac | H | Ph | H | Me | H | D | 40 | 0 | 20 d |
| ad | H | CO2Et | H | H | Me | A | 59 | 0 | 0 d |
| ae | H | H | Me | Et | H | A (C) | 0 | 28 (70) | 25 (8) f |
| af | H | H | Me | nPr | H | A (C) | 0 | 27 (62) | 65 (8) f |
| ag | H | H | Me | nBu | H | A (C) | 0 | 30 (65) | 50 (8) f |
| Comp. | R1 | R3 | R4 | R5 | R6 | Method a | Yield, % |
|---|---|---|---|---|---|---|---|
| 5g | H | H | Me | Me | H | A (B) | 88 (90) b |
| 5h | H | H | Me | Ph | H | A (B) | 83 (70) b |
| 5i | H | H | Me | 4-Me2NC6H4 | H | B | 80 |
| 5j | H | H | Me | Et | Me | A (B) | 94 (95) b |
| 5k | H | H | Me | nPr | Me | A | 94 |
| 5r | H | Me | Me | Me | H | C | 74 c |
| 5s | H | H | Et | Me | H | B | 82 |
| 5t | H | H | nPr | Me | H | A | 92 |
| 5u | H | Me | nPr | nPr | Me | A | 63 c |
| 5v | p-Tol | H | H | Me | H | A | 94 |
| Comp. | R1 | R4 | R6 | R7 | R8 | Conditions | Yield % |
|---|---|---|---|---|---|---|---|
| 7a | H | H | H | H | H | CHCl3, 95 °C, 2 h (sealed tube) | 79 a,d |
| 7b | Me | Me | H | H | H | AcOEt, 100 °C, 24 h (sealed tube) | 81 a |
| 7c | H | Me | Me | H | H | AcOEt, 120 °C, 24 h (sealed tube) | 64 a,c |
| 7d | H | Me | H | H | Me | AcOEt, 120 °C, 14 h (sealed tube) | 60 c |
| 7e | H | Me | Me | H | Me | AcOEt, 120 °C, 1 h, then xylene, 150 °C, 36 h | 57 a |
| 7f (marcanine A) | H | Me | H | H | H | AcOEt, 120 °C, 24 h (sealed tube) | 81 b |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Avendaño, C.; López-Alvarado, P.; Pérez, J.M.; Alonso, M.Á.; Pascual-Alfonso, E.; Ruiz-Serrano, M.; Menéndez, J.C. Structure-Antitumor Activity Relationships of Aza- and Diaza-Anthracene-2,9,10-Triones and Their Partially Saturated Derivatives. Molecules 2024, 29, 489. https://doi.org/10.3390/molecules29020489
Avendaño C, López-Alvarado P, Pérez JM, Alonso MÁ, Pascual-Alfonso E, Ruiz-Serrano M, Menéndez JC. Structure-Antitumor Activity Relationships of Aza- and Diaza-Anthracene-2,9,10-Triones and Their Partially Saturated Derivatives. Molecules. 2024; 29(2):489. https://doi.org/10.3390/molecules29020489
Chicago/Turabian StyleAvendaño, Carmen, Pilar López-Alvarado, José María Pérez, Miguel Ángel Alonso, Eva Pascual-Alfonso, Miriam Ruiz-Serrano, and J. Carlos Menéndez. 2024. "Structure-Antitumor Activity Relationships of Aza- and Diaza-Anthracene-2,9,10-Triones and Their Partially Saturated Derivatives" Molecules 29, no. 2: 489. https://doi.org/10.3390/molecules29020489
APA StyleAvendaño, C., López-Alvarado, P., Pérez, J. M., Alonso, M. Á., Pascual-Alfonso, E., Ruiz-Serrano, M., & Menéndez, J. C. (2024). Structure-Antitumor Activity Relationships of Aza- and Diaza-Anthracene-2,9,10-Triones and Their Partially Saturated Derivatives. Molecules, 29(2), 489. https://doi.org/10.3390/molecules29020489