


Six New Compounds from the Herbaceous Stems of Ephedra intermedia Schrenket C. A. Meyer and Their Lung-Protective Activity


Abstract
1. Introduction
2. Results and Discussion
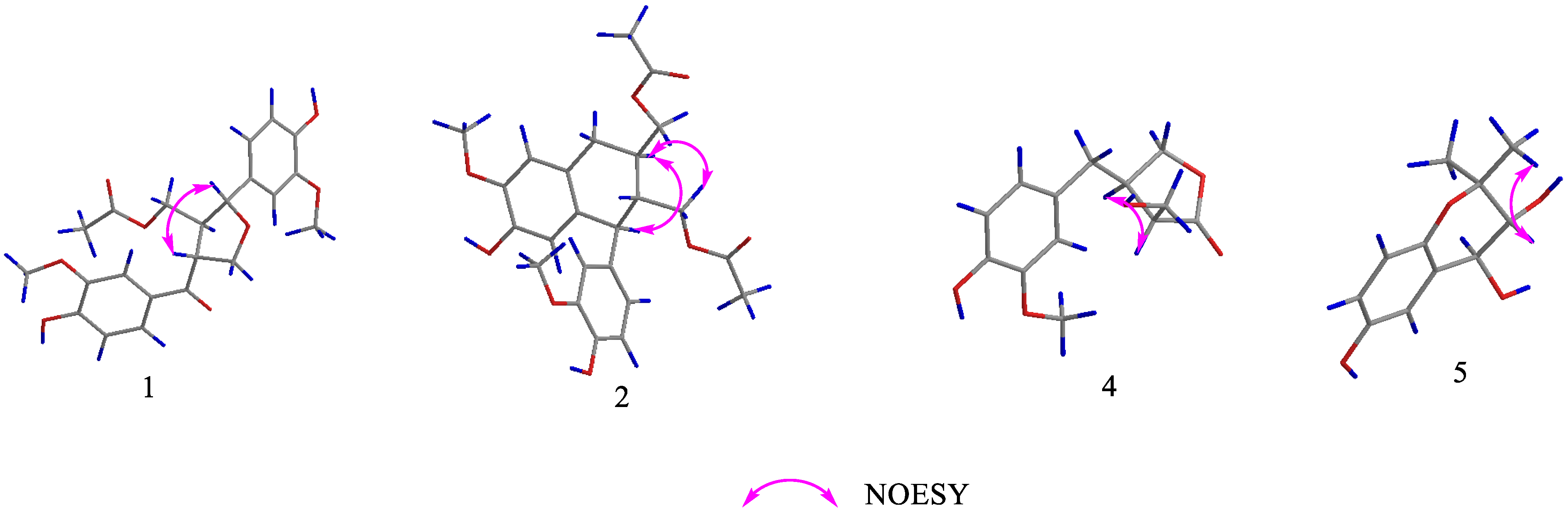
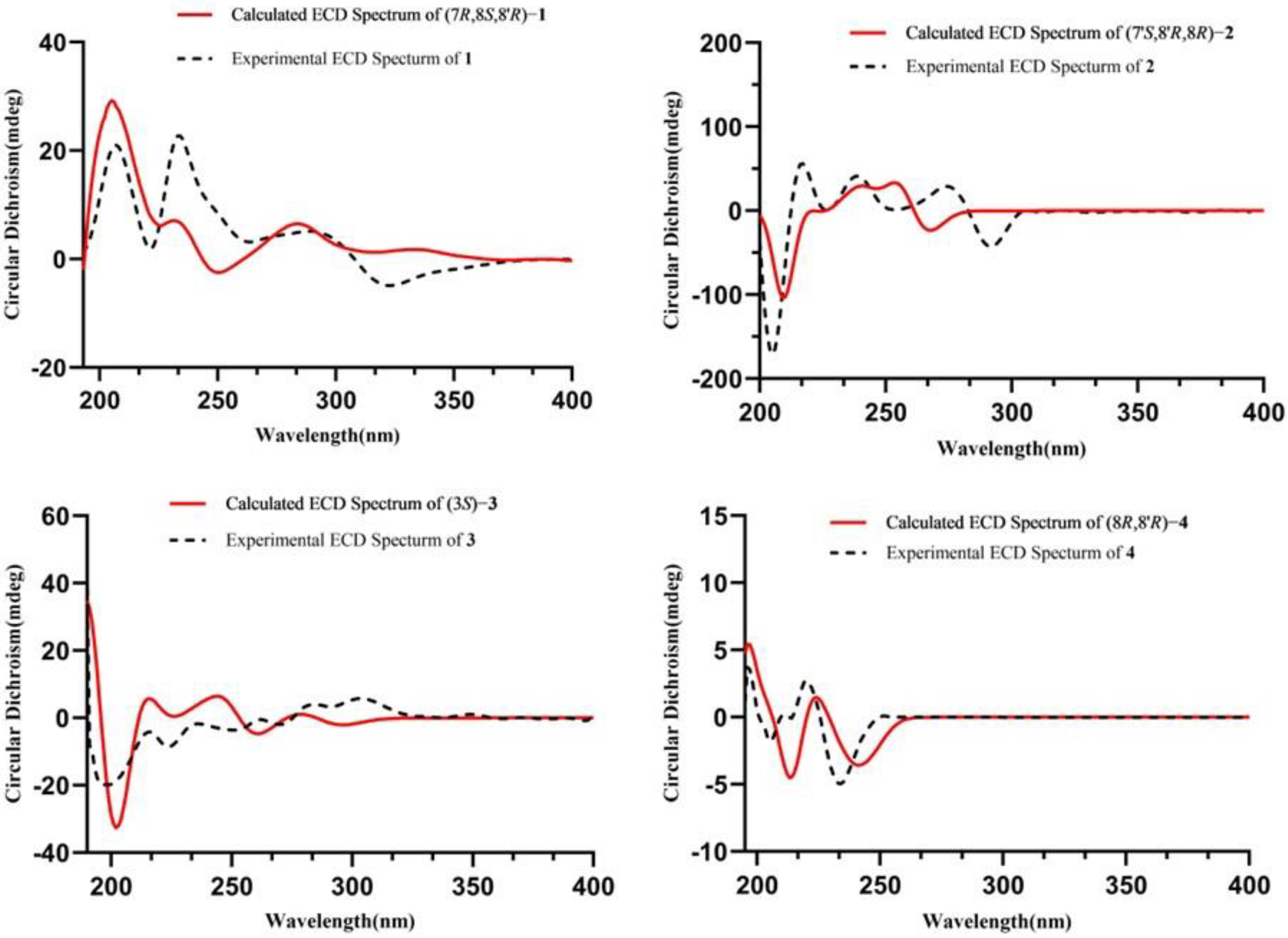
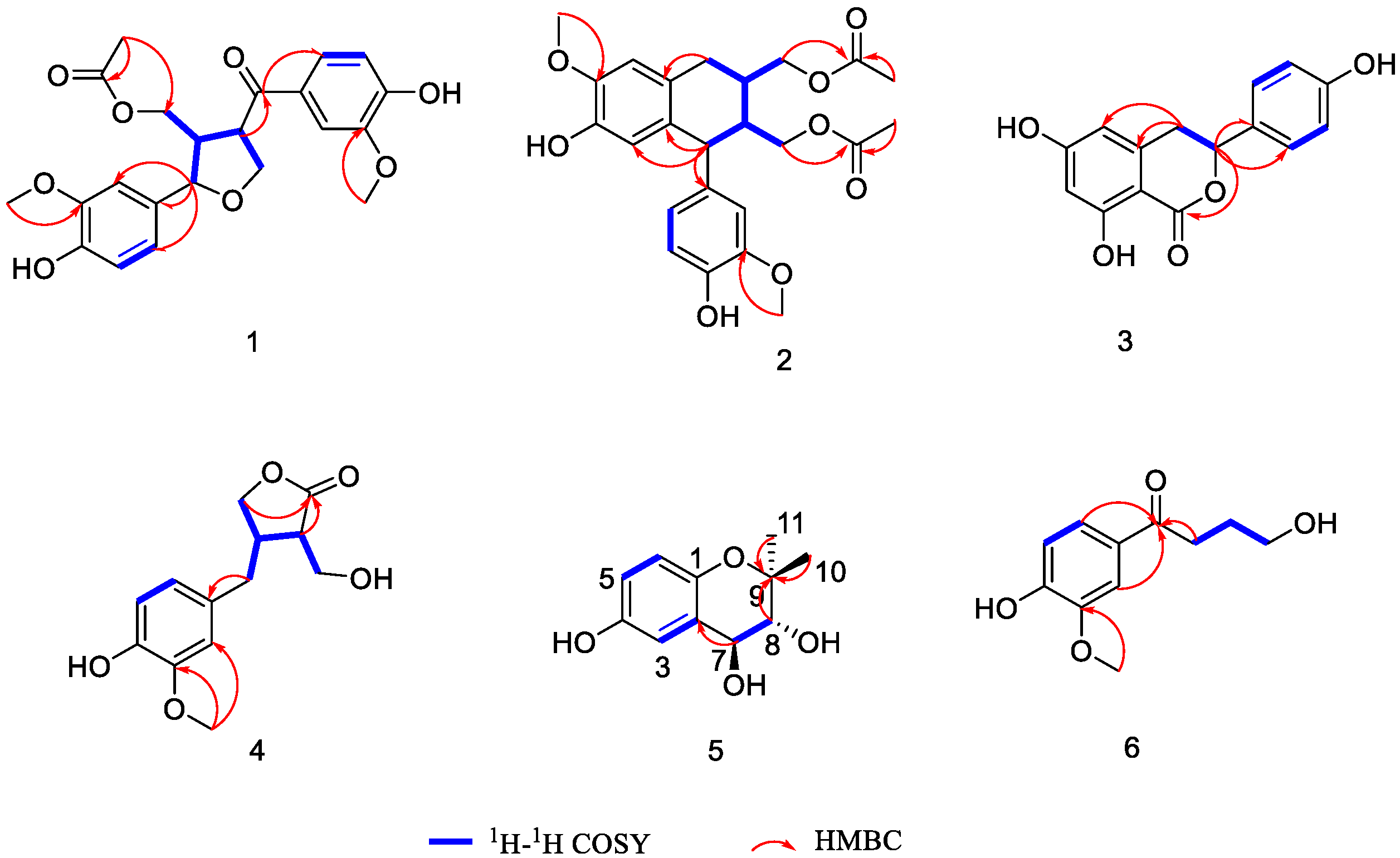
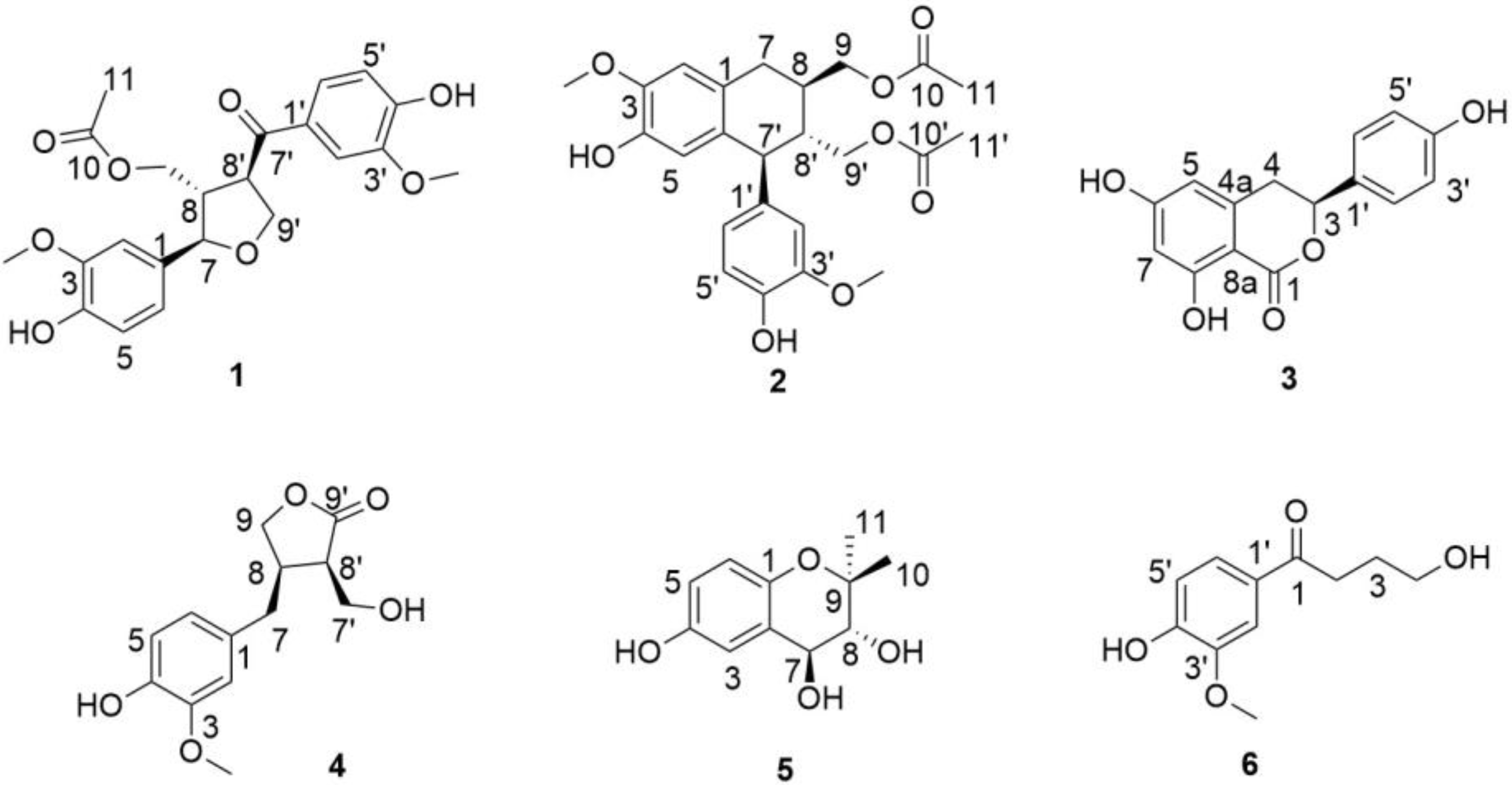
Structure Elucidation of the Isolated Compounds
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Plant Material
3.3. Extraction and Isolation
3.4. Biological Activity
3.4.1. Cell Culture
3.4.2. MTT Assay
3.4.3. Statistical Analysis
3.5. ECD Spectra Calculations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Li, H.Y.; Ding, X.Y.; Zhang, D.; An, Q.; Jin, Y.; Zhan, Z.L.; Zheng, Y.G. Herbal textual research on Ephedrae herba in famous classical formulas. Chin. J. Exp. Tradit. Med. Formulae 2022, 28, 102–110. [Google Scholar]
- Ren, H.B.; Wang, Y.C.; Ma, J.M.; Li, J.S.; Niu, L.Y. Active ingredients of Ephedra and their clinical application and contraindications. Chin. J. Pharmacovigil. 2021, 18, 396–399. [Google Scholar]
- Zhu, D.H.; Zhang, J.K.; Jia, J.F.; Liu, J.J.; Wei, J.J.; Yang, M.; Yang, Y.; Li, M.; Hao, Z.Y.; Zheng, X.K.; et al. Lignans and terpenoids from the stem of Ephedra equisetina Bunge. Phytochemistry 2022, 200, 113230. [Google Scholar] [CrossRef]
- Zang, X.Y.; Shang, M.Y.; Xu, F.; Liang, J.; Wang, X.; Mikage, M.; Cai, S.Q. A-type proanthocyanidins from the stems of Ephedra sinica (Ephedraceae) and their antimicrobial activities. Molecules 2013, 18, 5172–5189. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.L.; Cao, Y.G.; Zeng, M.N.; Wang, R.; Liu, Y.L.; Chen, X.; Ma, X.Y.; Ren, Y.J.; He, C.; Li, X.D.; et al. A New lignan from the herbaceous stems of Ephedra intermedia Schrenket CA Meyer. Rec. Nat. Prod. 2023, 17, 293–299. [Google Scholar]
- Long, C.; Kakiuchi, N.; Zhong, G.; Mikage, M. Survey on resources of Ephedra plants in Xinjiang. Biol. Pharm. Bull. 2005, 28, 285–288. [Google Scholar] [CrossRef]
- Tian, N.N.; Yang, Q.H.; Zhu, Y.X.; Zeng, X.S.; Yuan, J.Y.; Yang, J.L.; Jia, W.W.; Li, C. Mahuang (herbaceous stem of Ephedra spp.): Chemistry, pharmacodynamics, and pharmacokinetics. Chin. J. Chin. Mater. Med. 2022, 47, 3409–3424. [Google Scholar]
- Ma, X.H.; Lu, Y.Y.; Zhang, X.F.; Huang, D.D.; Zhu, T.T.; Jin, L. UPLC fingerprint and chemical components difference among Ephedra intermedia from different regions. J. Chin. Med. Mater. 2016, 39, 2217–2220. [Google Scholar]
- Jia, J.F.; Zeng, M.N.; Zhu, D.H.; Jiao, X.M.; Zhang, B.B.; Yang, R.L.; Feng, W.S.; Zheng, X.K. An amide alkaloid isolated from Ephedra sinica ameliorates OVA-induced allergic asthma by inhibiting mast cell activation and dendritic cell maturation. Int. J. Mol. Sci. 2022, 23, 13541. [Google Scholar] [CrossRef]
- Wang, X.; Fu, W.; Wang, Z.B.; Kuang, H.X.; Wang, Q.H. Research progress of Ephedrae herab polysaccharide. China J. Tradit. Chin. Med. 2019, 34, 3138–3139. [Google Scholar]
- Miao, S.M.; Zhang, Q.; Bi, X.B.; Cui, J.L.; Wang, M.L. A review of the phytochemistry and pharmacological activities of Ephedra herb. Chin. J. Nat. Med. 2020, 18, 321–344. [Google Scholar] [CrossRef] [PubMed]
- Nagata, Y.; Ando, H.; Sasaki, Y.; Suzuki, R. Ephedra herb, Mao, inhibits antigen-induced mast cell degranulation by induction of the affinity receptor for IgE internalization. Pharm. Res. 2021, 38, 569–581. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Deng, A.J.; Du, G.H.; Zhang, J.L.; Li, Z.H.; Qin, H.L. Chemical constituents of the stems of Ephedra sinica. J. Asian Nat. Prod. Res. 2009, 11, 168–171. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.G.; Mu, X.Y.; Pan, S.B.; Luan, R.Q.; Zhao, P. Ephedrae herba: A comprehensive review of its traditional uses, phytochemistry, pharmacology, and toxicology. J. Ethnopharmacol. 2023, 307, 116153. [Google Scholar] [CrossRef] [PubMed]
- Lv, Y.N.; Wang, S.S.; Liang, P.D.; Wang, Y.M.; Zhang, X.; Jia, Q.Q.; Fu, J.; Han, S.L.; He, L.C. Screening and evaluation of anti-SARS-CoV-2 components from Ephedra sinica by ACE2/CMC-HPLC-IT-TOF-MS approach. Anal. Bioanal. Chem. 2021, 413, 2995–3004. [Google Scholar] [CrossRef]
- Elhadef, K.; Smaoui, S.; Fourati, M.; Ben Hlima, H.; Chakchouk Mtibaa, A.; Sellem, I.; Ennouri, K.; Mellouli, L. A review on worldwide Ephedra history and story: From fossils to natural products mass spectroscopy characterization and biopharmacotherapy potential. Evid. Based Complement. Alternat. Med. 2020, 30, 1540638. [Google Scholar]
- Thakur, A.; Pathak, S.R. Introduction to medicinally important constituent from Chinese medicinal plants. Synthesis. Med. Agents Plants 2018, 333–349. [Google Scholar] [CrossRef]
- Duan, K.F.; Zang, X.Y.; Shang, M.Y.; Zhang, W.; Xie, B.B.; Wang, L.; Xu, F.; Cai, S.Q. Non-ephedrine constituents from the herbaceous stems of Ephedra sinica. Fitoterapia 2021, 153, 104998. [Google Scholar] [CrossRef] [PubMed]
- Lv, M.Y.; Chen, J.Q.; Gao, Y.Q.; Sun, J.B.; Zhang, Q.Q.; Zhang, M.H.; Xu, F.G.; Zhang, Z.J. Metabolomics based on liquid chromatography with mass spectrometry reveals the chemical difference in the stems and roots derived from Ephedra sinica. J. Sep. Sci. 2015, 38, 3331–3336. [Google Scholar] [CrossRef]
- Zhang, B.M.; Wang, Z.B.; Xin, P.; Wang, Q.H.; Bu, H.; Kuang, H.X. Phytochemistry and pharmacology of genus Ephedra. Chin. J. Nat. Med. 2018, 16, 811–828. [Google Scholar] [CrossRef] [PubMed]
- Kuang, H.X.; Xia, Y.G.; Yang, B.Y.; Wang, Q.H.; Wang, Y.H. Screening and comparison of the immunosuppressive activities of polysaccharides from the stems of Ephedra sinica Stapf. Carbohydr. Polym. 2011, 83, 787–795. [Google Scholar] [CrossRef]
- Pullela, S.V.; Takamatsu, S.; Khan, S.I.; Khan, I.A. Isolation of lignans and biological activity studies of Ephedra viridis. Planta Med. 2005, 71, 789–791. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Kim, Y.N.; Kwak, H.J.; Jeong, E.J.; Kim, S.H. Estrogenic activity of constituents from the rhizomes of Rheum undulatum Linné. Bioorg. Med. Chem. Lett. 2018, 28, 552–557. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.B.; Zhao, B.B.; Lu, C.H.; Lin, X.J.; Zheng, Z.H.; Su, W.J. Isolation, structure elucidation and apoptosis-inducing activity of new compounds from the edible fungus Lentinus striguellus. Nat. Prod. Commun. 2009, 4, 501–506. [Google Scholar] [CrossRef] [PubMed]
- Kis, Z.; Close, A.; Sigg, H.P. Structure of panepoxydone and related fungi metabolites. Helvetica Chimica Acta 1970, 53, 1577–1597. [Google Scholar] [CrossRef]
- Wei, J.; Zhang, X.Y.; Deng, S.; Cao, L.; Xue, Q.H.; Gao, J.M. α-glucosidase inhibitors and phytotoxins from Streptomyces xanthophaeus. Nat. Prod. Res. 2017, 31, 2062–2066. [Google Scholar] [CrossRef]
- Gao, H.X.; Ding, A.W.; Tang, Y.P.; Zhang, X.; Duan, J.A. Chemical Constituents from the rhizomas of Phragmites communis. Chin. J. Nat. Med. 2009, 7, 196–198. [Google Scholar] [CrossRef]
- Li, K.; Li, Q.L.; Ji, N.Y.; Liu, B.; Zhang, W.; Cao, X.P. Deoxyuridines from the marine sponge associated actinomycete streptomyces microflavus. Mar. Drugs 2011, 9, 690–695. [Google Scholar] [CrossRef]
- Wu, L.L.; Liu, J.E.; Yu, Z.L.; Zhang, L.Q.; Li, Y.M. Phenolic constituents of Sanguisorba officinalis and their Nrf2 agonistic effect. Acta Pharmacol. Sin. 2023, 41, 1–27. [Google Scholar]
- Wang, X.G.; Shen, L.T.; Zeng, Y.Y.; Shen, L.T.; Xu, H.H. Flavonoids from Ficus sarmentosa var. henryi. Chin. Tradit. Herbal. Drugs 2010, 41, 526–529. [Google Scholar]
- Xu, Q.Q.; Qiao, Y.B.; Zhang, Z.J.; Deng, Y.F.; Chen, T.Q.; Tao, L.; Xu, Q.X.; Liu, J.J.; Sun, W.G.; Ye, J.; et al. New polyketides with anti-inflammatory activity from the Fungus Aspergillus rugulosa. Front. Pharmacol. 2021, 12, 700573. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.F.; Zhang, N.; Zhou, Z.Y.; Yang, J.X.; Liu, J.S.; Wang, G.K. Phenolic constituents from Kalimeris shimadae and their antitumor activity. Chin. Tradil. Patent Med. 2020, 42, 2922–2926. [Google Scholar]
- Wen, D.X.; Chen, Z.L. Studies on thechemical constituents of Tacca chantrieri Andre. in Hainan. Guihaia 1995, 15, 371–373. [Google Scholar]
- Liu, Y.L.; Wang, M.N.; Cao, Y.G.; Zeng, M.N.; Zhang, Q.Q.; Ren, Y.J.; Chen, X.; He, C.; Fan, X.L.; Zheng, X.K.; et al. Chemical constituents from the flowers of Carthamus tinctorius L. and their lung protective activity. Molecules 2022, 27, 3573. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | 1 | 2 | ||
|---|---|---|---|---|
| δH | δC | δH | δC | |
| 1 | 133.4 | 128.1 | ||
| 2 | 6.98 (1H, d, 1.8) | 111.0 | 6.65 (1H, s) | 112.3 |
| 3 | 149.3 | 147.5 | ||
| 4 | 147.7 | 145.5 | ||
| 5 | 6.79 (1H, d, 8.0) | 116.1 | 6.16 (1H, s) | 117.2 |
| 6 | 6.83 (1H, dd, 8.0, 1.8) | 120.5 | 133.4 | |
| 7 | 4.71 (1H, d, 7.9) | 85.2 | 2.83 (1H, dd, 15.8, 5.2) 2.77 (1H, dd, 15.8, 10.4) | 33.6 |
| 8 | 2.88 (1H, m,) | 51.6 | 2.20 (1H, m) | 37.1 |
| 9 | 4.01 (1H, dd, 11.3, 8.1) 3.97 (1H, dd, 11.3, 6.1) | 63.3 | 4.20 (1H, dd, 11.2, 4.7) 4.07 (1H, dd, 11.2, 6.2) | 67.9 |
| 10 | 172.7 | 173.0 | ||
| 11 | 1.56 (3H, s) | 20.2 | 2.0 (3H, s) | 20.8 |
| 3-OCH3 | 3.87 (3H, s) | 56.4 | 3.80 (3H, s) | 56.4 |
| 1′ | 131.1 | 137.4 | ||
| 2′ | 7.56 (1H, d, 2.0) | 111.9 | 6.64 (1H, d, 2.0) | 113.7 |
| 3′ | 149.2 | 149.2 | ||
| 4′ | 153.7 | 146.3 | ||
| 5′ | 6.87 (1H, d, 8.4) | 115.9 | 6.74 (1H, d, 8.0) | 116.2 |
| 6′ | 7.66 (1H, dd, 8.4, 2.0) | 120.5 | 6.57 (1H, dd, 8.0, 2.0) | 123.1 |
| 7′ | 200.0 | 3.78 (1H, m) | 48.5 | |
| 8′ | 4.48 (1H, m) | 48.0 | 2.00 (1H, m) | 45.0 |
| 9′ | 4.33 (1H, dd, 8.6, 7.3) 4.21 (1H, dd, 8.6, 6.7) | 71.6 | 4.08 (1H, dd, 11.5, 3.2) 3.90 (1H, dd, 11.5, 3.8) | 64.9 |
| 10′ | 172.9 | |||
| 11′ | 2.0 (3H, s) | 20.6 | ||
| 3′-OCH3 | 3.90 (3H, s) | 56.4 | 3.76 (3H, s) | 56.4 |
| No. | 3 | 4 | 5 | 6 | ||||
|---|---|---|---|---|---|---|---|---|
| δH | δC | δH | δC | δH | δC | δH | δC | |
| 1 | 171.8 | 131.9 | 146.8 | 199.6 | ||||
| 2 | 6.81 (1H, d, 1.8) | 113.3 | 126.4 | 3.14 (2H, t, 6.2) | 41.6 | |||
| 3 | 5.48 (1H, dd, 12.2, 3.1) | 82.1 | 149.2 | 6.88 (1H, d, 2.7) | 114.8 | 1.89 (2H, brs) | 24.2 | |
| 4 | 3.25 (1H, dd, 16.5, 12.2) 3.01(1H, dd, 16.5, 3.1) | 35.9 | 146.2 | 151.2 | 3.92 (2H, t, 6.2) | 59.0 | ||
| 4a | 143.7 | |||||||
| 5 | 6.26 (1H, s) | 107.9 | 6.72 (1H, d, 8.0) | 116.3 | 6.59 (1H, dd, 8.7, 2.7) | 117.2 | ||
| 6 | 166.4 | 6.66 (1H, dd, 8.0, 1.8) | 122.2 | 6.55 (1H, d, 8.7) | 118.1 | |||
| 7 | 6.22 (1H, d, 2.0) | 102.3 | 2.83 (1H, dd, 14.9, 8.6) 2.55 (1H, dd, 14.9, 6.4) | 33.8 | 4.39 (1H, d, 8.4) | 70.4 | ||
| 8 | 165.7 | 3.00 (1H, m) | 40.7 | 3.48 (1H, d, 8.4) | 77.3 | |||
| 8a | 101.7 | |||||||
| 9 | 4.14 (1H, dd, 8.6, 7.3) 4.07 (1H, dd, 8.6, 6.0) | 73.2 | 79.2 | |||||
| 10 | 1.13 (3H, s) | 19.0 | ||||||
| 11 | 1.38 (3H, s) | 27.2 | ||||||
| 3-OCH3 | 3.84 (3H, s) | 56.4 | ||||||
| 1′ | 130.7 | 129.5 | ||||||
| 2′ | 7.31 (1H, d, 8.5) | 129.0 | 7.51 (1H, d, 1.6) | 111.7 | ||||
| 3′ | 6.81 (1H, d, 8.5) | 116.3 | 155.4 | |||||
| 4′ | 159.1 | 149.6 | ||||||
| 5′ | 6.81 (1H, d, 8.5) | 116.3 | 6.81 (1H, d, 8.3) | 116.2 | ||||
| 6′ | 7.31 (1H, d, 8.5) | 129.0 | 7.56 (1H, dd, 8.3, 1.6) | 125.1 | ||||
| 7′ | 3.96 (1H, dd, 11.1, 3.9) 3.89 (1H, dd, 11.1, 6.2) | 59.6 | ||||||
| 8′ | 2.97 (1H, m) | 47.0 | ||||||
| 9′ | 180.7 | |||||||
| 3′-OCH3 | 3.88 (3H, s) | 56.3 | ||||||
| Group | Dose (µM) | Cell Viability (%) |
|---|---|---|
| NC | - | 1.071 ± 0.051 |
| M | 0.5 µg mL−1 | 1.000 ± 0.048 ## |
| 1 | 0.5 µg mL−1, 10 µM | 0.964 ± 0.026 |
| 3 | 0.5 µg mL−1, 10 µM | 0.959 ± 0.020 |
| 4 | 0.5 µg mL−1, 10 µM | 0.931 ± 0.023 |
| 5 | 0.5 µg mL−1, 10 µM | 1.023 ± 0.042 |
| 6 | 0.5 µg mL−1, 10 µM | 1.047 ± 0.041 * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fan, X.; Cao, Y.; Zeng, M.; Ren, Y.; Zheng, X.; Feng, W. Six New Compounds from the Herbaceous Stems of Ephedra intermedia Schrenket C. A. Meyer and Their Lung-Protective Activity. Molecules 2024, 29, 432. https://doi.org/10.3390/molecules29020432
Fan X, Cao Y, Zeng M, Ren Y, Zheng X, Feng W. Six New Compounds from the Herbaceous Stems of Ephedra intermedia Schrenket C. A. Meyer and Their Lung-Protective Activity. Molecules. 2024; 29(2):432. https://doi.org/10.3390/molecules29020432
Chicago/Turabian StyleFan, Xiling, Yangang Cao, Mengnan Zeng, Yingjie Ren, Xiaoke Zheng, and Weisheng Feng. 2024. "Six New Compounds from the Herbaceous Stems of Ephedra intermedia Schrenket C. A. Meyer and Their Lung-Protective Activity" Molecules 29, no. 2: 432. https://doi.org/10.3390/molecules29020432
APA StyleFan, X., Cao, Y., Zeng, M., Ren, Y., Zheng, X., & Feng, W. (2024). Six New Compounds from the Herbaceous Stems of Ephedra intermedia Schrenket C. A. Meyer and Their Lung-Protective Activity. Molecules, 29(2), 432. https://doi.org/10.3390/molecules29020432