
Statistical Characterization of Food-Derived α-Amylase Inhibitory Peptides: Computer Simulation and Partial Least Squares Regression Analysis



Abstract
1. Introduction
2. Results and Discussion
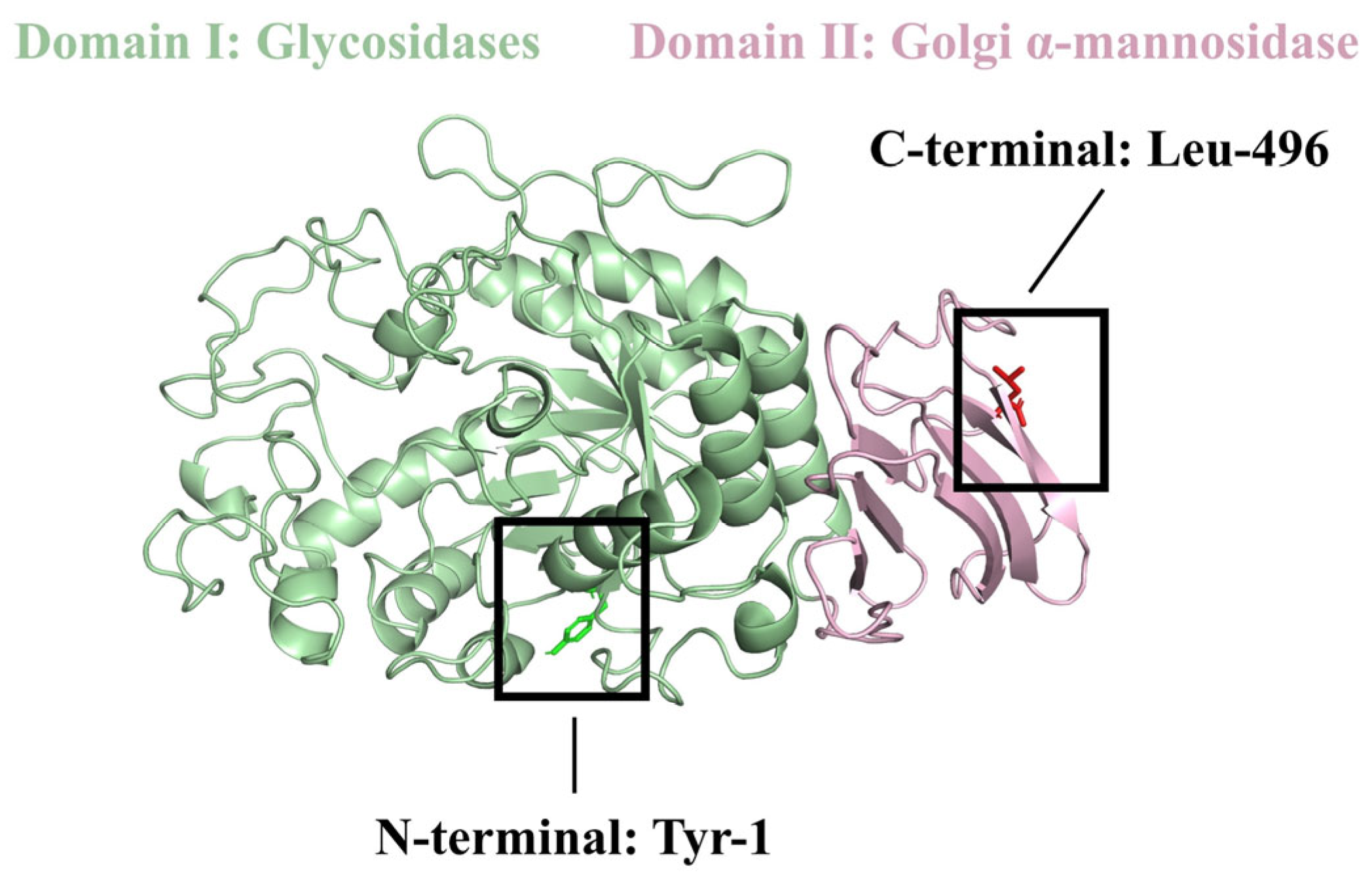
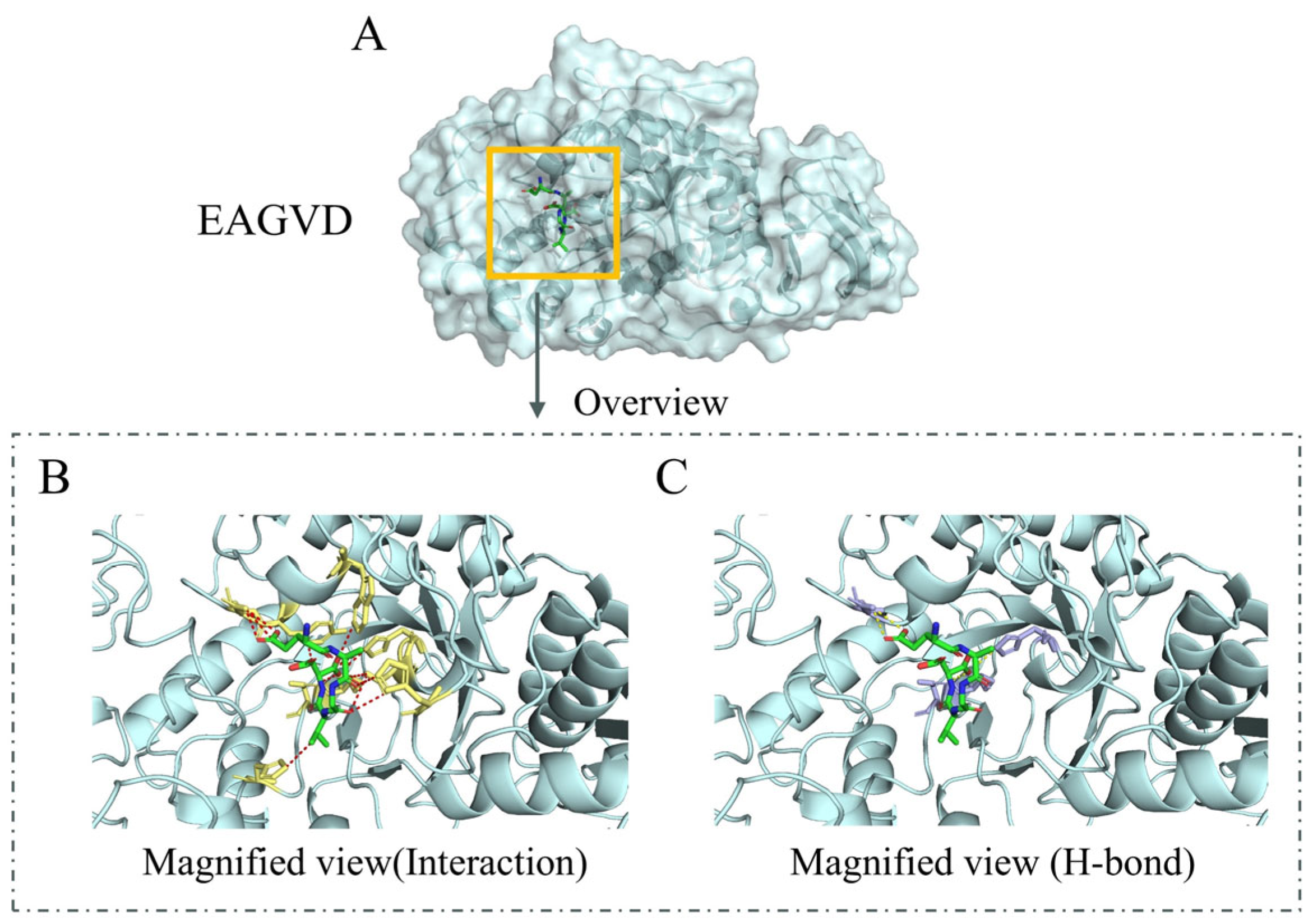
2.1. Molecular Docking Result Analysis of α-Amylase Molecule and Its Inhibitory Peptides
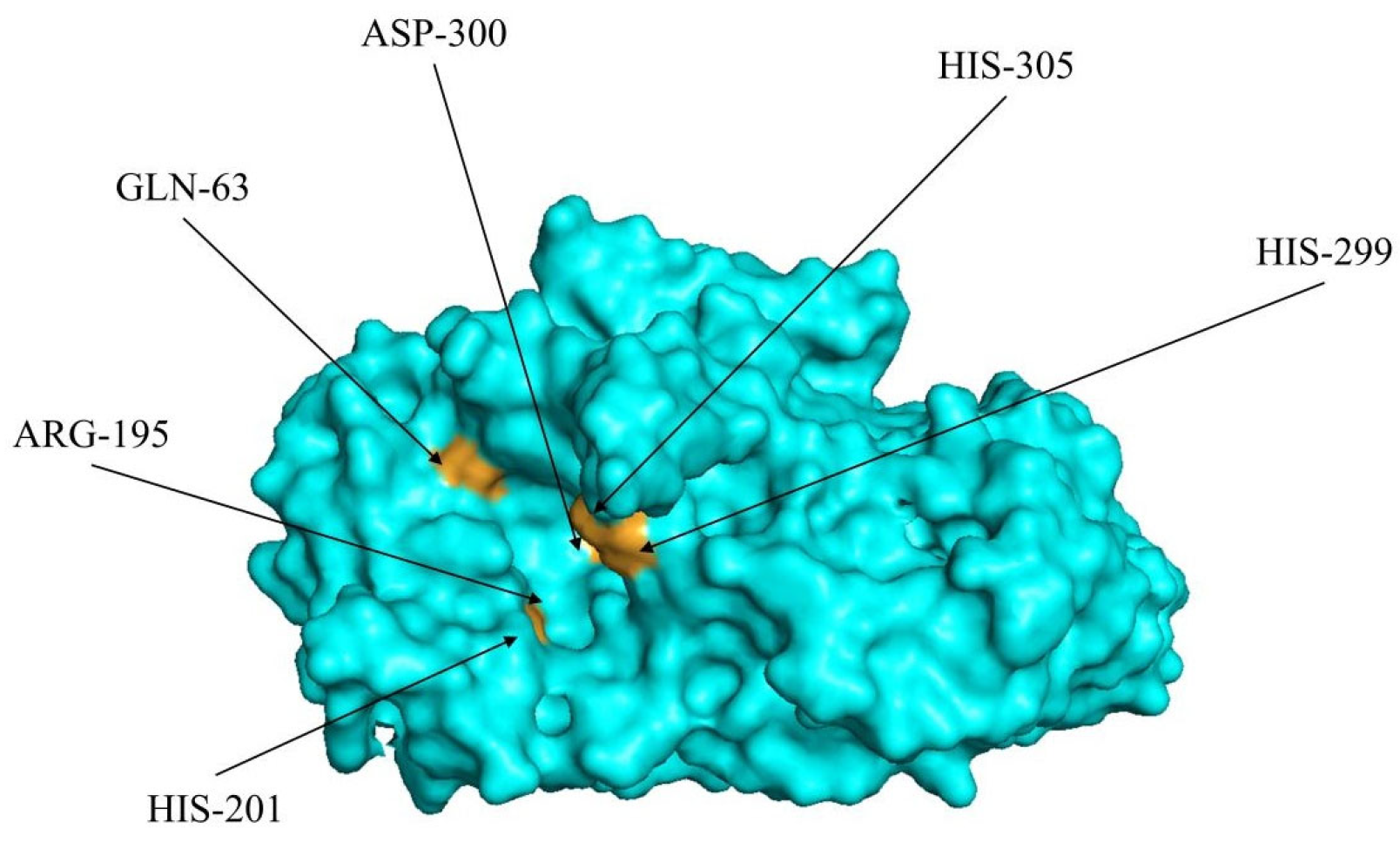
2.2. Analysis of the Key Sites on the α-Amylase Molecules
2.2.1. Analysis of the Docking Site Interaction Frequency on the α-Amylase Molecule
2.2.2. Analysis of the Docking Site Hydrogen Bond Qualities of α-Amylase Molecules
2.2.3. Combined IN and HBV Analysis of the Amino Acid Sites of the α-Amylase Molecule
2.3. Analysis of the Key Sites on the α-Amylase Inhibitory Peptides
2.4. Analysis of Hydrogen Bond Distribution Characteristics on α-Amylase Inhibitory Peptides
2.5. Molecular Dynamics Result Analysis of the α-Amylase Molecule and Its Inhibitory Peptides
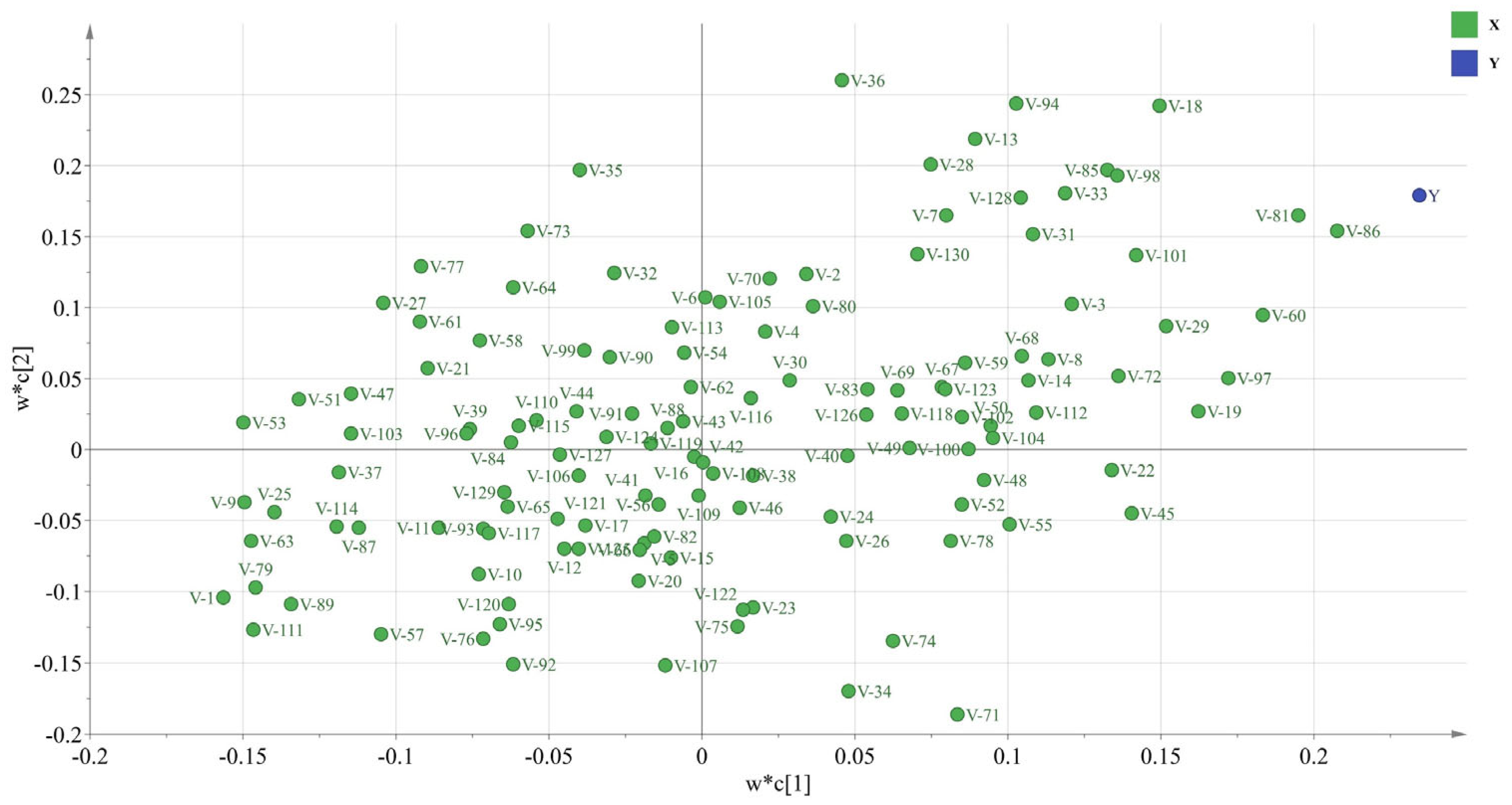
2.6. Analysis of the PLSR Results for the Physicochemical Properties and Inhibitory Peptide Activity
3. Materials and Methods
3.1. Materials
3.2. Molecular Docking of Food-Derived α-Amylase Inhibitory Peptides
3.3. Docking Site Interaction Frequency of the α-Amylase Molecules
3.4. Hydrogen Bonding Qualities of the Docking Sites on the α-Amylase Molecules and Inhibitory Peptides
3.5. Examination of the Hydrogen Bond Distribution Characteristics of the α-Amylase Inhibitory Peptides
3.6. Molecular Dynamics Simulation
3.7. PLSR of the Physicochemical Peptide Properties and Inhibitory Activity
3.8. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sun, H.; Saeedi, P.; Karuranga, S.; Pinkepank, M.; Ogurtsova, K.; Duncan, B.B.; Stein, C.; Basit, A.; Chan, J.C.N.; Mbanya, J.C.; et al. IDF Diabetes Atlas: Global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res. Clin. Pract. 2022, 183, 109119. [Google Scholar] [CrossRef]
- Wang, J.; Wu, T.; Fang, L.; Liu, C.; Liu, X.; Li, H.; Shi, J.; Li, M.; Min, W. Anti-diabetic effect by walnut (Juglans mandshurica Maxim.)-derived peptide LPLLR through inhibiting α-glucosidase and α-amylase, and alleviating insulin resistance of hepatic HepG2 cells. J. Funct. Food. 2020, 69, 103944. [Google Scholar] [CrossRef]
- Valencia-Mejia, E.; Batista, K.A.; Fernandez, J.J.A.; Fernandes, K.F. Antihyperglycemic and hypoglycemic activity of naturally occurring peptides and protein hydrolysates from easy-to-cook and hard-to-cook beans (Phaseolus vulgaris L.). Food Res. Int. 2019, 121, 238–246. [Google Scholar] [CrossRef]
- Ngoh, Y.Y.; Gan, C.Y. Enzyme-assisted extraction and identification of antioxidative and alpha-amylase inhibitory peptides from Pinto beans (Phaseolus vulgaris cv. Pinto). Food Chem. 2016, 190, 331–337. [Google Scholar] [CrossRef]
- Ramírez Fuentes, L.; Richard, C.; Chen, L. Sequential alcalase and flavourzyme treatment for preparation of α-amylase, α-glucosidase, and dipeptidyl peptidase (DPP)-IV inhibitory peptides from oat protein. J. Funct. Food. 2021, 87, 104829. [Google Scholar] [CrossRef]
- Park, B.Y.; Yoon, K.Y. Biological activity of enzymatic hydrolysates and the membrane ultrafiltration fractions from perilla seed meal protein. Czech. J. Food Sci. 2019, 37, 180–185. [Google Scholar] [CrossRef]
- De Matos, F.M.; de Lacerda, J.T.J.G.; Zanetti, G.; de Castro, R.J.S. Production of black cricket protein hydrolysates with α-amylase, α-glucosidase and angiotensin I-converting enzyme inhibitory activities using a mixture of proteases. Biocatal. Agric. Biotechnol. 2022, 39, 102276. [Google Scholar] [CrossRef]
- Evaristus, N.A.; Wan Abdullah, W.N.; Gan, C.Y. Extraction and identification of alpha-amylase inhibitor peptides from Nephelium lappacheum and Nephelium mutabile seed protein using gastro-digestive enzymes. Peptides 2018, 102, 61–67. [Google Scholar] [CrossRef]
- Tao, X.; Huang, Y.; Wang, C.; Chen, F.; Yang, L.; Ling, L.; Che, Z.; Chen, X. Recent developments in molecular docking technology applied in food science: A review. Int. J. Food Sci. Technol. 2020, 55, 33–45. [Google Scholar] [CrossRef]
- Bai, G.; Pan, Y.; Zhang, Y.; Li, Y.; Wang, J.; Wang, Y.; Teng, W.; Jin, G.; Geng, F.; Cao, J. Research advances of molecular docking and molecular dynamic simulation in recognizing interaction between muscle proteins and exogenous additives. Food Chem. 2023, 429, 136836. [Google Scholar] [CrossRef]
- Vidal-Limon, A.; Aguilar-Toalá, J.E.; Liceaga, A.M. Integration of Molecular Docking Analysis and Molecular Dynamics Simulations for Studying Food Proteins and Bioactive Peptides. J. Agric. Food Chem. 2022, 70, 934–943. [Google Scholar] [CrossRef]
- Lan, X.; Sun, L.; Muhammad, Y.; Wang, Z.; Liu, H.; Sun, J.; Zhou, L.; Feng, X.; Liao, D.; Wang, S. Studies on the Interaction between Angiotensin-Converting Enzyme (ACE) and ACE Inhibitory Peptide from Saurida elongata. J. Agric. Food Chem. 2018, 66, 13414–13422. [Google Scholar] [CrossRef]
- Hu, J.; Wang, H.; Weng, N.; Wei, T.; Tian, X.; Lu, J.; Lyu, M.; Wang, S. Novel angiotensin-converting enzyme and pancreatic lipase oligopeptide inhibitors from fermented rice bran. Front. Nutr. 2022, 9, 1010005. [Google Scholar] [CrossRef]
- Cai, B.; Wan, P.; Chen, H.; Huang, J.; Ye, Z.; Chen, D.; Pan, J. Purification and Identification of Novel Myeloperoxidase Inhibitory Antioxidant Peptides from Tuna (Thunnas albacares) Protein Hydrolysates. Molecules 2022, 27, 2681. [Google Scholar] [CrossRef]
- Ibrahim, M.A.; Bester, M.J.; Neitz, A.W.H.; Gaspar, A.R.M. Structural properties of bioactive peptides with alpha-glucosidase inhibitory activity. Chem. Biol. Drug Des. 2018, 91, 370–379. [Google Scholar] [CrossRef]
- Yu, Z.; Yin, Y.; Zhao, W.; Liu, J.; Chen, F. Anti-diabetic activity peptides from albumin against alpha-glucosidase and alpha-amylase. Food Chem. 2012, 135, 2078–2085. [Google Scholar] [CrossRef]
- Ngoh, Y.Y.; Gan, C.Y. Identification of Pinto bean peptides with inhibitory effects on alpha-amylase and angiotensin converting enzyme (ACE) activities using an integrated bioinformatics-assisted approach. Food Chem. 2018, 267, 124–131. [Google Scholar] [CrossRef]
- Na, S.; Li, Y. Jirimutu, Preparation and Identification of α-Amylase Inhibitory Peptides from Camel Milk Protein. Food Sci. 2020, 41, 148–157. [Google Scholar]
- Esfandi, R.; Seidu, I.; Willmore, W.; Tsopmo, A. Antioxidant, pancreatic lipase, and alpha-amylase inhibitory properties of oat bran hydrolyzed proteins and peptides. J. Food Biochem. 2021, 46, e13762. [Google Scholar]
- Zlotek, U.; Jakubczyk, A.; Rybczynska-Tkaczyk, K.; Cwiek, P.; Baraniak, B.; Lewicki, S. Characteristics of New Peptides GQLGEHGGAGMG, GEHGGAGMGGGQFQPV, EQGFLPGPEESGR, RLARAGLAQ, YGNPVGGVGH, and GNPVGGVGHGTTGT as Inhibitors of Enzymes Involved in Metabolic Syndrome and Antimicrobial Potential. Molecules 2020, 25, 2492. [Google Scholar] [CrossRef]
- Hu, S.; Fan, X.; Qi, P.; Zhang, X. Identification of anti-diabetes peptides from Spirulina platensis. J. Funct. Food. 2019, 56, 333–341. [Google Scholar] [CrossRef]
- De Sales, P.M.; de Souza, P.M.; Simeoni, L.A.; Magalhaes, P.D.; Silveira, D. alpha-Amylase Inhibitors: A Review of Raw Material and Isolated Compounds from Plant Source. J. Pharm. Pharm. Sci. 2012, 15, 141–183. [Google Scholar] [CrossRef]
- Payan, F. Structural basis for the inhibition of mammalian and insect alpha-amylases by plant protein inhibitors. Biochim. Biophys. Acta. 2004, 1696, 171–180. [Google Scholar] [CrossRef]
- Liu, F.F.; Wang, T.; Dong, X.Y.; Sun, Y. Rational design of affinity peptide ligand by flexible docking simulation. J. Chromatogr. A 2007, 1146, 41–50. [Google Scholar] [CrossRef]
- Li, M.; Liang, Q.; Song, X. Using Molecular Docking to Investigate the Alpha-amylase Inhibitory Activity of Bitter Peptides RK7 and KQ7 Derived from Yak Cheese. Food Sci. 2023, 44, 132–138. [Google Scholar]
- Zheng, Y.; Tian, J.; Yang, W.; Chen, S.; Liu, D.; Fang, H.; Zhang, H.; Ye, X. Inhibition mechanism of ferulic acid against alpha-amylase and alpha-glucosidase. Food Chem. 2020, 317, 126346. [Google Scholar] [CrossRef]
- Mojica, L.; de Mejia, E.G. Optimization of enzymatic production of anti-diabetic peptides from black bean (Phaseolus vulgaris L.) proteins, their characterization and biological potential. Food Funct. 2016, 7, 713–727. [Google Scholar] [CrossRef]
- Abe, J.; Sidenius, U.; Svensson, B. Arginine is essential for the alpha-amylase inhibitory activity of the alpha-amylase/subtilisin inhibitor (BASI) from barley seeds. Biochem. J. 1993, 293, 151–155. [Google Scholar] [CrossRef]
- Ishibashi, N.; Sadamori, K.; Yamamoto, O.; Kanehisa, H.; Kouge, K.; Kikuchi, E.; Okai, H.; Fukui, S. Bitterness of Phenylalanine- and Tyrosine-containing Peptides. Agric. Biol. Chem. 2016, 51, 3309–3313. [Google Scholar] [CrossRef]
- Ishibashi, N.; Arita, Y.; Kanehisa, H.; Kouge, K.; Okai, H.; Fukui, S. Bitterness of Leucine-containing Peptides. Agri. Biol. Chem. 1987, 51, 2389–2394. [Google Scholar] [CrossRef]
- Li, Y.W.; Li, B.; He, J.; Qian, P. Structure-activity relationship study of antioxidative peptides by QSAR modeling: The amino acid next to C-terminus affects the activity. J. Pept. Sci. 2011, 17, 454–462. [Google Scholar] [CrossRef]
- Chen, X.; Liu, W.; Zhang, J.; Li, H.; Liu, X. Selenium-enriched peptides identified from selenium-enriched soybean protein hydrolysate: Protective effects against heat damage in Caco-2 cells. Food Funct. 2023, 14, 7882–7896. [Google Scholar] [CrossRef]
- Du, X.; Jiang, C.; Wang, S.; Jing, H.; Mo, L.; Ma, C.; Wang, H. Preparation, identification, and inhibitory mechanism of dipeptidyl peptidase IV inhibitory peptides from goat milk whey protein. J. Food Sci. 2023, 88, 3577–3593. [Google Scholar] [CrossRef]
- Riyad, P.; Purohit, A.; Sen, K.; Panwar, A.; Ram, H. HMG-CoA reductase inhibition mediated hypocholesterolemic potential of myricetin and quercetin: In silico and in vivo studies. CyTA J. Food 2023, 21, 115–125. [Google Scholar] [CrossRef]
- Li, H.; Zhang, Y.; Liu, Z.; Guo, C.; Battino, M.; Cai, S.; Yi, J. Exploring antioxidant activities and inhibitory effects against α-amylase and α-glucosidase of Elaeocarpus braceanus fruits: Insights into mechanisms by molecular docking and molecular dynamics. Int. J. Food Sci. Technol. 2023, 59, 343–355. [Google Scholar] [CrossRef]
- Bello, M. Binding free energy calculations between bovine beta-lactoglobulin and four fatty acids using the MMGBSA method. Biopolymers 2014, 101, 1010–1018. [Google Scholar] [CrossRef]
- Du, J.; Hu, S.; Dong, J.; Wu, R.; Yu, J.; Yin, H. Exploring the factors that affect the themostability of barley limit dextrinase—Inhibitor complex. J. Mol. Graph. Model. 2021, 109, 108043. [Google Scholar] [CrossRef]
- Shen, Y.; Maupetit, J.; Derreumaux, P.; Tuffery, P. Improved PEP-FOLD Approach for Peptide and Miniprotein Structure Prediction. J. Chem. Theory Comput. 2014, 10, 4745–4758. [Google Scholar] [CrossRef]
- Burley, S.K.; Berman, H.M.; Christie, C.; Duarte, J.M.; Feng, Z.; Westbrook, J.; Young, J.; Zardecki, C. RCSB Protein Data Bank: Sustaining a living digital data resource that enables breakthroughs in scientific research and biomedical education. Protein Sci. 2018, 27, 316–330. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Valdes-Tresanco, M.S.; Valdes-Tresanco, M.E.; Valiente, P.A.; Moreno, E. gmx_MMPBSA: A New Tool to Perform End-State Free Energy Calculations with GROMACS. J. Chem. Theory Comput. 2021, 17, 6281–6291. [Google Scholar] [CrossRef]
- Liu, J.; Guan, X.; Peng, J.Q. QSAR Study on ACE Inhibitory Peptides Based on Amino Acids Descriptor SHVHES. Acta Chim. Sin. 2012, 70, 83–91. [Google Scholar] [CrossRef]
- Qi, T.; Yaowang, L.; Bo, L. Development of a Peptide Length Differential Characterization Method for Peptide QSAR. J. Chin. Inst. Food Sci. Technol. 2021, 21, 28–38. [Google Scholar]
- Chong, I.-G.; Jun, C.-H. Performance of some variable selection methods when multicollinearity is present. Chemom. Intell. Lab. Syst. 2005, 78, 103–112. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Sequence of Peptide | Peptide Length | IC50 Value (mmol/U) | Source | References |
|---|---|---|---|---|---|
| 1 | EAGVD | 5 | 110.00 | Egg | [16] |
| 2 | KLPGF | 5 | 120.00 | Egg | [16] |
| 3 | PPHMLP | 6 | 103.69 | Pinto bean | [17] |
| 4 | PPHMGGP | 7 | 88.13 | Pinto bean | [17] |
| 5 | LPLPLPLR | 8 | 446.54 | Camel milk | [18] |
| 6 | PLPLHMLP | 8 | 70.22 | Pinto bean | [17] |
| 7 | PLPWGAGF | 8 | 69.91 | Pinto bean | [17] |
| 8 | RALPIDVL | 8 | 24.27 | Oat bran | [19] |
| 9 | NINAHSVVY | 9 | 22.43 | Oat bran | [19] |
| 10 | RLARAGLAQ | 9 | 69.88 | Millet grain | [20] |
| 11 | IPLPLPLPLP | 10 | 364.70 | Camel milk | [18] |
| 12 | LRSELAAWSR | 10 | 527.80 | Spirulina platensis | [21] |
| 13 | YFDEQNEQFR | 10 | 12.50 | Oat bran | [19] |
| 14 | YGNPVGGVGH | 10 | 80.28 | Millet grain | [20] |
| 15 | GQLGEHGGAGMG | 12 | 53.00 | Millet grain | [20] |
| 16 | EQGFLPGPEESGR | 13 | 51.09 | Millet grain | [20] |
| 17 | GNPVGGVGHGTTGT | 14 | 50.01 | Millet grain | [20] |
| 18 | GEHGGAGMGGGQFQPV | 16 | 44.58 | Millet grain | [20] |
| 19 | HAGPTWNPISIGISFM | 16 | 387.74 | Camel milk | [18] |
| 20 | LSSLEMGSLGALFVCM | 16 | 44.58 | Pinto bean | [17] |
| Residue | Residue Type | IN | Residue | Residue Type | IN | Residue | Residue Type | IN |
|---|---|---|---|---|---|---|---|---|
| His201 | Positive | 15 | Asp402 | Negative | 3 | Val50 | Nonpolar | 1 |
| Gln63 | Uncharged | 15 | Asn53 | Uncharged | 3 | Val51 | Nonpolar | 1 |
| Asp300 | Negative | 14 | Ser289 | Uncharged | 3 | Pro54 | Nonpolar | 1 |
| His305 | Positive | 14 | Arg252 | Positive | 3 | Glu149 | Negative | 1 |
| His299 | Positive | 12 | Thr6 | Uncharged | 2 | Gly164 | Uncharged | 1 |
| Val163 | Nonpolar | 11 | Ser105 | Uncharged | 2 | Leu165 | Nonpolar | 1 |
| Trp59 | Nonpolar | 10 | Ala198 | Nonpolar | 2 | Ser226 | Uncharged | 1 |
| Asp197 | Negative | 10 | His331 | Positive | 2 | Arg227 | Positive | 1 |
| Tyr151 | Uncharged | 10 | Glu352 | Negative | 2 | Ala241 | Nonpolar | 1 |
| Glu233 | Negative | 9 | Arg398 | Positive | 2 | Thr264 | Uncharged | 1 |
| Lys200 | Positive | 9 | Phe406 | Nonpolar | 2 | Ser270 | Uncharged | 1 |
| Arg195 | Positive | 9 | Arg421 | Positive | 2 | Glu272 | Negative | 1 |
| His101 | Positive | 8 | Glu282 | Negative | 2 | Asp290 | Negative | 1 |
| Asp356 | Negative | 8 | Gly9 | Uncharged | 2 | Phe335 | Nonpolar | 1 |
| Leu162 | Nonpolar | 7 | Gly106 | Uncharged | 2 | Gly403 | Uncharged | 1 |
| Tyr62 | Uncharged | 6 | Tyr2 | Uncharged | 2 | Asn152 | Uncharged | 1 |
| Ile235 | Nonpolar | 6 | Glu240 | Negative | 2 | Pca1 | — | 1 |
| Ala307 | Nonpolar | 6 | Gln161 | Uncharged | 2 | Val354 | Nonpolar | 1 |
| Ile148 | Nonpolar | 4 | Trp280 | Nonpolar | 2 | Ala107 | Nonpolar | 1 |
| Gly306 | Uncharged | 4 | Arg291 | Positive | 2 | Ser310 | Uncharged | 1 |
| Gly308 | Uncharged | 4 | Gln5 | Uncharged | 1 | Ser311 | Uncharged | 1 |
| Trp58 | Nonpolar | 3 | Ser8 | Uncharged | 1 | Ala3 | Nonpolar | 1 |
| Gly309 | Uncharged | 3 | Thr11 | Uncharged | 1 | Thr52 | Uncharged | 1 |
| Residue | Residue Type | HBV | Residue | Residue Type | HBV |
|---|---|---|---|---|---|
| Gln63 | Uncharged | 11.50 | Trp280 | Nonpolar | 2.12 |
| His201 | Positive | 10.33 | Gln161 | Uncharged | 2.06 |
| Arg252 | Positive | 10.00 | Glu233 | Negative | 2.06 |
| Arg195 | Positive | 6.06 | Glu240 | Negative | 2.03 |
| His299 | Positive | 5.19 | Asn53 | Uncharged | 2.00 |
| Asp356 | Negative | 5.18 | Tyr2 | Uncharged | 1.75 |
| Gly308 | Uncharged | 4.72 | Ala3 | Nonpolar | 1.67 |
| Arg291 | Positive | 4.45 | Gly106 | Uncharged | 1.59 |
| Gly306 | Uncharged | 4.34 | Ser311 | Uncharged | 1.59 |
| His305 | Positive | 4.30 | Gly9 | Uncharged | 1.52 |
| Thr52 | Uncharged | 4.20 | Ser310 | Uncharged | 1.40 |
| Asp300 | Negative | 4.19 | Ala107 | Nonpolar | 1.25 |
| Tyr151 | Uncharged | 3.85 | Pca1 | — | 1.06 |
| Lys200 | Positive | 3.59 | Val354 | Nonpolar | 1.06 |
| Asp197 | Negative | 3.29 | Asp402 | Negative | 1.06 |
| Ser289 | Uncharged | 3.05 | Ile148 | Nonpolar | 1.03 |
| Trp59 | Nonpolar | 2.87 | Asn152 | Uncharged | 1.03 |
| Side Chain Type | IN | HBV | ||||
|---|---|---|---|---|---|---|
| Quantity of Residues | Total IN | Mean Value | Quantity of Residues | Total IN | Mean Value | |
| Nonpolar | 19 | 62 | 3.26 | 6 | 10.00 | 1.67 b |
| Polar uncharged | 26 | 72 | 2.77 | 14 | 44.60 | 3.19 ab |
| Positively charged | 12 | 79 | 6.58 | 8 | 46.67 | 5.83 a |
| Negatively charged | 11 | 63 | 5.73 | 7 | 18.81 | 2.69 ab |
| Amino Acid Kind | Residue Type | Serial Number of Residues | HBV | Number of Amino Acids | Total HBV | Mean Value |
|---|---|---|---|---|---|---|
| Alanine | Nonpolar | 7-6 | 1.40 | 13 | 7.81 | 0.60 ab |
| 19-2 | 3.42 | |||||
| 20-11 | 2.99 | |||||
| Aspartate | Negative | 1-5 | 6.00 | 3 | 10.24 | 3.41 a |
| 8-6 | 1.40 | |||||
| 13-3 | 2.84 | |||||
| Glutamate | Negative | 1-1 | 1.59 | 10 | 10.55 | 1.06 ab |
| 12-4 | 1.59 | |||||
| 13-4 | 1.84 | |||||
| 16-1 | 1.59 | |||||
| 18-2 | 2.94 | |||||
| 20-5 | 1.00 | |||||
| Phenylalanine | Nonpolar | 2-5 | 5.35 | 8 | 11.69 | 1.46 ab |
| 7-8 | 6.34 | |||||
| Glycine | Uncharged | 14-6 | 1.52 | 37 | 12.55 | 0.34 ab |
| 15-10 | 1.52 | |||||
| 15-8 | 1.59 | |||||
| 15-12 | 2.12 | |||||
| 17-8 | 1.40 | |||||
| 17-10 | 1.35 | |||||
| 18-4 | 1.30 | |||||
| 20-7 | 1.75 | |||||
| Histidine | Positive | 3-3 | 1.35 | 9 | 11.23 | 1.25 ab |
| 4-3 | 1.46 | |||||
| 6-5 | 2.70 | |||||
| 14-10 | 2.86 | |||||
| 15-6 | 1.52 | |||||
| 17-9 | 1.35 | |||||
| Isoleucine | Nonpolar | 11-1 | 1.00 | 6 | 1.00 | 0.17 ab |
| Lysine | Positive | 2-1 | 1.09 | 1 | 1.09 | 1.09 ab |
| Leucine | Nonpolar | 6-2 | 1.46 | 26 | 6.00 | 0.23 b |
| 6-7 | 1.30 | |||||
| Methionine | Nonpolar | 19-16 | 1.06 | 8 | 4.71 | 0.59 ab |
| 20-16 | 3.65 | |||||
| Asparagine | Uncharged | 13-6 | 1.00 | 6 | 4.15 | 0.69 ab |
| 14-3 | 3.15 | |||||
| Proline | Nonpolar | 2-3 | 1.59 | 28 | 18.06 | 0.65 ab |
| 3-6 | 3.78 | |||||
| 3-6 | 3.78 | |||||
| 4-7 | 1.06 | |||||
| 6-1 | 1.06 | |||||
| 6-8 | 1.59 | |||||
| 11-10 | 5.20 | |||||
| Arginine | Positive | 5-6 | 1.84 | 8 | 12.06 | 1.51 a |
| 5-8 | 1.03 | |||||
| 8-1 | 3.09 | |||||
| 10-4 | 1.06 | |||||
| 12-2 | 1.94 | |||||
| 12-10 | 1.00 | |||||
| 13-10 | 3.93 | |||||
| Serine | Uncharged | 12-9 | 1.35 | 9 | 6.80 | 0.76 ab |
| 19-14 | 2.86 | |||||
| 20-2 | 1.35 | |||||
| 20-3 | 1.25 | |||||
| Threonine | Uncharged | 17-14 | 3.82 | 4 | 3.82 | 0.96 ab |
| Valine | Nonpolar | 17-4 | 1.30 | 10 | 1.30 | 0.13 ab |
| Tryptophan | Nonpolar | 12-8 | 1.94 | 3 | 3.01 | 1.00 ab |
| 19-6 | 1.06 | |||||
| Tyrosine | Uncharged | 13-1 | 1.00 | 3 | 4.65 | 1.55 ab |
| 14-1 | 3.65 |
| Side Chain Type | Quantity of Residues | Total HBV | Mean Value |
|---|---|---|---|
| Nonpolar | 102 | 50.32 | 0.49 b |
| Polar uncharged | 59 | 31.97 | 0.54 b |
| Positively charged | 18 | 26.22 | 1.46 a |
| Negatively charged | 13 | 20.79 | 1.60 a |
| Peptides | RN | F |
|---|---|---|
| EAGVD | 0.814 | 1.000 |
| KLPGF | 0.741 | 0.867 |
| PPHMLP | 0.864 | 0.774 |
| PPHMGGP | 0.719 | 0.719 |
| LPLPLPLR | 0.898 | 0.761 |
| PLPLHMLP | 0.495 | 0.563 |
| PLPWGAGF | 0.957 | 0.901 |
| RALPIDVL | 0.141 | 0.869 |
| RLARAGLAQ | 0.375 | 0.250 |
| IPLPLPLPLP | 0.760 | 1.000 |
| LRSELAAWSR | 0.684 | 0.693 |
| YFDEQNEQFR | 0.540 | 0.666 |
| YGNPVGGVGH | 0.347 | 0.780 |
| GQLGEHGGAGMG | 0.815 | 0.628 |
| EQGFLPGPEESGR | 0.000 | 1.000 |
| GNPVGGVGHGTTGT | 0.735 | 0.592 |
| GEHGGAGMGGGQFQPV | 0.115 | 0.754 |
| HAGPTWNPISIGISFM | 0.572 | 0.722 |
| LSSLEMGSLGALFVCM | 0.583 | 0.662 |
| Mean value | 0.587 | 0.747 a |
| Test value | 0.5 | 0.5 b |
| Energy Component (kcal/mol) | KLPGF | PLPLHMLP | GNPVGGVGHGTTGT |
|---|---|---|---|
| ΔGvdw | −41.24 | −39.93 | −60.12 |
| ΔGele | −375.03 | −118.30 | −78.05 |
| ΔGpolar | 372.90 | 138.72 | 117.78 |
| ΔGnonpolar | −6.90 | −4.83 | −7.19 |
| ΔGgas | −416.27 | −158.23 | −138.17 |
| ΔGsolv | 366.00 | 133.89 | 110.59 |
| ΔGtotal | −50.26 | −24.34 | −27.58 |
| Position End | 1 | 2 | 3 | 4 | 5 | Sum |
|---|---|---|---|---|---|---|
| N | 6 | 6 | 9 | 3 | 7 | 31 |
| C | 1 | 3 | 5 | 6 | 6 | 21 |
| Sum | 7 | 9 | 14 | 9 | 13 | 52 |
| Properties | Hydrophobic Properties | Electrical Properties | Hydrogen Bond Contributory Properties | Steric Properties |
|---|---|---|---|---|
| Descriptor quantity | 2 | 4 | 2 | 5 |
| Total value | 6 | 13 | 14 | 19 |
| Mean value | 3.00 | 3.25 | 7.00 | 3.80 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, W.; Yang, S.; An, J.; Wang, M.; Li, H.; Liu, X. Statistical Characterization of Food-Derived α-Amylase Inhibitory Peptides: Computer Simulation and Partial Least Squares Regression Analysis. Molecules 2024, 29, 395. https://doi.org/10.3390/molecules29020395
Li W, Yang S, An J, Wang M, Li H, Liu X. Statistical Characterization of Food-Derived α-Amylase Inhibitory Peptides: Computer Simulation and Partial Least Squares Regression Analysis. Molecules. 2024; 29(2):395. https://doi.org/10.3390/molecules29020395
Chicago/Turabian StyleLi, Wenhui, Shangci Yang, Jiulong An, Min Wang, He Li, and Xinqi Liu. 2024. "Statistical Characterization of Food-Derived α-Amylase Inhibitory Peptides: Computer Simulation and Partial Least Squares Regression Analysis" Molecules 29, no. 2: 395. https://doi.org/10.3390/molecules29020395
APA StyleLi, W., Yang, S., An, J., Wang, M., Li, H., & Liu, X. (2024). Statistical Characterization of Food-Derived α-Amylase Inhibitory Peptides: Computer Simulation and Partial Least Squares Regression Analysis. Molecules, 29(2), 395. https://doi.org/10.3390/molecules29020395