
Support Effect of Boron Nitride on the First N-H Bond Activation of NH3 on Ru Clusters

Abstract
1. Introduction
2. Results and Discussion
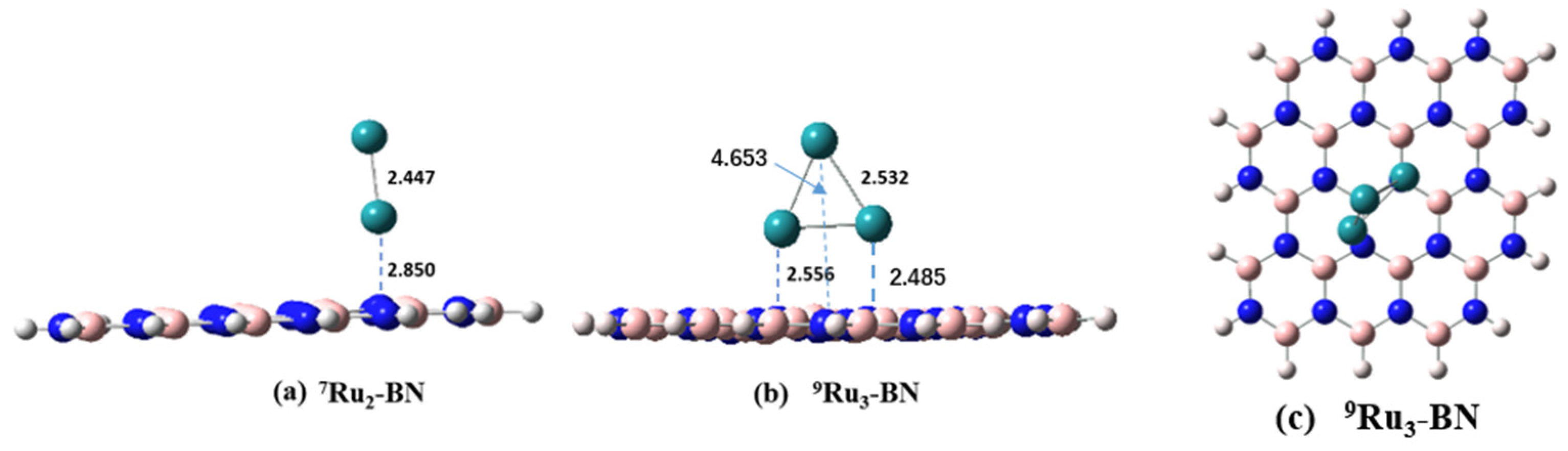
2.1. Adsorption Energy of Run Atom/Clusters on the BN Support
2.2. Support Effect on the First N-H Bond Activation Process of NH3 on One Ru Atom
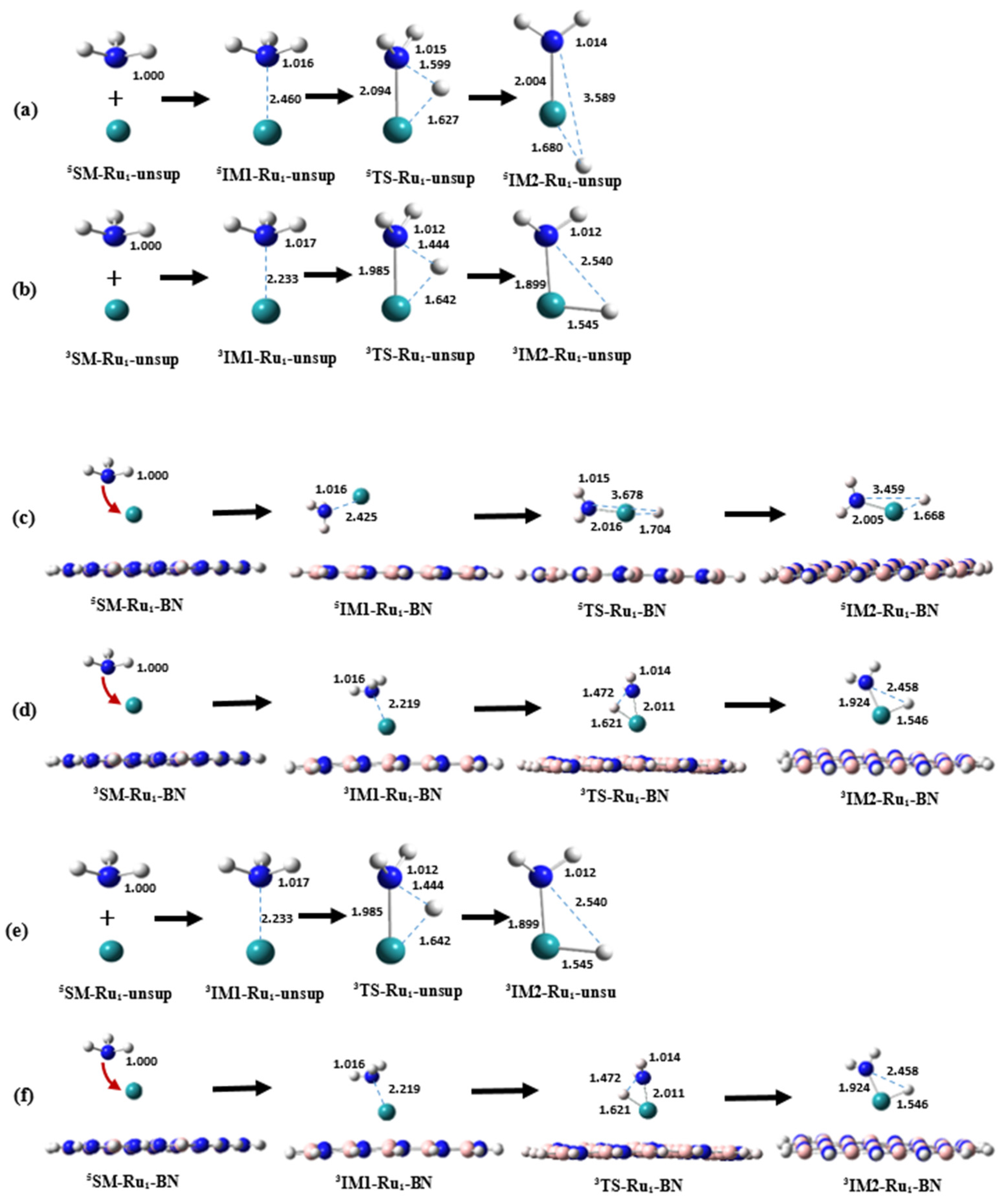
2.2.1. Structure
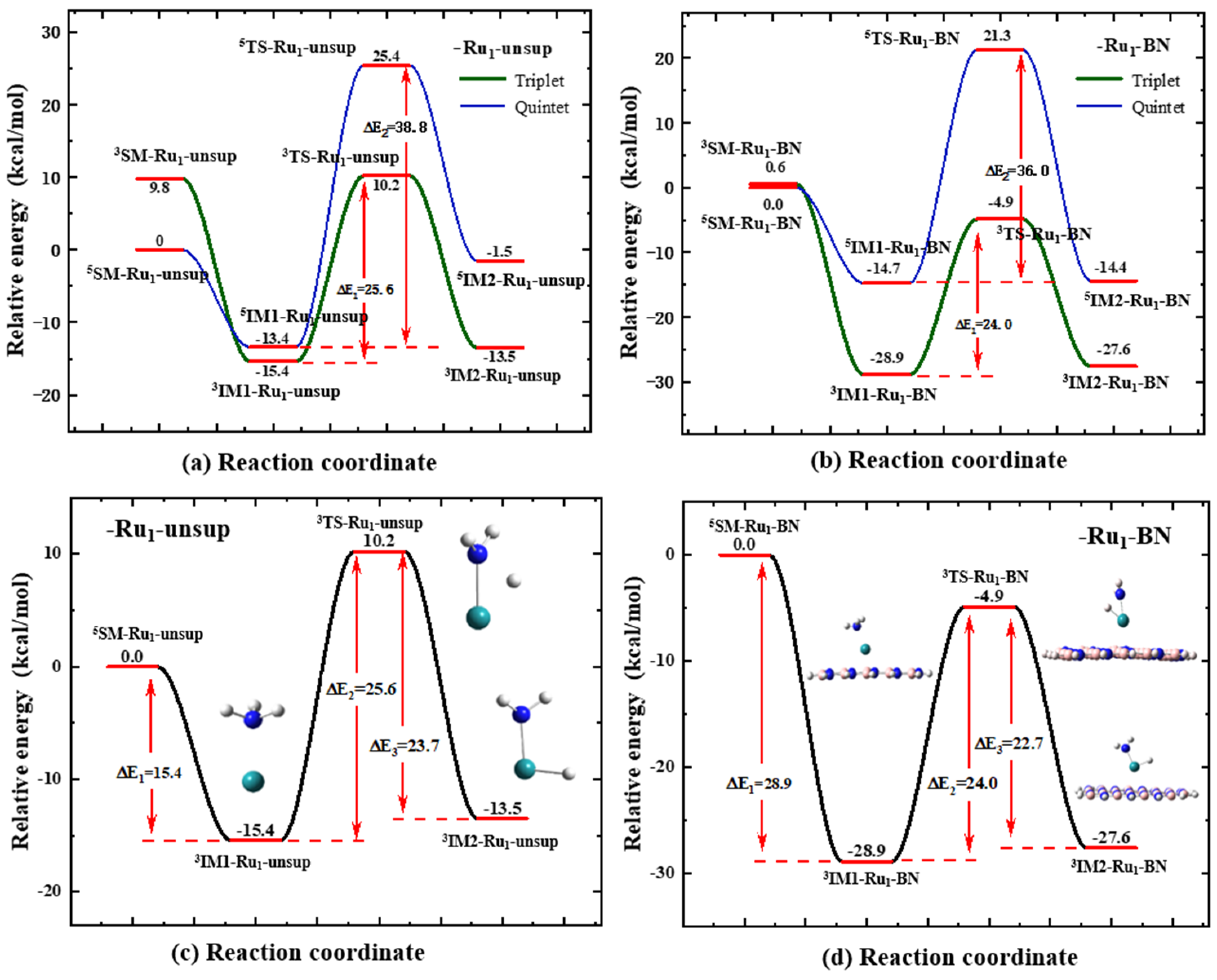
2.2.2. Energy Profiles
2.2.3. Effect of the BN Model Size
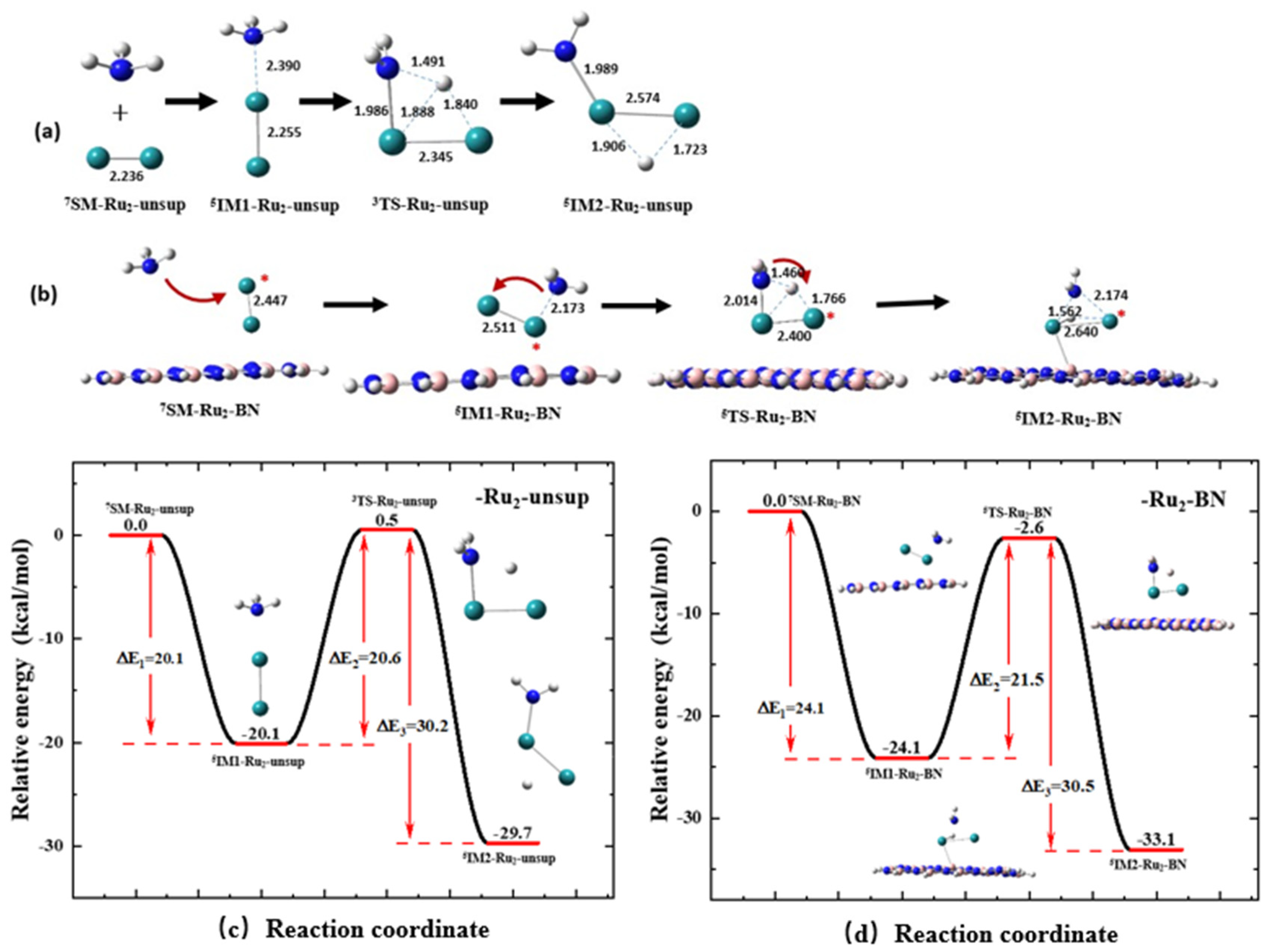
2.3. Support Effect on the First N-H Bond Activation Process of NH3 on an Ru2 Cluster
2.4. Support Effect on the First N-H Bond Activation Process of NH3 on an Ru3 Cluster
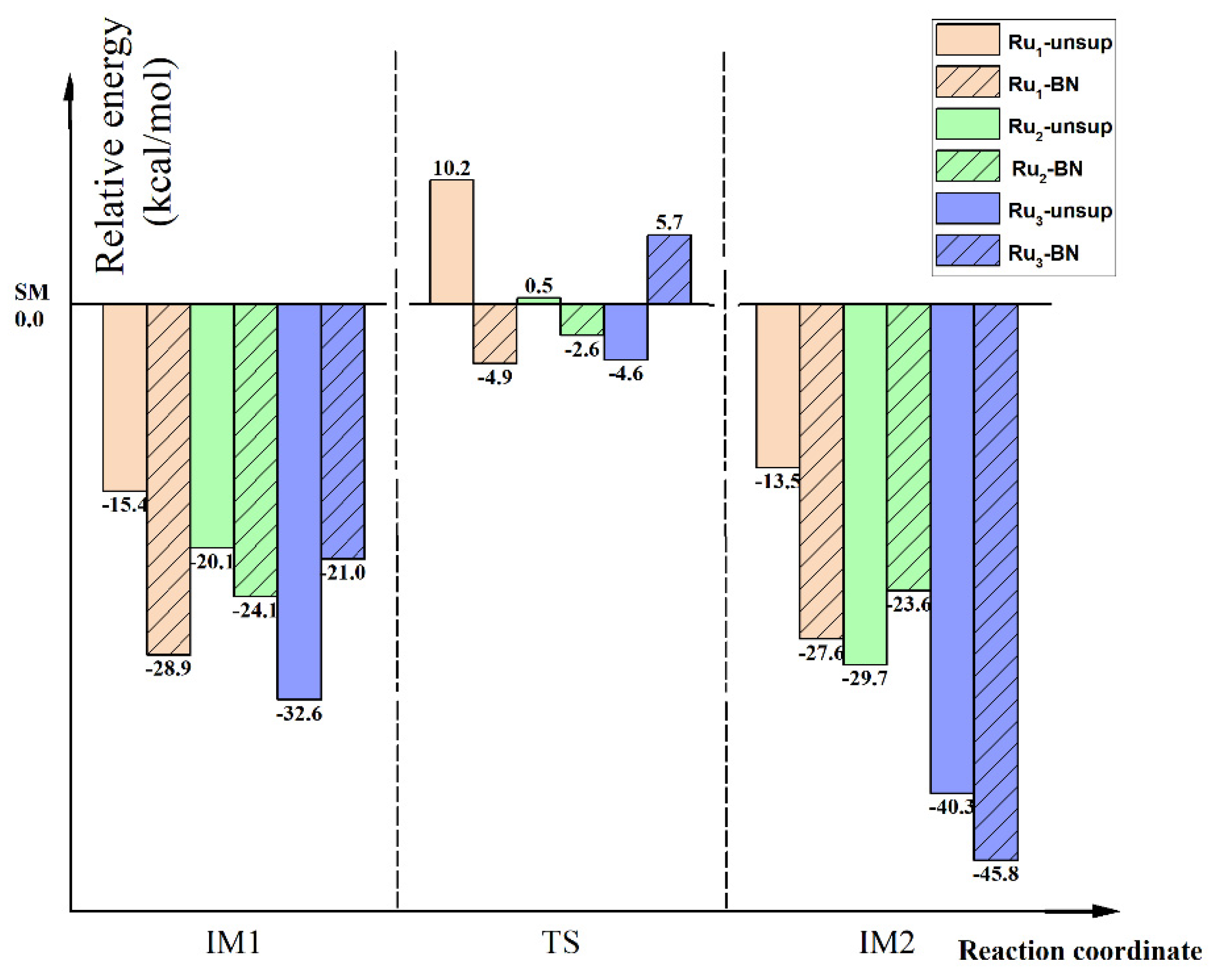
2.5. Further Discussion on the Support Effect for the First N-H Bond Activation of NH3 on Run (n = 1, 2, 3) Clusters
2.5.1. Thermodynamic Aspect of Support Effect
2.5.2. Kinetic Aspect of Support Effect
2.5.3. Cluster Size Effect
2.5.4. Support Effect on the Electron Transfer from IM1 to TS
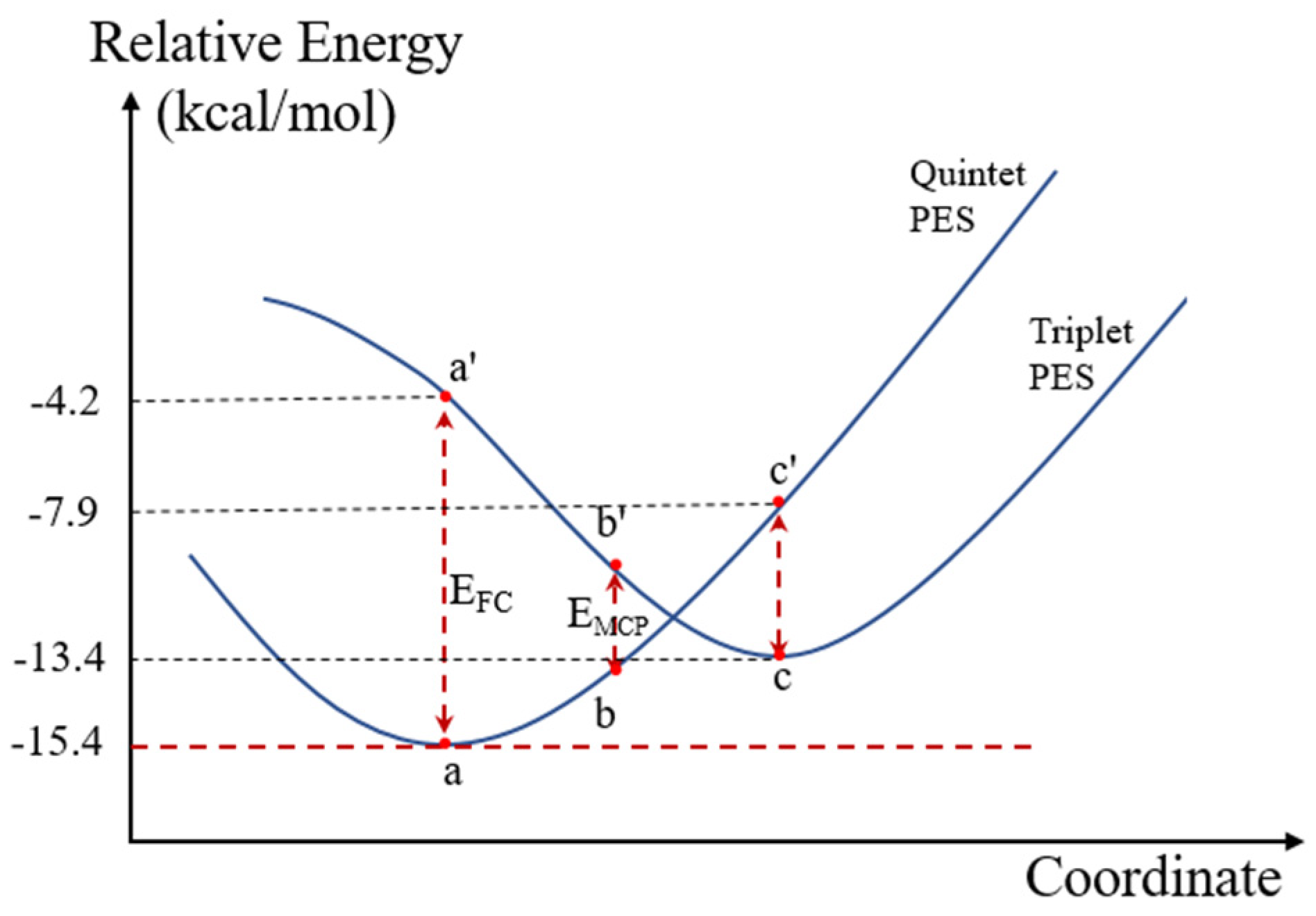
2.5.5. Support Effect on the Spin Conversion Behavior for the MFPs
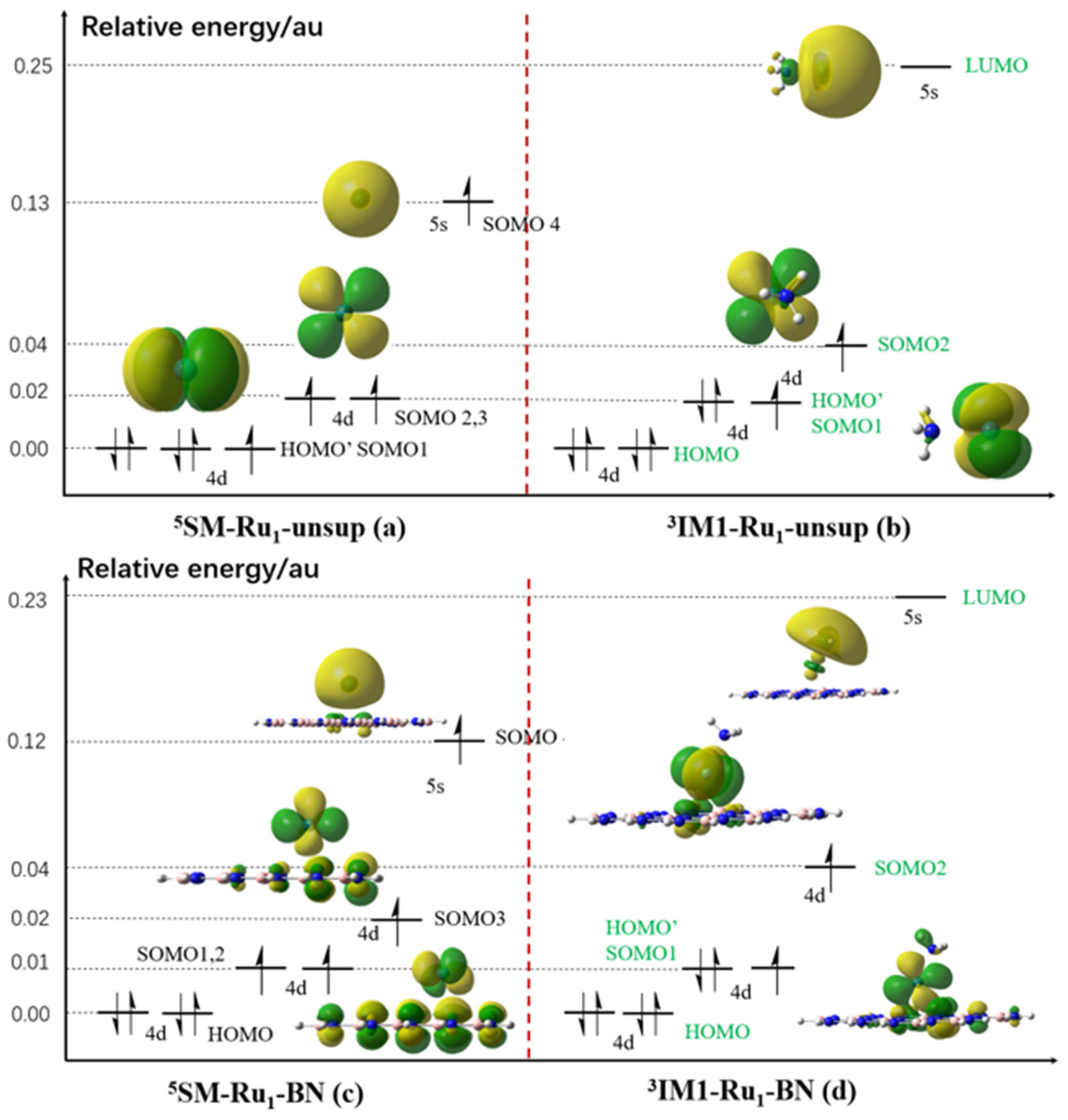
2.5.6. Preliminary Orbital Analysis
3. Computational Methods and Reactant Models
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bockris, J.O.M. The hydrogen economy: Its history. Int. J. Hydrogen Energy 2013, 38, 2579–2588. [Google Scholar] [CrossRef]
- Staffell, I.; Scamman, D.; Abad, A.V.; Balcombe, P.; Dodds, P.E.; Ekins, P.; Shah, N.; Ward, K.R. The role of hydrogen and fuel cells in the global energy system. Energy Environ. Sci. 2019, 12, 463–491. [Google Scholar] [CrossRef]
- Pandev, M.; Lucchese, P.; Mansilla, C.; Le Duigou, A.; Abrashev, B.; Vladikova, D. Hydrogen Economy: The future for a sustainable and green society. Bulg. Chem. Commun. 2017, 49, 84–92. [Google Scholar]
- Lucentini, I.; Garcia, X.; Vendrell, X.; Llorca, J. Review of the Decomposition of Ammonia to Generate Hydrogen. Ind. Eng. Chem. Res. 2021, 60, 18560–18611. [Google Scholar] [CrossRef]
- Yi, Y.; Wang, L.; Guo, Y.; Sun, S.; Guo, H. Plasma-Assisted ammonia decomposition over Fe–Ni alloy catalysts for COx-Free hydrogen. AIChE J. 2019, 65, 691–701. [Google Scholar] [CrossRef]
- Hu, Z.-P.; Weng, C.-C.; Chen, C.; Yuan, Z.-Y. Two-dimensional mica nanosheets supported Fe nanoparticles for NH3 decomposition to hydrogen. Mol. Catal. 2018, 448, 162–170. [Google Scholar] [CrossRef]
- Hu, Z.-P.; Chen, L.; Chen, C.; Yuan, Z.-Y. Fe/ZSM-5 catalysts for ammonia decomposition to COx-free hydrogen: Effect of SiO2/Al2O3 ratio. Mol. Catal. 2018, 455, 14–22. [Google Scholar] [CrossRef]
- Pinzón, M.; Ruiz-López, E.; Romero, A.; de la Osa, A.R.; Sánchez, P.; de Lucas-Consuegra, A. Electrochemical activation of Ru catalyst with alkaline ion conductors for the catalytic decomposition of ammonia. Mol. Catal. 2021, 511, 111721. [Google Scholar] [CrossRef]
- Li, G.; Yu, X.; Lei, Z.; Yin, F.; Zhang, H.; He, X. Preparation of Lanthanum Hexaaluminate Supported Nickel Catalysts for Hydrogen Production by Ammonia Decomposition. Catal. Lett. 2023, 153, 3148–3158. [Google Scholar] [CrossRef]
- Maleki, H.; Bertola, V. Co–Ce–Al–O mesoporous catalysts for hydrogen generation via ammonia decomposition. Int. J. Hydrogen Energy 2022, 51, 267–275. [Google Scholar] [CrossRef]
- Qiu, Y.; Fu, E.; Gong, F.; Xiao, R. Catalyst support effect on ammonia decomposition over Ni/MgAl2O4 towards hydrogen production. Int. J. Hydrogen Energy 2022, 47, 5044–5052. [Google Scholar] [CrossRef]
- Liu, P.; Sun, L.; Zhang, Z.; Wang, X.; Zhang, Y.; Yang, X. Hydrogen production from ammonia decomposition catalyzed by Ru nano-particles in alkaline molecular sieves under photothermal conditions. Mol. Catal. 2023, 543, 113160. [Google Scholar] [CrossRef]
- Chellappa, A.S.; Fischer, C.M.; Thomson, W.J. Ammonia decomposition kinetics over Ni-Pt/Al2O3 for PEM fuel cell applications. Appl. Catal. A Gen. 2002, 227, 231–240. [Google Scholar] [CrossRef]
- Yue, C.; Zhang, K.; Wang, J.; Zhang, Z.; Bian, H. N-H bond activation of ammonia for catalytic organic reactions (chinese). Chem. World 2019, 60, 553–560. [Google Scholar]
- Almquist, C.C.; Removski, N.; Rajeshkumar, T.; Gelfand, B.S.; Maron, L.; Piers, W.E. Spontaneous Ammonia Activation through Coordination-Induced Bond Weakening in Molybdenum Complexes of a Dianionic Pentadentate Ligand Platform. Angew. Chem. Int. Ed. Engl. 2022, 61, e202203576. [Google Scholar] [CrossRef]
- Jian, Z.; Pan, Q.; Li, M.; Yan, T.; Fang, T. Density functional theory study on direct catalytic decomposition of ammonia on Pd (111) surface. Appl. Surf. Sci. 2014, 292, 494–499. [Google Scholar] [CrossRef]
- Duan, X.; Ji, J.; Qian, G.; Fan, C.; Zhu, Y.; Zhou, X.; Chen, D.; Yuan, W. Ammonia decomposition on Fe(110), Co(111) and Ni(111) surfaces: A density functional theory study. J. Mol. Catal. A Chem. 2012, 357, 81–86. [Google Scholar] [CrossRef]
- Jiang, Z.; Qin, P.; Fang, T. Mechanism of ammonia decomposition on clean and oxygen-covered cu (1 1 1) surface: A dft study. Chem. Phys. 2014, 445, 59–67. [Google Scholar] [CrossRef]
- Jiang, Z.; Fang, T. Probing the effect of Pd coverage towards NH3 decomposition on Cu(100) surface. Chem. Phys. Lett. 2019, 729, 30–31. [Google Scholar] [CrossRef]
- Rao, X.; Lou, Y.; Zhou, Y.; Zhang, J.; Zhong, S. First-principles insights into ammonia decomposition on WC (0001) surface terminated by W and C. Appl. Surf. Sci. 2021, 566, 150635. [Google Scholar]
- Takahashi, A.; Fujitani, T. Kinetic Analysis of Decomposition of Ammonia over Nickel and Ruthenium Catalysts. J. Chem. Eng. Jpn. 2016, 49, 22–28. [Google Scholar] [CrossRef]
- Nakamura, I.; Fujitani, T. Role of metal oxide supports in NH3 decomposition over Ni catalysts. Appl. Catal. A Gen. 2016, 524, 45–49. [Google Scholar] [CrossRef]
- Garcia-Garcia, F.R.; Guerrero-Ruiz, A.; Rodriguez-Ramos, I.; Goguet, A.; Shekhtman, S.O.; Hardacre, C. TAP studies of ammonia decomposition over Ru and Ir catalysts. Phys. Chem. Chem. Phys. 2011, 13, 12892–12899. [Google Scholar] [CrossRef]
- Sun, S.; Jiang, Q.; Zhao, D.; Cao, T.; Sha, H.; Zhang, C.; Song, H.; Da, Z. Ammonia as hydrogen carrier: Advances in ammonia decomposition catalysts for promising hydrogen production. Renew. Sustain. Energy Rev. 2022, 169, 112918. [Google Scholar] [CrossRef]
- Zielinski, M.; Janiszewska, E.; Drewniak, A.; Pietrowski, M. Methanation of CO2 over Ruthenium Supported on Alkali-Modified Silicalite-1 Catalysts. Molecules 2023, 28, 6376. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Chen, C.; Bian, C.; Ren, J.; Liu, J.; Huang, W. Enhanced Ammonia Decomposition by Tuning the Support Properties of Ni/GdxCe1-xO2-δ at 600 °C. Molecules 2023, 28, 2750. [Google Scholar] [CrossRef] [PubMed]
- Anota, E.C.; Cocoletzi, G.H.; Tapia, A.M.G. Armchair Boron Nitride nanotubes—Heterocyclic molecules interactions: A computational description. Open Chem. 2015, 13, 734–742. [Google Scholar] [CrossRef]
- Bautista, M.C.F.; Cortés-Arriagada, D.; Shakerzadeh, E.; Anota, E.C. Acetylsalicylic acid interaction with Boron nitride nanostructures—A density functional analysis. J. Mol. Liq. 2022, 355, 118980. [Google Scholar] [CrossRef]
- Anota, E.C. 2D boron nitride incorporating homonuclear boron bonds: Stabilized in neutral, anionic and cationic charge. SN Appl. Sci. 2022, 4, 295. [Google Scholar] [CrossRef]
- Harvey, J.N.; Aschi, M. Spin-forbidden dehydrogenation of methoxy cation: A statistical view. Phys. Chem. Chem. Phys. 1999, 1, 5555–5563. [Google Scholar] [CrossRef]
- Poli, R.; Harvey, J.N. Spin forbidden chemical reactions of transition metal compounds. New ideas and new computational challenges. Chem. Soc. Rev. 2002, 32, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Lin, S.; Guo, H. First-Principles Insights into Ammonia Decomposition Catalyzed by Ru Clusters Anchored on Carbon Nanotubes: Size Dependence and Interfacial Effects. J. Phys. Chem. C 2018, 122, 9091–9100. [Google Scholar] [CrossRef]
- Larijani, H.T.; Jahanshahi, M.; Ganji, M.D.; Kiani, M.H. Computational studies on the interactions of glycine amino acid with graphene, h-BN and h-SiC monolayers. Phys. Chem. Chem. Phys. 2017, 19, 1896–1908. [Google Scholar] [CrossRef] [PubMed]
- Hussain, R.; Saeed, M.; Mehboob, M.Y.; Khan, S.U.; Usman Khan, M.; Adnan, M.; Ahmed, M.; Iqbal, J.; Ayub, K. Density functional theory study of palladium cluster adsorption on a graphene support. RSC Adv. 2020, 10, 20595–20607. [Google Scholar] [CrossRef] [PubMed]
- Atkins, P.; de Paula, J. Atkins’ Physical Chemistry, 7th ed.; Chapter 27, molecular reaction dynamics; Oxford University Press: Oxford, UK, 2002; pp. 944–976. [Google Scholar]
- Lin, X.; Xi, Y.; Phillips, D.L.; Guo, W. The effect of a silica support: A density functional theory study of the C-H bond activation of ethane on a nickel oxide cluster. J. Phys. Org. Chem. 2016, 29, 134–144. [Google Scholar] [CrossRef]
- Cao, Y.; Ge, X.; Li, Y.; Si, R.; Sui, Z.; Zhou, J.; Duan, X.; Zhou, X. Structural and Kinetics Understanding of Support Effects in Pd-Catalyzed Semi-Hydrogenation of Acetylene. Engineering 2021, 7, 103–110. [Google Scholar] [CrossRef]
- Su, T.; Guan, B.; Zhou, J.; Zheng, C.; Guo, J.; Chen, J.; Zhang, Y.; Yuan, Y.; Xie, W.; Zhou, N.; et al. Review on Ru-Based and Ni-Based Catalysts for Ammonia Decomposition: Research Status, Reaction Mechanism, and Perspectives. Energy Fuels 2023, 37, 8099–8127. [Google Scholar] [CrossRef]
- Pinzón, M.; Avilés-García, O.; de la Osa, A.R.; de Lucas-Consuegra, A.; Sánchez, P.; Romero, A. New catalysts based on reduced graphene oxide for hydrogen production from ammonia decomposition. Sustain. Chem. Pharm. 2022, 25, 100615. [Google Scholar] [CrossRef]
- Yin, S.-F.; Xu, B.-Q.; Ng, C.-F.; Au, C.-T. Nano Ru/CNTs: A highly active and stable catalyst for the generation of COx-free hydrogen in ammonia decomposition. Appl. Catal. B Environ. 2004, 48, 237–241. [Google Scholar] [CrossRef]
- Gütlich, P.; Garcia, Y.; Goodwin, H.A. Spin crossover phenomena in Fe(II) complexes. Chem. Soc. Rev. 2000, 29, 419–427. [Google Scholar] [CrossRef]
- Terasaki, I.; Shibasaki, S.; Yoshida, S.; Kobayashi, W. Spin State Control of the Perovskite Rh/Co Oxides. Materials 2010, 3, 786–799. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2007, 120, 215–241. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Granatier, J.; Lazar, P.; Prucek, R.; Šafářová, K.; Zbořil, R.; Otyepka, M.; Hobza, P. Interaction of Graphene and Arenes with Noble Metals. J. Phys. Chem. C 2012, 116, 14151–14162. [Google Scholar] [CrossRef]
- Shakourian-Fard, M.; Kamath, G.; Jamshidi, Z. Trends in Physisorption of Ionic Liquids on Boron-Nitride Sheets. J. Phys. Chem. C 2014, 118, 26003–26016. [Google Scholar] [CrossRef]
- Kozuch, S.; Shaik, S. How to conceptualize catalytic cycles? The energetic span model. Acc. Chem. Res. 2017, 44, 101–110. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Gonzalez, C.; Schlegel, H.B. An improved algorithm for reaction path following. J. Chem. Phys. 1989, 90, 2154–2161. [Google Scholar] [CrossRef]
- Schuchardt, K.L.; Didier, B.T.; Elsethagen, T.; Sun, L.; Gurumoorthi, V.; Chase, J.; Li, J.; Windus, T.L. Basis set exchange: A community database for computational sciences. J. Chem. Inf. Model. 2007, 47, 1045–1052. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Adsorption Process | Ead (kcal/mol) | Gad (kcal/mol, 298 K) |
|---|---|---|
| 5Ru1 + 1BN → 5Ru1-BN | −11.7 | −4.4 |
| 7Ru2 + 1BN → 7Ru2-BN | −29.0 | −6.4 |
| 9Ru3 + 1BN → 9Ru3-BN | −32.2 | −10.6 |
| N-H Activation of NH3 on | Unsupported, 298.15 K | BN-Supported, 298.15 K | Unsupported, 673.15 K | BN-Supported, 673.15 K |
|---|---|---|---|---|
| Ru1 atom | 26.0 | 24.1 | 25.8 | 16.9 |
| Ru2 cluster | 21.8 | 21.1 | 24.0 | 21.0 |
| Ru3 cluster | 29.8 | 28.9 | 32.5 | 25.0 |
| NBO Charge Change of Different Moieties | |||||
|---|---|---|---|---|---|
| Ha | N | NH2 | Run | B19N19H16 | |
| Ru1-unsup | −0.214 | −2.207 | −2.204 | 0.418 | 0.000 |
| Ru1-BN | −0.190 | 0.021 | −0.055 | 0.086 | 0.159 |
| Ru2-unsup | −0.283 | −0.037 | −0.056 | 0.339 | 0.000 |
| Ru2-BN | −0.251 | −0.098 | −0.146 | 0.372 | 0.025 |
| Ru3-unsup | −0.274 | −0.024 | −0.058 | 0.332 | 0.000 |
| Ru3-BN | −0.240 | −0.087 | −0.130 | 0.417 | −0.047 |
| Most Favorable Spin State | ||||
|---|---|---|---|---|
| SM | IM1 | TS | IM2 | |
| Ru1-unsup | 5 | 3 | 3 | 3 |
| Ru1-BN | 5 | 3 | 3 | 3 |
| Ru2-unsup | 7 | 7 | 5 | 7 |
| Ru2-BN | 7 | 5 | 5 | 5 |
| Ru3-unsup | 9 | 9 | 9 | 9 |
| Ru3-BN | 9 | 9 | 9 | 9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, L.; Zhuang, H.; Zhang, Y.; Ma, L.; Xi, Y.; Lin, X. Support Effect of Boron Nitride on the First N-H Bond Activation of NH3 on Ru Clusters. Molecules 2024, 29, 328. https://doi.org/10.3390/molecules29020328
Zhao L, Zhuang H, Zhang Y, Ma L, Xi Y, Lin X. Support Effect of Boron Nitride on the First N-H Bond Activation of NH3 on Ru Clusters. Molecules. 2024; 29(2):328. https://doi.org/10.3390/molecules29020328
Chicago/Turabian StyleZhao, Li, Huimin Zhuang, Yixuan Zhang, Lishuang Ma, Yanyan Xi, and Xufeng Lin. 2024. "Support Effect of Boron Nitride on the First N-H Bond Activation of NH3 on Ru Clusters" Molecules 29, no. 2: 328. https://doi.org/10.3390/molecules29020328
APA StyleZhao, L., Zhuang, H., Zhang, Y., Ma, L., Xi, Y., & Lin, X. (2024). Support Effect of Boron Nitride on the First N-H Bond Activation of NH3 on Ru Clusters. Molecules, 29(2), 328. https://doi.org/10.3390/molecules29020328