Abstract
Since Mallouk’s earliest contribution, dye-sensitized photoelectrochemical cells (DSPECs) have emerged as a promising class of photoelectrochemical devices capable of storing solar light into chemical bonds. This review primarily focuses on metal complexes outlining stabilization strategies and applications. The ubiquity and safety of water have made its splitting an extensively studied reaction; here, we present some examples from the outset to recent advancements. Additionally, alternative oxidative pathways like HX splitting and organic reactions mediated by a redox shuttle are discussed.
1. Introduction
Owing to the acceleration of the economic development, the world’s total energy consumption is rapidly increasing and it has been predicted that the demand will reach more than 25 TW by 2050 [1]. Nowadays, fossil fuels such as coal, crude oil, and natural gas provide more than 80% of the request [2] but it is predicted that their reserves will last just for the next 50–60 years. Furthermore, greenhouse gasses such as carbon dioxide, which are produced by the combustion of the fossil fuels, will reach >1300 ppm CO2eq by the end of 2100 (460 ppm in 2010), leading to increases in the global mean temperature of up to 5 °C [3]. The scientific community is committed to using carbon-neutral energy sources, including biomass, geothermal, wind, and sun. The latter is distinguished by the benefit of being free, plentiful, and accessible to all the population, as well as by having a spectrum that spans a broad range of wavelengths from 280 nm (4.43 eV) to 2500 nm (0.5 eV), with a peak at about 2.5 eV. At high noon on a cloudless day, the surface of the Earth at average latitudes receives 1000 watts of solar power per square meter (1 kW m−2). Such standard irradiance is expressed as the Air Mass 1.5 (AM 1.5 G) condition. Due to its seasonal, day/night, and weather cycles, the sun has also the significant flaw of being intermittent. The most effective approach to store solar energy for an extended period of time is still under research but a number of photovoltaic (PV) technologies have been successfully developed to transform solar power into electricity [4]. The energy produced by the PVs can be temporarily stored into Li-batteries but it can also be used to create high-value products. With the technology at our disposal, creating high-density energy molecular bonds is likely the most effective method. For example, 3 kg of hydrogen produces 100 KWh of chemical energy, while the same amount of energy could be provided by 450 kg of lithium-ion batteries [5]. PVs can provide the bias to split water into O2 and H2 in the electrolyzers but multiple junctions are needed to satisfy the required overpotential. Electrolyzers are also constrained by the use of expensive electrodes [6]. These limitations can be sorted out by employing photoelectrochemical cells (PECs), devices able to obtain storable solar fuels as a result of water splitting, organic oxidations (e.g., halogen oxidation, formation of new C-C bonds, etc.), or CO2 reduction. As shown in Figure 1, in a typical PEC device, an n-type semiconductor absorbs the sunlight, generating electron-hole pairs. Subsequently, the holes will reach the semiconductor/electrolyte interface to perform oxidation reactions, while the photogenerated electrons in the conduction band (CB) of the semiconductor material will migrate to the counterelectrode in which the target substrate is reduced at the interface with the electrolyte.
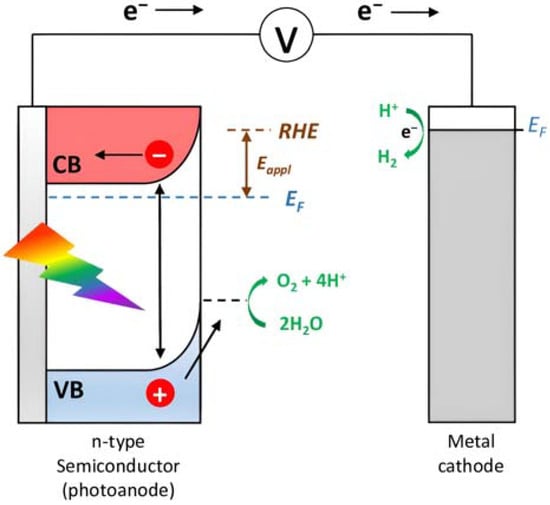
Figure 1.
Schematic illustration of the basic functioning of a PEC.
Artificial photosynthesis takes inspiration from nature, where both plants and bacteria can perform it, converting the sunlight into chemical energy. Bacterial photosynthesis, probably, represents the simplest form of light conversion being used to perform photophosphorylation, i.e., conversion of adenosine diphosphate (ADP) into adenosine trisphosphate (ATP). The oxygenic photosynthesis, performed for example by cyanobacteria, algae, and higher plants, instead, is characterized by a net chemical reaction leading to the conversion of water into oxygen, protons, and electrons. Protons are employed in the formation of ATP, while electrons for the reduction of carbon dioxide into carbohydrates. The two-photon architecture can be simply illustrated in terms of energy with the “Z-scheme”, in which the two photosystems (i.e., PSI and PSII) are reported (Figure 2) [7]. A first photon is absorbed by the chlorophyll P680 in PSII leading to the formation of its excited state P680*, from which a chain of electron transfer takes place, and electrons can diffuse through the plastocyanin (PC) to the second reaction center PSI, in which a second series of photoinduced charge separation occurs from P700. The resulted P680+ is characterized by an oxidation potential close to 1.2 V vs. NHE able to drive the water oxidation in conjunction with an oxygen evolving complex catalyst (OEC). The hole localized on P680 is filled by electron transfer through a tyrosine residue (Yz) relay from the Mn4Ca cluster catalyst that will perform the considerably demanding water oxidation (four protons and four electrons). On the other side, the reduced form of P700 can transfer electrons through ferroxidin (Fd) and ferroxin-NADP(+) (FNR) to NADP+, generating its reduced form NADPH. This, together with the generated ATP, will be used in the Calvin cycle for CO2 reduction into carbohydrates.
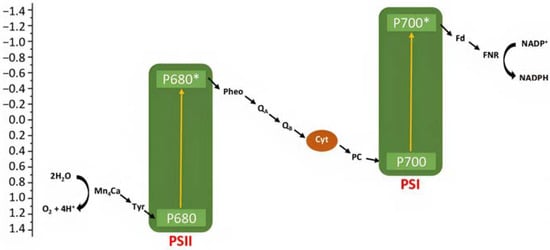
Figure 2.
Simplified representation of the Z-scheme for a photosynthetic process.
Artificial photosynthesis was first demonstrated by Honda e Fujishima in 1972 by employing ultraviolet (UV) irradiation using a TiO2-based PEC for water splitting [8]. Based on this observation, attempts to develop a dispersion of Pt/RuO2 catalyst for water splitting have been started [9,10]. A further step forward was realized by Grätzel who, being fascinated by the outstanding performances of TiO2 substrate, realized a titania-based Pt/RuO2 water oxidation catalyst (WOC) [11]. From here, several efforts have been made to try to synthesize different materials for the photoanodic compartment with the aim of extending the spectral response to the visible part of the solar spectrum. In this contest, narrow band-gap semiconductors (SCs), like WO3 [12,13], Fe2O3 [14,15], BiVO4 [16], WO3/BiVO4 [17], CuWO4 [18], and others, are very useful and promising tools that enable high photocurrent densities and incident photon to current conversion efficiencies (IPCE%). Nevertheless, some drawbacks should be thought over. For instance, it is difficult to prepare narrow band-gap material able to match both the oxidation and reduction potential of water; these substrates are also stable just over a short range of pH and, more generally, all the semiconductors are characterized by a deep valence band (VB) that does not allow selective oxidations. Beside this technology, since the pioneering work of Mallouk [19], dye-sensitized photoelectrochemical cells (DPECs) represent a promising approach to the traditional PECs, which offer flexibility in design and the possibility to fine tune both the light absorption and the oxidizing capabilities [20]. A myriad of chromophore–catalyst has been synthesized in the past but the performances of the resulted devices are relatively low compared to the narrow band-gap-based semiconductors. Although water represents an interesting target owing to its natural abundance and safety, its oxidation is relatively demanding (four protons and four electrons) and it requires a high oxidation potential of 1.23 V vs. NHE pH 0. Furthermore, the resulting oxygen is poor in terms of industrial attraction, and it is generally catalyzed by expensive noble metal-based catalysts [21,22]. Besides water, other inexpensive storage mechanisms for solar energy have been developed, one of which is the hydrohalic acid (HX) splitting [23]. Among this category, hydrobromic acid (HBr) is characterized by the highest theoretical solar-hydrogen efficiency and its energy can be extracted by combustion [24], fuel cells [25,26], and redox flow batteries [27]. Water could be also replaced by the selective oxidation of organic molecules, where, for instance, the transformation of alcohols to high-valued carbonyl compounds is currently under investigations [28]. The emphasis in this review is on the use of metal complexes (i.e., sensitizers and/or catalysts) owing to their outstanding performances in performing oxidation reaction when supported onto the semiconductor surface.
2. Basic Concepts of DSPECs
Dye-sensitized photoelectrochemical cells are based on a molecular approach; the main processes involved in the standard n-type configuration are depicted in Figure 3a. The absorption of sunlight results in the formation of the dye’s excited state from which the oxidative quenching occurs as consequence of the electron injection into the CB of the n-type semiconductor. This leads to a temporary localization of the holes onto the oxidized dye (D+) and eventually, hole transfer to a proper catalyst (e.g., water oxidation catalyst (WOC)). Consequently, the photoinjected electrons and protons migrate to the cathodic compartment due to the free energy gradient. Lastly, the holes and the electrons are consumed to promote oxidation (H2O, HX, etc.) and reduction (H+, CO2, etc.) pathways. One of the greatest advantages of the DSPECs is the employment of a sensitizer able to collect the visible photons precluded to the wide band-gap n-type semiconductors, such as TiO2 [29], SnO2 [30], and ZnO [31] (some examples will be described in Paragraph 4). Considering that the typical semiconductor nanoparticles dimensions are on the order of 20–30 nm, with an overall thickness ranging between 3–8 µm and sensitized with a monolayer of dye (ε ≈ 104–105 M−1 cm−1), this translates into a light-harvesting capacity higher than 90% on the maximum absorption [32,33].
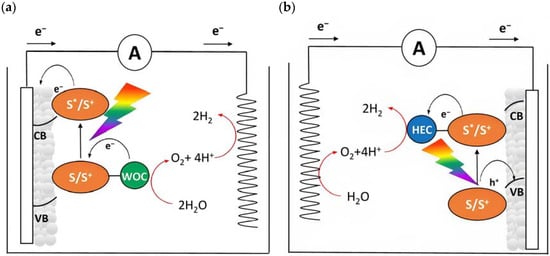
Figure 3.
Schematic representation of a DSPEC in the framework of water splitting in an (a) n-type or in (b) p-type configuration in which the semiconductors, the sensitizers, the catalysts (WOC or HEC), and target reactions are depicted.
An opposite situation (Figure 3b) can be realized in the presence of a photocathode in which the electrode is decorated with a sensitized p-type based-semiconductor like NiO or, more recently, the promising Cu(In,Ga)Se2 (CIGS) [34]. After the absorption of sunlight with the consequent generation of the dye’s excited state, the holes will be injected into the valence band (VB) of the mesoporous substrate and the reduced sensitizer is restored to its ground state by transferring an electron to promote the hydrogen evolution. The photoinjected holes, instead, migrate through the back contact to the anodic counterpart to promote the oxidation reaction.
The quasi-Fermi level is an important parameter that determines the thermodynamics of the cell and describes the capability to lead to the desired fuels. For instance, in order to promote hydrogen evolution without applying an external bias, the quasi-Fermi level of the photoanode, which is in equilibrium with the Fermi level of the cathode, should be more negative than the formal potential for the hydrogen evolution ( vs. RHE). If this requirement is not met, an external bias is needed to enable the desired reaction (Figure 4). In particular, this voltage is applied by an external source, and it should also provide the cathodic overvoltage necessary to sustain the current flow. In a first approximation, this external bias does not affect the redox potential of the sensitized surface and thus, the photoproduced holes are able to prompt for the anodic reaction, such as water oxidation ( vs. RHE).
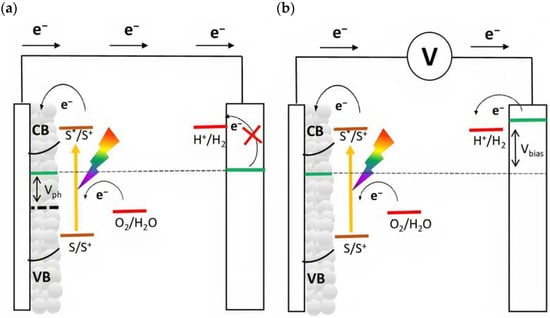
Figure 4.
Schematic representation of energy levels for an n-type-based DSPEC for water splitting. The quasi-Fermi level (dotted line) of the n-type material is approximated with the flat band potential (Vfb) of the semiconductor in condition of (a) zero bias and (b) under the application of a bias.
In order to avoid the external bias, a DSPEC should be realized by coupling an n-type semiconductor to a p-type substrate, realizing an n-p junction (Figure 5), in which the two compartments are able to promote reactions in consideration of their own photovoltage. This setup can be represented as a photochemical diode [35]. In all the above-described situations, an effective approach to collect the products is to physically separate the anodic and cathodic compartments with an ion-permeable membrane (PEM), the most common of these is the Nafion membrane, which is able to exchange protons from the anode to the cathode. This separation is required to avoid the reduction in the oxidized products at the cathode and vice versa (Figure 5).
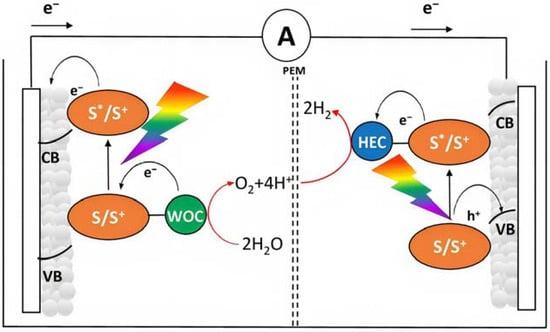
Figure 5.
Tandem n-p DSPEC for solar water splitting. The photoanode (left) and photocathode (right) compartments are depicted.
3. Efficiency Parameters
The most important parameter that describes the efficiency of a DSPEC exposed to a broadband solar irradiance, such as the Air Mass 1.5 Global illumination (AM 1.5 G), is the solar-to-hydrogen conversion efficiency (STH). This parameter should be employed in zero-bias conditions, meaning when the applied voltage between the working and the counter electrode is zero, and the process is sustained solely by sunlight. Both the working and the counter electrode should be maintained in the same pH conditions to avoid a Nernstian gradient and the electrolyte must not contain any sacrificial agent that could affect the water-splitting performances [36]. STH can be defined as follows:
where Jsc is the short circuit photocurrent density, 1.23 V represents the thermodynamic water splitting potential at 25 °C and pH 0, ηF is the faradic efficiency related to the hydrogen production, and Ptotal represents the incident illumination power intensity in mW cm−2. The STH is no longer valid when a bias between the working and the counter electrode is applied because the energy loss due to the need or the required overvoltage should also be taken into account. The applied bias photon-to-current efficiency is defined as follows:
where Jph represents the photocurrent density obtained under an applied bias Vb. Certain precautions are still applicable, such as avoiding the use of a sacrificial donor as well as maintaining the same pH in the two compartments of the cell. Another important figure of merit of the DSPECs is the incident photon-to-current conversion efficiency (IPCE%) that describes the photocurrent recorded per incident photon flux as a function of the monochromatic illumination (action spectrum) and can be described as follows:
where h is the Planck’s constant, c is the speed of light in vacuo (m s−1), e is the electron charge (C), J is the photocurrent density (A m−2), λ is the photon wavelength (m), and P(λ) is the radiant power density (W m−2). Generally, it is important to express the IPCE as a function of the key steps of the device working principle as follows:
where LHE is the light harvesting efficiency, Φinj is the injection quantum yield of the photogenerated electrons into the CB, and ηc represents the electron collection efficiency at the photoanode. This former parameter can be expressed as follows:
where the absorbance A can be made explicit as 1000 ε Γ with ε the molar extinction coefficient (M−1 cm−1) and Γ the dye surface concentration (mol cm−2). The photocurrent density (Jph) generated by a DSPEC device can be extrapolated by integrating the IPCE over the entire set of wavelengths as in Equation (6):
LHE = 1 − 10−A(λ)
The photocurrent density extrapolable from a DSPEC device can be also derived from the diode equation [37] as follows:
where Jph is the limiting current density, Vj = V + iR represents the iR drop of the applied potential across the cell, and m is the ideality factor of the diode characterized by the J0 dark current. In particular, it should be highlighted that J0, m, and R depend on the specific chemical nature of the system, and they also describe the quality of the cell components, such as the mesoporous semiconductor characteristics, the ohmic contacts, and the conductivity of the solution.
4. Sensitizers and Surface Binding and Stabilization Strategies
To collect the visible light precluded by the wide band-gap semiconductors, the sensitizers represent one of the most important components of the DSPECs. To ensure the optimal functioning of the device, thereby translating into higher efficiencies, an ideal chromophore should exhibit some strict requirements. For instance, a good red response is enormously welcome since it can lead to efficient light harvesting by collecting the low-energy photons of the visible spectrum [33]. Large values for the driving force for the charge injection (ΔGinj = |Eox* − Efb|, where Efb represents the flat band potential of the semiconductor) are essential to maximizing the injection quantum yield (Φinj). Thus, it is imperative that the excited state oxidation potential (Eox*) of the sensitizer is more negative with respect to the metal oxide’s Fermi level, as depicted in the scheme of Figure 6 [32]. Efficient hole transfer to the target substrate is another important aspect and, from this perspective, the ground state oxidation potential of the sensitizer (Eox(s/s+)) must be larger than the oxidation potential of the reduced target molecule (Eox(substrate)) (e.g., water, HX, organic substrates, etc.), being ΔE = Eox(s/s+) − Eox(substrate) > 0 [38]. Additionally, stable anchoring groups are necessary to avoid hydrolytic detachment of the dye from the surface and, as expected, the sensitizers must be chemically and electrochemically stable to face several working cycles [39].
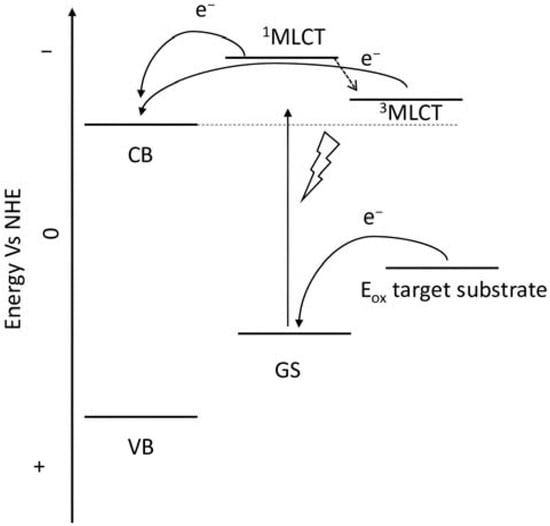
Figure 6.
Representative scheme of the energetic levels of the semiconductor, a general ruthenium complex, and the target substrate.
Since the pioneering contribution of Mallouk, ruthenium-polypyridine complexes [19] exhibit the best characteristics as light harvesters owing to their rather broad absorption extending between the blue-green portion of the visible spectrum and their relatively high molar extinction coefficient (ε ≈ 104 M−1 cm−1). Ruthenium complexes are also characterized by a lifetime on the order of hundreds of ns, by electrochemical stability, and by ground and excited state oxidation potential, which matches the required thermodynamics (see Figure 6). Spectroscopic and electrochemical properties can be fine-tuned by introducing, for instance, different electron donating or electron-withdrawing functional groups on the ancillary ligands, typically bipyridines or terpyridines [40,41]. Electron injection is thermodynamically feasible from both the lowest singlet (1MLCT) and triplet (3MLCT) states but, in order to maximize the charge injection rate, the electronic coupling between the sensitizer and the substrate should be established through the formation of covalent bonds. Due to its relative stability and easy synthesis, the typical anchoring group adopted for classic DSSC devices consists of carboxylic moieties directly linked to the aromatic system of the ligand [32]. However, the resulting ester covalent bond between the anchoring moieties and the metal oxide can be easily cleaved in alkaline or even neutral conditions, leading to the desorption of the dye and the consequent decay of performance [42,43,44,45,46]. Among several alternative strategies that have been explored, studies that have focused on comparing the carboxylic (–COOH) and phosphonic (–PO3H2) anchoring groups are worth noting. For instance, a contribution realized by the Group of T. Meyer [47] highlighted a 90% of desorption in a water medium at pH 5.7 for complexes bearing -COOH against only 30% when –PO3H2 are employed. The group of Choi [42,43] investigated the effect of the number and the kind of grafting units in two families of ruthenium complexes bearing carboxylic (entries 1, 2, and 3 in Table 1) and phosphonic (4, 5, and 6) moieties, respectively. In particular, –PO3H2 bearing dyes resulted in a stable absorption over higher pH and the number of anchoring groups resulted in being less crucial than in the case of the -COOH-based analogs, owing to the strong bond exhibited by phosphonic groups with the TiO2. Despite the fact that the -PO3H2 anchoring group seems to be a better choice, the long-term stability of Ru-based complexes was achieved by employing dyed-surface passivation strategies, such as hydrophobic polymer overlayers [48], electro-assembly [49,50,51,52], siloxane [53,54] or silatrane surface bindings [55,56,57] or, more in general, hydrophobic environments [58]. Atomic layer deposition (ADL) to realize an aluminum oxide (Al2O3) or a titanium dioxide (TiO2) overlayer, starting from Al(CH3)3 or TiCl4 precursors, respectively, was found to represent an efficient way to prevent the hydrolysis of the ester binding mode. T. Meyer analyzed the stability of a water oxidation catalyst [Ru(Mebimpy)(4,4′-(PO3H2CH2)2bpy)-(OH2)]2+ (Mebimpy = 2,6-bis(1-methyl-1H-benzo[d]-imidazol-2-yl)pyridine) (7) with and without ALD overlayer on an ITO electrode, obtaining a stable film over 2.8 h in a pH 11 water medium [59]. The same group employed a “mummy” strategy to stabilize chromophore–catalyst structures (Figure 7a), obtaining light-assisted water oxidation stable over 6 h photolysis (at pH 8) [60]. This approach consisted in loading 4 onto nanoTiO2 or nanoITO followed by an ALD overlayer of a 1.5 nm thick Al(CH3)3/H2O film. The catalyst 7 was then loaded onto the ALD overlayer and, subsequently, a final coating was realized in a similar fashion by increasing the total thickness to ~3 nm by “mummifying” the chromophore–catalyst assembly.
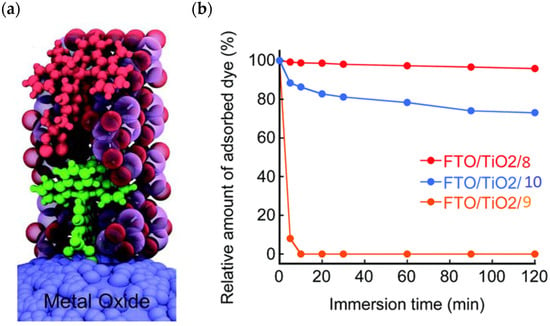
Figure 7.
(a) Visualization of the ALD mummy-protected surface assembly with the dye 4 (green molecule) and the catalyst 7 (red molecule) embedded in ~3 nm of Al2O3 (purple and red spheres). (b) Plots of the relative amount of dye (8, 9, and 10) adsorbed on the TiO2 electrode as a function of immersion time in an aqueous acetate buffer solution (0.1 M, pH 5.0). Reprinted and adapted with permission of the Royal Chemical Society from refs. [60,61].
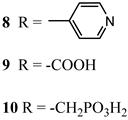
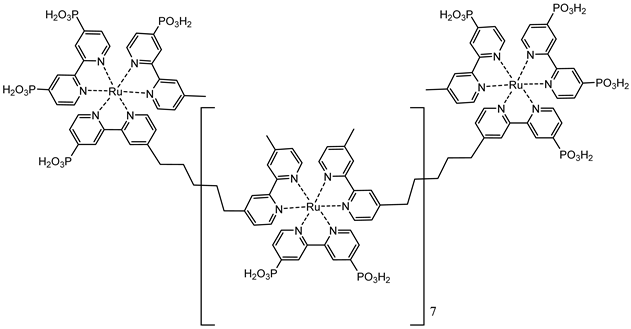
An interesting strategy to achieve a stable linkage in a basic medium consists of employing pyridyl (py) anchoring moieties [61] that are known to form coordination bonds with surface Ti4+ ions, unlike the -COOH and -PO3H2 which interact with the Bronsted acid sites (i.e., the exposed OH groups) [62,63,64]. Indeed, stability tests in the electrolysis medium at pH 5 yielded negligible desorption of 8 (pyridyl linkage) at least for 2 h whereas, in the same conditions, 9 (-COOH linkage) and 10 (PO3H2 linkage) (Figure 7b) resulted in almost quantitative or 30% detachment, respectively, after only ≈10 min. In addition, 8 was stable in a neutral (pH 7) or basic (pH 9) aqueous buffer solution, clearly suggesting that the pyridyl anchoring moiety is likely a more suitable choice for photocatalytic applications under a water medium. A recent strategy was achieved by Mallouk by employing a phosphonate oligomeric ruthenium dye (11) [65], which resulted in a dramatically improved electrode adhesion in the range of pH between 4 and 7.8. The authors highlighted a faster cross-surface hole diffusion for the multinuclear species (≈ one order of magnitude) if compared to the monomeric one. It is important to note that this parameter was identified as one of the limiting factors to reach a higher turnover number [66,67,68].
Another approach to reach a stable system in a basic medium was proposed by the group of G. Meyer, who compared the diazonium group (12) with the classic -COOH (13) and -PO3H2 (14) sites as grafting agents for ruthenium complexes, as depicted in Figure 8 [69]. As for 12, it was stable over 6 h in saturated NaOH while 14 desorbed by more than 50% in 1 min at pH 7 and 13 detached immediately at pH > 5. In addition, 12 exhibited a stable photocurrent response at pH 12 for 4 h confirming the stability of this new anchoring group in a basic medium. The same experiment was extended in acid conditions where 14 is highly stable: the photocurrent response was significantly higher for the phosphonic dye with respect to 12; this is ascribable to a less efficient electronic injection of the latter, which arises from the weaker electronic coupling with the Ti sites and/or a fast-back electron transfer.
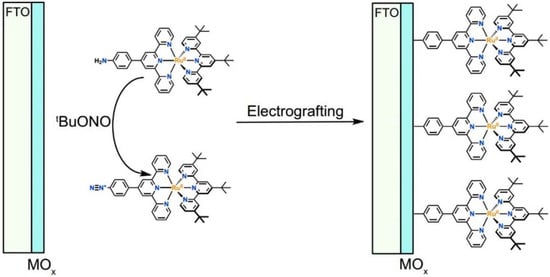
Figure 8.
General strategy developed for electrografting diazonium-substituted sensitizer molecules on metal oxide (MOx) surfaces. Reprinted with permission of the American Chemical Society from ref. [69].
Other transition elements were explored as metal centers in coordination compounds in the framework of n-type sensitization such as Ni(II) [70,71], Cu(I) [72], Zn(II) [71], or, more recently, Fe(II) [73,74].
Porphyrins represent another promising sensitizers family [75,76] owing to their high molar extinction coefficient (>105 M−1 cm−1 for the Soret band) over the high and low energy portion of the visible spectrum (i.e., Soret and Q bands) and owing to their redox properties well suited for DSPECs applications. In fact, the singlet ground state (S0) is generally higher in energy than the corresponding state of Ru complexes, resulting in stronger oxidizing capabilities by the photo-oxidized sensitizer (S+). The electron injection into the semiconductor’s conduction band is favorable from the Singlet excited states (S1 and S2) because the Triplet state (T1) is closer in energy or even lower than that of the CB (Figure 9). As in the case of the Ru-based sensitizers, the electrochemical properties can be tuned by varying the peripheral substituents [77].
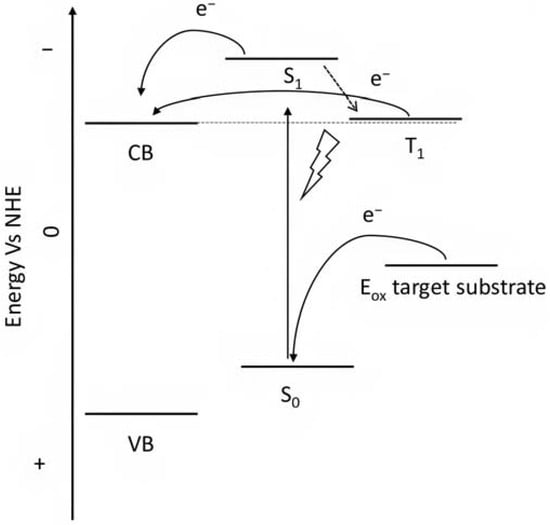
Figure 9.
Representative scheme of the energetic levels of the semiconductor, a general porphyrin, and the target substrate.
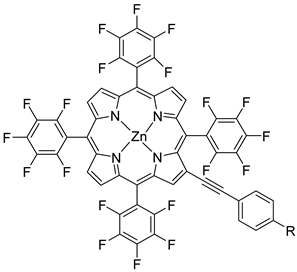
Regarding the covalent attachment of porphyrin sensitizers, -COOH [78], -PO3H2 [79], -CNCOOH [75], and -py [80] have been explored to reach high stability, excited state directionality, and good electronic coupling. In particular, T. Meyer investigated the absorbance of electrodes sensitized with -COOH- and -PO3H2-based porphyrins every 15 min for 16 h in 0.1 M HClO4 [79]. A desorption of 80% and just 10% for -COOH or -PO3H2 grafting moieties, respectively, was observed, further confirming the improved adhesion provided by the phosphonate groups with respect to carboxylic functions. In the framework of HBr oxidation, Orbelli et al. [75] synthesized three perfluorinated Zn porphyrins (15, 16, and 17) bearing pentafluorophenyl electron-withdrawing groups to increase the ground-state oxidation potential. Moreover, sensitizers differed for the nature and position of the π–conjugate linker between the core and anchoring group, tasked to bind the metal oxide characterized by both carboxylic and cyanoacrylic groups. The latter resulted in the best performer among the series owing to the strongest electron-withdrawing properties that led to the best coupling and directionality for the electron injection. Since they worked in acidic medium, the photocatalytic performances were not affected by the stability of both the employed anchoring groups, which is not critical in these conditions. Among the macrocycles-based chromophores, phthalocyanines (Pcs) are robust and intensely colored (blue pigments) with high chemical, thermal, and light stability. These interesting features suggested this class of compounds as promising light harvester in the framework of solar cells [81,82,83].
However, despite the high performances of the described classes of compounds, ruthenium polypyridyl complexes are characterized by the main disadvantages of being rare, expensive, and toxic and by having a molar extinction coefficient that steeply decreases for λ > 500 nm. On the other hand, a relevant drawback when it comes to porphyrin dyes is related to the fact that their synthesis involves a multi-step route with a relatively low overall yield [84,85]. These limitations can be sorted out by employing metal-free organic dyes owing to their low cost, easiness of tuning their molecular orbital energetics by employing electron donating or withdrawing moieties, and also considering their extremely high molar extinction coefficient. The most widespread structure employs a donor–π bridge–acceptor (D-π-A) design which represents a class of compounds successfully analyzed in the framework of the regenerative cells [86]. After a few contributions related to the employment of perylene diimide (PDI), F. Li et al. reported one of the first applications of an organic-based sensitizer tandem cell. In detail, 4-(Diphenylamino)phenylcyanoacrylic acid (18), a D-π-A dye, was applied for the photoanodic compartment coupled with the well-known (4-(Bis-(4-[5-(2,2-dicyano-vinyl)-thiophene-2-yl]-phenyl)-amino)-benzoic acid) (19) for the photocathode side in which the photoanode was able to produce 300 µA cm−2 at pH 7 [87]. It is important to highlight the attempts to prompt the water splitting by employing 4,4-difluoro-4-bora-3a,4a-diaza-s-indacene (BODIPY) dyes given their high molar extinction coefficient and photostability, which translates into promising performances [88,89]. After having adopted a π-extended BODIPY with a near infrared response for DSSC applications [90,91] and for bulk heterojunction solar cells [92], Kubo et al. synthesized 20, which is characterized by a λmax at around 687 nm (λonset ≈ 728 nm) with an ε of 1.09 × 105 M−1 cm−1. This molecular system was used for water-splitting applications [93]. Thanks to the spectroscopic, electrochemical, and photoelectrochemical analysis, they were able to demonstrate the capability of 20 to collect the NIR region and the ability to provide the water oxidation by applying a low overpotential. A similar approach was followed by T. Meyer that sensitized a SnO2/TiO2 photoanode with a series of BODIPY for the oxidation of hydroquinone and benzyl alcohol. The best performing, namely 21 (N,N′-Difluoroboryl-1,7-dimethyl-3,5-bis(4-(3,6-di-tert-butylcarbazol-9-ylstyryl)phenyl)-8-[4-(dihydroxyphosphoryl)phenyl]dipyrrin), was able to reach a stable photocurrent of 200 µA cm−2 in the case of the oxidation of the former substrate and a current of 35 µA cm−2 in the case of the latter (Figure 10) [94].
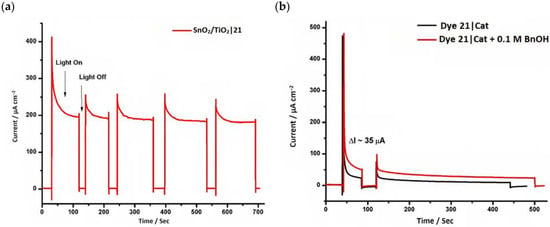
Figure 10.
(a) Chopped current–time (I–t) profiles for SnO2/TiO2|21 at pH 4.5, 0.1 M acetate buffer with 0.4 M LiClO4 and 20 mM H2Q, Eapp = 0.2 V (b) Current–time (I–t) profile for 21 in 0.1 M acetate buffer with 0.4 M LiClO4 with and without 0.1 M BnOH. Reprinted and adapted with permission of the American Chemical Society from ref. [94].

Table 1.
Structures of the molecules reported in the main text. S and C refer to a sensitizer or a catalyst, respectively.
Table 1.
Structures of the molecules reported in the main text. S and C refer to a sensitizer or a catalyst, respectively.
| Structures | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
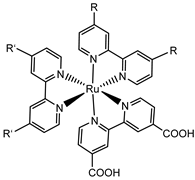 |  | 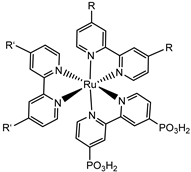 | 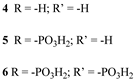 |  | 7 | |||||||
| S [42,43,95] | S [42,43,60,96,97,98,99] | C [59,60,97] | ||||||||||
 |  |  | 11 | |||||||||
| S [61] | S [65] | |||||||||||
 | 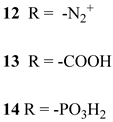 |  |  | |||||||||
| S [69] | S [75] | |||||||||||
 | 17 | 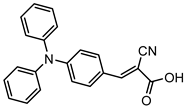 | 18 | 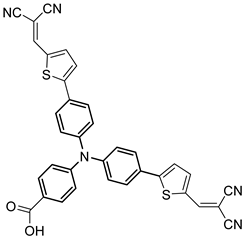 | 19 | |||||||
| S [75] | S [87] | S [87] | ||||||||||
 | 20 |  | 21 | |||||||||
| S [93] | S [94] | |||||||||||
5. Water Splitting
The overall water splitting process can be described by the following reactions:
5.1. Water Oxidation Catalyst (WOC)
The complex kinetic route that drives the water oxidation involves multi-electron steps and would result in exceedingly high activation barriers for the direct hole transfer from the photo-oxidized chromophore to water molecules, thereby resulting in predominant back electron recombination. Thus, the water oxidation catalyst (WOC) plays an important role in boosting the performances of DSPECs. Molecular complexes offer advantages such as a fast rate for electron transfer (ET), redox and synthetic tunability, a relatively fast rate for water oxidation, and improved catalytic efficiency, especially when scarce metals are used. Among the catalysts, ruthenium complexes are widely studied owing to their well-known synthetic route, strong metal-to-ligand bonds, rapid rates of oxygen evolution ensuring system stability, and well-defined oxidation states (RuII, RuIII, RuIV, and RuV) able to facilitate the water oxidation. Some representative Ru-based WOC are depicted in Table 2.
Table 2.
Historical overview of the main breakthroughs toward the realization of efficient Ru-based WOC.
In 1982, T. Meyer and co-workers introduced the first homogeneous WOC knows as blue dimer, which is a ruthenium polypyridyl dinuclear complex characterized by a Ru-O-Ru bridge and the coordination of two H2O molecules to each Ru center (Table 2-I) [105,106]. The μ-oxo bridge plays an important role in stabilizing the complexes in their high oxidation states through electronic delocalization, facilitated by the strong coupling between the two metal centers. The proposed mechanism involves four proton coupled electron transfer (PCET) steps that give access to a high-valent Ru(V,V) intermediate (intermediate 2, Figure 11), which is subsequently attacked by H2O. This yields the formation of a hydroperoxo species (intermediate 4, Figure 11) that is intramolecularly oxidized by the second metal core, ultimately resulting in the release of O2 (intermediate 6, Figure 11) [107].
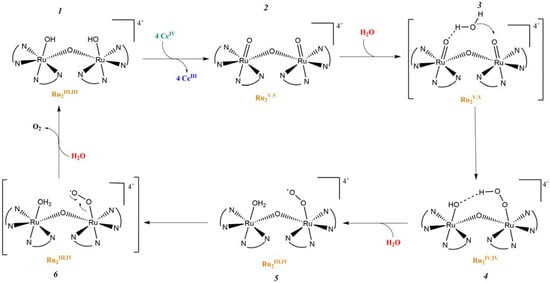
Figure 11.
Proposed mechanism of oxygen evolution in μ-oxo bridged blue dimer-type Ru (II) complexes.
The instability of this class of WOCs is mainly due to the cleavage of the μ-oxo bridge, forming the single non-active monomeric system, which generally led to relatively low performances towards water oxidation. To sort out this problem, Liobet et al. [108] reported the first example of a dinuclear ruthenium complex (Ru-Hbpp = [Ru2(OH2)2(bpp)(tpy)2]2+ where Hbpp = 2,2′-(1H-pyrazole-3,5-diyl)dipyridine) capable of driving water oxidation in the absence of the Ru-O-Ru bridge (Table 2-II). The oxygen evolution rate of this complex was found to be three times larger than that of the blue dimer. Mimicking the multimetallic core of the natural OEC, all the first examples of WOC were based on multiple metal cores, spreading the idea that at least two metal centers were needed to prompt the water oxidation. In 2005, Thummel’s group reported the first evidence of a single-site catalyst capable of driving the OER (Table 2-III) [109]. This study was then corroborated by Meyer and co-workers in 2008 who proposed a well-defined mechanism involving the Ru(V) state, paving the way for a series of stable and efficient single site catalysts (Table 2-IV) [110,111]. One of the main drawbacks of the employment of the aforementioned neutral and π-accepting ligands-based WOC was found in the high oxidation potential necessary to reach the highly oxidized Ru(IV) and Ru(V) states, which are generally poorly aligned with the chromophore ground state level. In fact, Ce(IV) is often required as a triggering agent for oxygen production. Inspired by studies mimicking the OEC in PSII, which showed that phenolate and carboxylate ligands can efficiently stabilize high-valent Mn states, Sun and co-workers were the first to introduce carboxylate moieties to access high-valent Ru=O under lower potential. Their first example reported on a trans-dimeric complex coordinated by a 3,6-bis(6-carboxypyrid-2-yl)pyridazine (H2cppd) ligand, which is a modified pyridazine-based polypyridyl equatorial backbone containing two carboxylate groups (Table 2-V) [112]. This was followed by the cis-conformation (Table 2-VI) [113] and by a Ru(bda)(pic)2 monomeric system (where bda = 2,2′-bipyridine-6,6′-dicarboxylic acid) bearing two carboxylate groups in the equatorial ligands (Table 2-VII) [114]. Thanks to the strong electron-donating capabilities of the carboxylate moieties, the Ru(bda)(pic)2 exhibited a significantly lower oxidation potential compared to Ru complexes prepared with neutral ligands. The promising performances, coupled with the easy tunability of the axial ligands of this latter class of compounds, attracted an extensive attention and efforts from the scientific community, leading the application of the Ru-bda WOCs for efficient DSPECs [107,115,116].
5.2. Examples of Water splitting
As pioneered by Mallouk and co-workers [19], Ru(II) polypyridine complexes emerged as a very promising class of sensitizers for photoelectrochemical cells. Mallouk employed a Ru(II) tris(bipyridine) heteroleptic derivative (22 inTable 3) to oxidize water (Figure 12) in conjunction with an IrO2·nH2O WOC, which was synthesized and characterized in detail by Harriman et al. many years before [117]. Prior to supporting the sensitizer–catalyst system onto TiO2, it was analyzed in contact with a 1 M Na2S2O8 solution, acting as an electron acceptor. Upon laser excitation at 532 nm, an electron transfer from the excited state of the dye (22*) to the acceptor S2O82− takes place. The regeneration of the reduced form of 22 is thus driven by an electron transfer from the nanoparticles of IrO2·nH2O WOC, with a time constant of about 2 ms. When the system was supported onto TiO2, the oxidative quenching of 22 occurred by injecting electrons into the CB of the semiconductor. However, it was observed that the back-electron transfer was approximately one order of magnitude faster compared to the regeneration process facilitated by IrO2·nH2O, thereby limiting the performances of the device.
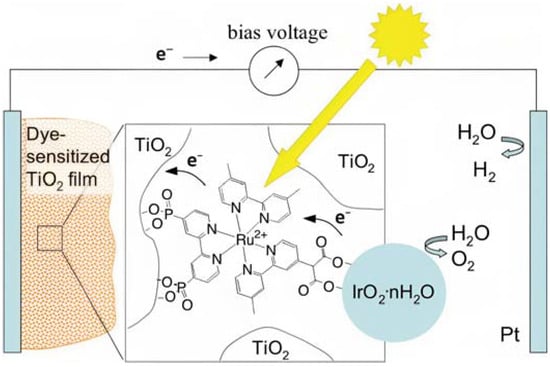
Figure 12.
Schematic representation of the main components of the DSPEC, namely the TiO2 semiconductor, the sensitizer (22), and the IrO2·nH2O WOC. Reprinted and adapted with permission from the American Chemical Society from ref. [19].
The photocurrent density–voltage profile in a buffered solution (pH 5.75) containing 30 mM Na2SiF6 and 500 mM Na2SO4 revealed an open circuit potential of −325 mV vs. Ag/AgCl along with a stable photocurrent of 30 μA. In the presence of an undyed TiO2, only a marginal current of ≈1–2 μA was observed. The authors were able to successfully describe the water-splitting reaction in the presence of visible light. However, they recorded a photoanodic oxygen generation of roughly 20%, which was mainly attributed to the slow electron transfer from the WOC to 22*. Based on the Mn4Ca active site into the PSII, Rüttinger et al. synthesized a [Mn4O4L6]+ (or “cubium”, L = (MeOPh)2PO2−) (23) WOC [118,119]. To ensure water solubility, the WOC was then supported onto a thin Nafion membrane [120]. Subsequently, the WOC has also been investigated in DSPEC in combination with the sensitizer 1 (Figure 13a), which is characterized by a 2–300 meV driving force for the oxidation of the WOC (23) (the system is reported as: 23-Nafion/1-TiO2) [95].
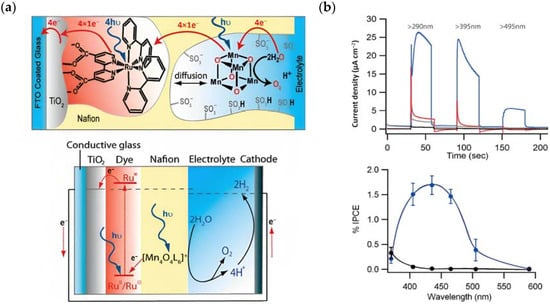
Figure 13.
(a) Top: Schematic representation of the main component of the DSPEC (TiO2, the sensitizer (1) and the Mn-based WOC (23) and bottom: the main operating principles. (b) Top: chopped photocurrent density–time profile for 23- Nafion/TiO2 (gray), Nafion/1-TiO2 (red), and 23-Nafion/1-TiO2 (blue), illuminated at 100 mW cm−2 through a series of long pass light filters, and bottom: average incident photon to current efficiency (IPCE) plot for 23-Nafion/1-TiO2 (blue) and TiO2 (black) electrodes. Reprinted and adapted with permission of American Chemical Science from ref. [95].
The undyed TiO2 generated a photocurrent lower than 1 µA cm−2 (Figure 13b, black line), a slightly higher photocurrent of ≈5 µA cm−2 (Figure 13b, red line) for the Nafion/1-TiO2 system, a comparable value for 23-Nafion/TiO2 and a significantly higher and sustained value for the complete 23-Nafion/1-TiO2 of ≈15 µA cm−2. An IPCE of 1.7% was observed (Figure 13b bottom) and it was consistent with the shape of the MLCT band of 1, confirming its capabilities as absorbing and injecting species. A similar approach was then realized by Li et al., leading to a significantly higher photocurrent density [96], by employing the sensitizer 4 and a 24-based WOC [114].
Since the catalytic oxidation driven by 24 is pH dependent (the onset for water oxidation was found to be 1.5 V at pH 1 and 0.98 V at pH 7); to prevent the catalytic process of 24 from shifting to a higher potential than the E1/2 of 4, the sulfonic acid groups in the Nafion membrane were neutralized before immobilizing the complex. The cationic WOC was generated with CeIV before intercalating the species into the membrane. The transient photocurrent densities are reported in Figure 14a. In 0.1 M Na2SO4 medium, the TiO2-Nafion-complex 24 system generated a negligible photocurrent (blue curve). When the WOC was replaced by the sensitizer, an initial photocurrent was detected which rapidly decayed. However, the entire TiO2-4-Nafion-complex 24 system led to a significant enhancement in the current, which was ascribed to the electron transfer from the catalyst to the photogenerated oxidized sensitizer. However, the classical overshootings ascribable to electron recombination were highlighted. The authors also analyzed the performances of the DSPEC under different pH conditions of the membrane, from 7 to 9.8, as reported in Figure 14b. In the latter condition, the current decay was lower and this was attributed to the rapid proton release occurring during the OER, thereby affecting the catalytic capabilities of 24 [121]. The current decay could also be ascribed to possible charge recombination of the photoinjected electrons onto the CB with the oxidized 24 [95]. The catalytic properties of 24 were analyzed by examining the produced oxygen: in the absence of the WOC, no O2 was detected, while ca. 9-fold higher quantity was found for the complete system compared to that characterized by the absence of the sensitizer (140 nmoml−1:16 nmol ml−1 TiO2-4-Nafion-complex 24:TiO2-Nafion-complex 24), resulting in a turnover number (TON) of 16 and a turnover frequency (TOF) of 27/h.
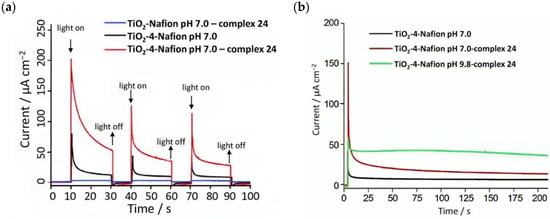
Figure 14.
(a) Chopped photocurrent density–time profile for the main components and the whole system and (b) photocurrent decays under different pH conditions. Reprinted and adapted with permission of the Royal Chemical Society from ref. [96].
Based on earlier evidence of Mallouk regarding the capabilities of the Ru(II) polypyridyl-IrOx system in performing the water splitting, Michaux et al. described the employment of 5 in a FTO/nanoITO/TiO2-5-IrOx system where IrOx acted as a nanoparticles (NPs)-based WOC for the OER [98]. The performances of the entire system and of the components themselves on a mesoporous TiO2 electrode have been analyzed by means of the current–time profile. The approximate three-fold enhancement of the photocurrent when moving from the single components to the semiconductor-dye-WOC assembly pointed out the efficient synergetic catalytic effect in promoting the water splitting. At the time of the contribution, the current was three times larger than the value previously reported in the literature owing to the uncapped nature of the IrOx NPs, which were shown to enhance the water oxidation catalysis [69,122,123]. When the dye-NP system was supported onto nanoITO/TiO2 core shell electrode, the photocurrent was significantly improved, primarily due to the decrease in back electron transfer of the core/shell material. Figure 15a reports the extracted photocurrent density of two families of the TiO2 shell, with different thicknesses, as a function of the applied bias. In particular, the two different TiO2 thicknesses were obtained by varying the number of ALD TiO2 cycles, from 50 (red dots) to 100 (blue dots). The 6.6 nm thick TiO2 shell (100 cycles—blue dots) exhibited the best performance. Specifically, the higher the applied bias, the more consistent the extracted photocurrent, suggesting a fast electron transfer along the system.
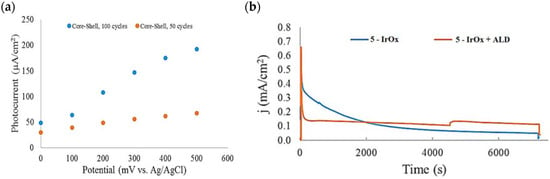
Figure 15.
(a) Photocurrent density–voltage profile of the 5–IrOX NP assembly on two different core/shell electrodes (50 (red) and 100 (blue) cycles) at pH 5.8. (b) Photocurrent density–time curves for the nanoITO/TiO2 core/shell-based electrodes with and without the ALD overlayer. Reprinted and adapted with permission from the American Chemical Society from ref. [98].
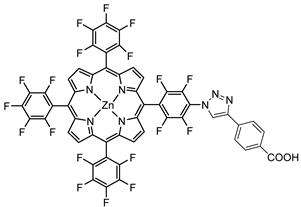
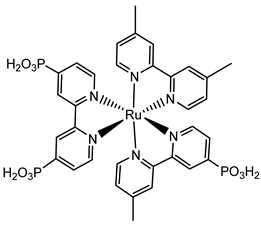
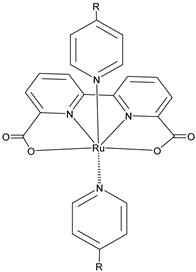
The 2 h photolysis experiments (Figure 15b) highlighted a significant loss in the performance over time, likely due to desorption of the -PO3H2-based sensitizer because of the hydrolysis of the ester bond in the pH conditions of the NaSiF6 buffer (pH 5.8). This problem was sorted out, or at least minimized, by employing an ALD overlayer carried out by depositing 10 TiO2 cycles, with a resulted thickness of less than 1 nm, before incorporating the iridium-based NPs onto the photoanodes. While the non-stabilized photoanode exhibited a photocurrent lower than the dark current (−97 µA cm−2), the ALD-based electrode demonstrated a sustained current of 110 µA cm−2, indicating a significant improvement in the stability. Promising results were achieved by Takijiri et al. with the aforementioned strategy (i.e., pyridyl (py) anchoring moieties) [61], employing the sensitizer 8 to facilitate water reduction in the presence of EDTA as sacrificial agent. The chopped photocurrent density–voltage profile in a standard three-electrode cell, filled with 0.1 M acetate buffer/30 mM EDTA/0.1 NaClO4 aqueous solution, revealed an anodic photocurrent of 1.1 mA cm−2. Long-term electrolysis demonstrated a stable photocurrent with only a 7% loss over 100 min, accompanied by nearly quantitative Faradaic efficiency (FE (%)) for the H2 production. Recently, based on the improved stability highlighted by the -py-based anchoring moieties, T. Meyer has reported an extensive study concerning the role of the grafting moieties of a series of derivatized Ru(bda)-based WOC (bda = 2,2-bipyridine-6,6-dicarboxylic acid) in conjunction with the sensitizer 4 (Table 1), employed to drive the water oxidation with outstanding performances [99]. As depicted in Table 3, the bda-based WOC molecular systems 25, 26 exhibit two different couple of ligands in their respective axial positions. In particular, two 4,4′-bipyridine units are used in 25 whereas the phosphonic acid-based ligands 4-pyridyl phosphonic acid was adopted to construct the complex 26.
The photoelectrochemical performances carried out in aqueous 0.5 M NaClO4 buffered at pH 5.8 revealed a 12-fold higher photocurrent density when the electrode was equipped with 25, reaching 1.7 mA cm−2 (with an IPCE of 25%) compared to the 140 µA cm−2 exhibited by 26 (Figure 16a). To gain insight into the lower performances of 26 and to better clarify the role of the bridging moieties, the authors performed photoelectrochemical impedance spectroscopy measurements (Figure 16b). The typical Nyquist plot of a DSPEC is characterized by a semicircle arising from the recombination charge transfer resistance (Rrec) at the interface between the TiO2|sensitizer and the WOC [124,125,126]. This parameter can be used to directly estimate the capability to counteract the back electron transfer, taking into account that the same amount of WOC is deposited onto the dyed electrode. Indeed, a slower recombination process was observed for 25 if compared to 26, as confirmed by the larger recombination rate constant of 146 s−1 exhibited by the latter, unlike the former which yielded a value of 56 s−1 and highlighted the important role of the pyridyl anchoring moieties in inhibiting the back electron transfer. Based on these results, Meyer’s group introduced a third Ru-bda which bears two axial diethyl 3-(pyridin-4-yloxy)decyl-phosphonic acid ligands to construct the WOC 27 (see Table 3) with the aim to further separate the catalytic core from the oxidized surface and, thus, to reduce the back electron transfer. In fact, the recombination rate decreased to the smallest value among the series of 35 s−1. This translated into a photocurrent of 0.8 mA cm−2 for 27, approximately a 6-fold difference from than that of 26, but surprisingly this value was only half of the photocurrent exhibited by 25 (insert of Figure 16a). This result was ascribed to the formation of surface-bound μ-oxo-bridged dimers by 26 and 27 phosphonate-based catalysts, which exhibited a decreased reactivity towards water oxidation [127,128].
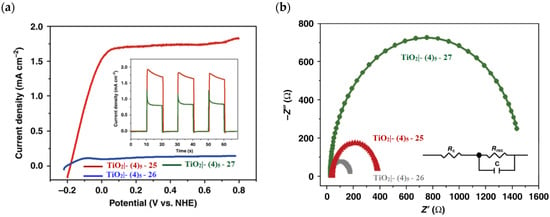
Figure 16.
(a) Photocurrent density–voltage curves of TiO2|-(4)5,25 and TiO2|-(4)5,26 in a 0.1 M acetate buffer/0.5 M NaClO4. Insert: chopped photocurrent density–time curves for TiO2|-(4)5,25 and TiO2|-(4)5,27 at a constant bias of 0.2 V vs. NHE. (b) Nyquist plot for TiO2|-(4)5,25, TiO2|-(4)5,26, and TiO2|-(4)5,27 with the equivalent circuit used for data fitting. Reprinted and adapted with permission of Nature from ref. [99].
Molecular Sensitizer–Catalyst Assembly
The development of covalently linked sensitizer–WOC assemblies is one of the most important breakthroughs, primarily due to the favorable electron transfer kinetics arising from the closeness of the two components and the optimal stoichiometric ratio between them [129]. In 2015, T. Meyer reported a [(PO3H2)2bpy)2Ru(4-Mebpy-4′-bimpy)Ru(tpy)(OH2)]4+ chromophore–catalyst assembly (28) onto nanoITO–TiO2 (ALD deposited) core-shell, capable of driving the water oxidation (Figure 17a). To highlight both the ruthenium cores, the whole system can be shortened as FTO|nanoITO/TiO2-[RuaII-RubII-OH2]4+ [130]. The MLCT transition of the sensitizer unit in 28 generates the chromophore excited state followed by the charge injection into the CB of the semiconductor, resulting in the charge-separated state (semiconductor(e−)-Dye(+)). This triggers a series of electron transfer events between the two ruthenium cores. Transient absorption kinetics on the nanosecond timescale revealed an injection occurring within 20 ns after laser pulse; the bleach recovery, owing to the back electron transfer, was noticeably longer for 28 compared to the sensitizer alone, consistent with hole transfer from the chromophore to the WOC (Figure 17b). The artificial photosynthesis prompts three single-photon-single-electron excitation events for each oxidative cycle to reach the (RuaIII–RubIV=O)5+ active intermediate, which is able to undergo attacks by water. However, due to the back electron transfer on a μs to ms timescale and the requirement for three injection–oxidation cycles to activate the WOC, the active species is present only in trace amounts under ambient solar illumination conditions. Consequently, when 28 is loaded onto TiO2, the resulting photocurrent is negligible. To minimize the recombination losses, core-shell particles have been developed. Under monochromatic illumination, the FTO|nanoITO/TiO2-[RuaII-RubII-OH2]4+ exhibited a photocurrent of ≈200 μA cm−2, which decreased to 20 μA cm−2 after 30 min illumination. In pH 4.6 acetate buffer medium, short-term experiments exhibited a stable current of 100 μA cm−2 under 91 mW cm−2 spectral irradiance with an applied bias of 0.2 V vs. NHE.
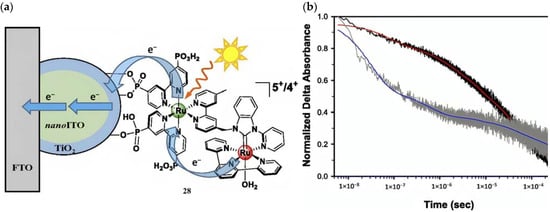
Figure 17.
(a) Schematic representation of the FTO|nanoITO/TiO2-[RuaII-RubII-OH2]4+ assembly and (b) 450 nm decays for the chromophore- (dark gray) and 28- (black) derivatized FTO|nanoITO/TiO2 in 0.5 M LiClO4/10 mM acetate buffer pH 4.6. Reprinted and adapted with permission of PNAS from ref. [130].
A slightly different approach was achieved by employing a SnO2/TiO2 core/shell structure, where the SnO2 core benefits from a CB more positive than TiO2 of ca. 400 meV, in conjunction with the molecular assembly 28 above described [131]. Once the electrons reach the core, the internal gradient at the SnO2/TiO2 interface prevents back electron transfer (Figure 18a).
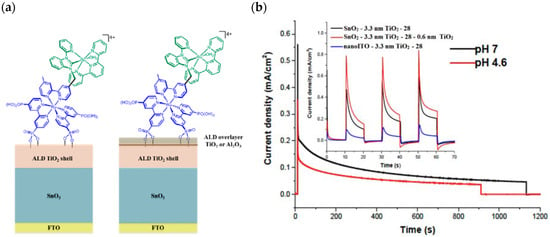
Figure 18.
(a) Representation of an ALD core/shell electrode surface with and without ALD overlayer. (b) Photocurrent–time curves for FTO|SnO2/TiO2(6.6 nm)|-[RuIIa-RuIIb-OH2]4+ with the TiO2 overlayer in 0.5 M LiClO4/20 mM acetate buffer at pH 4.6 (red) and in a 0.1 M phosphate buffer at pH 7/0.5 M NaClO4 (black). Insert: chopped photocurrent density–time curves comparisons between SnO2 and nanoITO core/TiO2 photoanodes, FTO|SnO2/TiO2|-[RuIIa-RuIIb-OH2]4+ (black), and FTO|nanoITO/TiO2|-[RuIIa-RuIIb-OH2]4+ (blue) with ALD TiO2 shells (3.3 nm) in 0.5 M LiClO4/20 mM acetate buffer. The red curve shows the impact of the TiO2 overlayer on the photocurrent profile of the SnO2-based electrode. Reprinted and adapted with permission of PNAS from ref. [131].
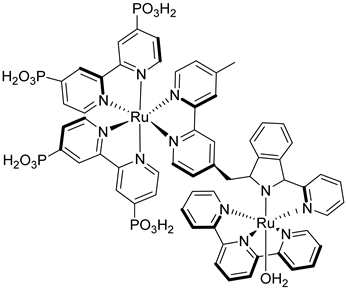
In Figure 18b (insert), the photocurrent–density curves of the short-term experiments conducted on nanoITO/TiO2 and SnO2/TiO2 electrodes are depicted. Peaks of current on the order of 480 μA cm−2 were observed for SnO2/TiO2 (3.3 nm) core/shell photoanodes, which then dropped to 100 μA cm−2 after 10 sec electrolysis in 0.5 M LiClO4/20 mM acetate buffer at pH 4.6. With the employment of a 0.66 nm TiO2 overlayer, deposited by the ALD method, the maximum photocurrent density increased to 790 μA cm−2. The role of the overlayer was ascribed to the stabilization of the phosphonate groups on the surface without affecting the charge injection, which was facilitated by quantum mechanical tunneling from the excited state of the dye. It is noteworthy that the replacement of nanoITO/TiO2 with the SnO2/TiO2 system led to a five-fold increase in photocurrent. Long-term experiments revealed an impressive stabilization toward hydrolysis until pH 7 when the photoanode is equipped with the ALD overlayer (FTO|SnO2/TiO2(6.6 nm)|-[RuaII-RubII-OH2]4+-(0.6 nm)TiO2) (Figure 18b). The gradual decrease in photocurrent was mainly ascribed to ligand loss by the Ru(III) form of the sensitizer in the assembly [45]. Generally, the realization of the sensitizer–WOC dyad suffers from multi-step synthesis, relatively low surface loading, and scarce versatility. Based on the contribution of Mallouk [132,133] and Haga [134,135] concerning the realization of multilayer alkyl diphosphonate films and multilayer Ru/polypyridyl assemblies, respectively, T. Meyer exploited a facile “layer-by-layer” approach by phosphonate/ZrIV coordination linkages to realize sensitizer–WOC self-assembly, as displayed in Figure 19 [136].
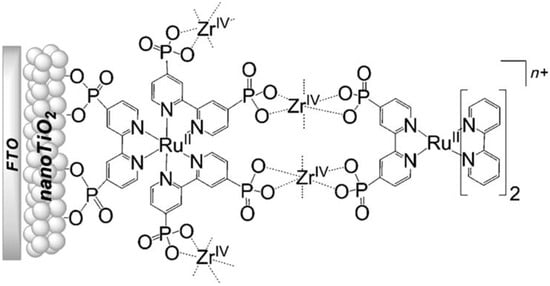
Figure 19.
Schematic representation of the bi-layer film assembly. Reprinted with permission of Wiley from ref. [136].
The bi-layer molecular structure (29) (Figure 19) can be easily achieved in a stepwise manner by subsequently immersing the photoanode in solutions of the sensitizer, ZrOCl2, and either another sensitizer or the WOC. Transient absorption spectroscopy revealed the capability of the self-assembly system to undergo inside-to-outside electron transfer, facilitating hole migration from the sensitizer to the WOC for water splitting. The back electron transfer from the photoinjected electrons to the WOC+ was significantly slowed down in the case of the whole system, which is consistent with an efficient charge separation along the assembly. A similar approach was explored by Sun e co-workers, who realized a self-assembly dye-catalyst characterized by a Ru(bpy)3 sensitizer coordinated to a Ru-bda unit through a Zr(IV) bridge (30, see Table 3) [137]. Under 3 SUN irradiation and in pH 6.4 buffered aqueous media, this assembly was able to drive the OER with a stable photocurrent of 500 μA cm−2, corresponding to an IPCE of around 4%. Later, T. Meyer combined the effect of the SnO2/TiO2 core/shell structure with the efficient Ru-bda WOC (31, Table 3) linked to a series of Ru(II) polypyridyl chromophores (entries 5 and 6 in Table 1) via the Zr(IV) bridge [138]. The study compared the performances of these assemblies with that of co-loaded WOC on the surface. Under the best conditions, the performances of the two systems were essentially aligned. In the presence of the chromophore/assembly 6-Zr-31, the plateau photocurrent of 1.45 mA cm−2 was found after 20 s photolysis, with an FE (%) of 74% for the O2 production, as determined by the collector–generator (C-G) technique. By combining all the knowledge described, a relatively simple mimetic of the components of natural PSII was recently reported (32) (Figure 20). It includes a mediator that reproduces the tyrosine-histidine redox couple, which is not commonly reported in water splitting-based systems [139]. This role was fulfilled by a triphenylamine (TPA) derivative acting as an electron transfer mediator between the oxidized dye and the WOC. In nature, tyrosine inhibits the back electron transfer and stabilizes the system by storing oxidative equivalents close to the WOC. In this contribution, the aim of the TPA was to act as a one-electron transfer mediator. TPA was covalently bonded to a phosphonate polypyridyl Ru(II) complex, which acted as the chromophore. The sensitizer was also linked to a Ru(II)-bda WOC, while the semiconductor was primarily covered by the SnO2/TiO2 core-shell. The system was analyzed with and without the TPA-based mediator.
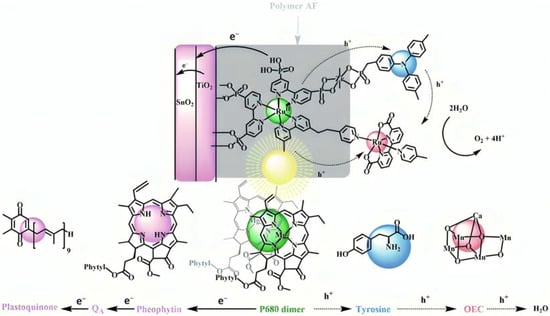
Figure 20.
Illustrative representation of the DSPEC and its electron flow for a fully assembled system and their analog in the natural PSII. Reprinted and adapted with permission of the Royal Chemical Society of Chemistry from ref. [139].
Comparable photocurrent responses were found for both the systems; however, the chopped photocurrent density–time profiles (J-t) (Figure 21a) showed a higher initial current for the TPA-free electrode, which then decreased by over two factors. A similar behavior was found in the long-term experiments in pH 7.0 solution at 0.6 V vs. NHE (Figure 21b), in which in the initial stage the current was 40% higher than in the absence of the TPA moieties as a consequence of the faster hole transfer to the catalyst. However, after 3 h of electrolysis, the TPA-based systems yielded a photocurrent of 120 μA cm−2 while after 1 h, the control system without the mediator reported 85 μA cm−2. These data clearly highlight the importance of employing TPA-based moieties for system stability. By means of the C-G experiments, the authors reported an FE (%) of 83% and 75% for O2 production in the systems with and without the TPA mediator. All the photoelectrochemical details such as the type of sensitizer and catalyst, the photocurrent density, and the applied potential as well as the experimental conditions are summarized in Table 4.
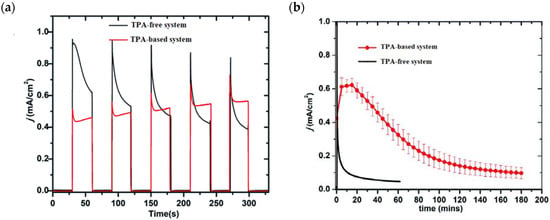
Figure 21.
(a) Chopped photocurrent density–time (j–t) curves for TPA-free (black) TPA-based (red) systems at an applied bias of 0.6 V vs. NHE in 0.1 M phosphonate buffers/0.4 M NaClO4 pH 7.0. (b) Electrolysis of 3 h duration of the two systems described in (a). Reprinted and adapted with permission of the Royal Chemical Society of Chemistry from ref. [139].
Table 4.
Experimental details for the DSPECs described in the main text in the field of water splitting.
Table 3.
Structures of the molecules reported in the main text to drive water splitting. S represents a sensitizer and C a catalyst, while A represents an assembly.
Table 3.
Structures of the molecules reported in the main text to drive water splitting. S represents a sensitizer and C a catalyst, while A represents an assembly.
| Structures | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
 | 22 |  | 23 |  | 24 | |||||
| S [19] | C [95,118,119,120] | C [96,114] | ||||||||
 |  |  | 28 | |||||||
| C [99] | A [130] | |||||||||
 | 29 |  | 30 | |||||||
| A [136] | A [137] | |||||||||
 | 31 |  | 32 | |||||||
| C [138] | A [139] | |||||||||
6. Hydrobromic Acid Oxidation (HBr)
Since oxygen generated from water splitting results in a product with poor economic value, several attractive alternatives have been found. For instance, the hydrohalic acid (HX, X = Cl− or Br−) splitting results in a promising inexpensive storage way for solar energy, which involves the reduction of H+ to H2 at the cathode and the concomitant oxidation of X− to X2 at the anode, generating Cl2 or Br2 [23]. Among the HX family, hydrobromic acid revealed the largest theoretical solar-to-hydrogen efficiency. Once the solar energy is stored in Br2, it can be earned back using fuel cells [25,140,141,142] or redox flow batteries [27]. The electrochemistry of Br- in aqueous medium has been extensively investigated [143] and the most relevant redox processes can be summarized by the following equations [143,144]:
From this series, the small equilibrium constant between Br2 and Br3− in the presence of the Br− anion, should be highlighted [145] as well as the fast recombination as a consequence of the back electron transfer kinetics of Br2 with the metal oxide surface [146]. The relatively high oxidation potential (1.05 V or 1.09 V) (Figure 22) of the Br- species can also activate a [Ru(bda)(L2)] catalyst for water oxidation, resulting in the so-called bromine-assisted water splitting [144] where the most challenging step is the RuIV/V oxidation, with a potential close to 1 V for pH higher than 5.5 [147,148].
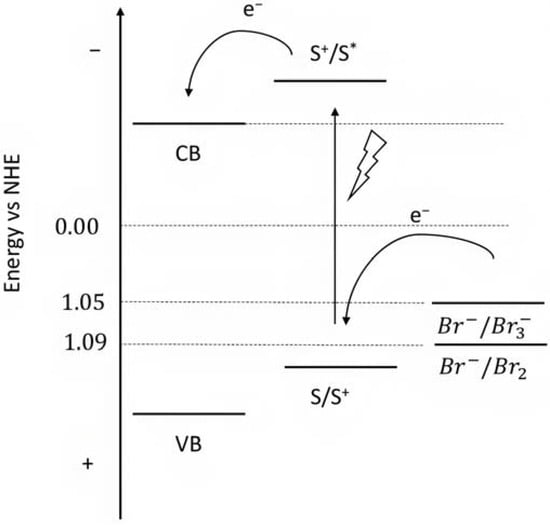
Figure 22.
Representative scheme of the energetics levels for the Br− photooxidation.
It should not be surprising that ruthenium-based complexes have also found interesting applications in bromide oxidation owing to their suitable redox and spectroscopic properties [149,150]. Concerning the heterogeneous catalysis, Li et al. reported one of the earliest examples of Br− homogenous catalysis driven by Ru(II) complexes in acetone medium [149,150,151]. Later, Tsai et al. found an Ru(II) complex able to perform the Br− oxidation in acetonitrile medium, generating the Ru(III) species by employing a sacrificial oxidant [152,153]. In 2017, T. and G. Meyer published a communication on heterogeneous catalysis, reporting the splitting of HBr driven by a [Ru(btfmb)2P]2+ species (btfmb = 4,4′-bis(trifluoromethyl)-2,2′-bipyridine and P = 2,2′-bipyridyl-4,4′-diphosphonic acid) (33 Table 5) loaded onto TiO2 (33|TiO2) or SnO2/TiO2 core-shell (33|CS)-based electrodes [154]. The photophysical properties of 33 were investigated by means of the nanosecond transient absorption measurements. A of 57% was quantified by employing a comparative actionometry [155,156] using 4 as an actinometric standard, 50 ns after the laser excitation. Owing to the distribution of recombination rate constants motivated by the inherent inhomogeneity of the nanocrystalline mesoporous thin film, the 400 nm recombination dynamics was accurately modeled by the Kohlrausch–Williams–Watts (KWW) stretched exponential function. For 33|CS systems, the recombination rate was found to be two orders of magnitude smaller if compared to the TiO2 counterpart 33|TiO2. The introduction of Br− into the electrolyte resulted in a regeneration efficiency () of 99% and 41% for the CS and TiO2 systems, respectively, which is consistent with the slow recombination provided by the electron gradient arising in the core-shell structures.
The photocurrent density–voltage curves for 33 supported onto core-shell or TiO2 in contact with 1 M HBr are reported in Figure 23. In both cases, the onset of the photocurrent density was around 0.4 V and the core-shell system revealed an 8-fold higher photocurrent response compared to the TiO2 analog, reaching to a maximum photocurrent of 1.5 mA cm−2 for the former device. This is consistent with the expectation based on the transient absorption data concerning the lower charge recombination exhibited by the CS structure. This translated into an IPCE of 20% and an APCE of 24% at 460 nm under an applied potential of +0.6 V. The Faradaic efficiency measurement for the Br- oxidation was carried out by means of the C-G (collector–generator) and absorption spectroscopy experiments. The latter was employed to determine the concentration of the Br3− species through its high relativity molar extinction coefficient of 40,900 M−1 cm−1 at 266 nm. On the other hand, the C-G measurements were adopted to quantify the Br2 species, characterized by an ε of only 175 M−1 cm−1 at 390 nm. The C-G measurement revealed a Faradaic efficiency of 71 ± 5%, in perfect agreement with the value of 72%, which was spectroscopically recorded. Slama-Schwok et al. [157] indicated that Br• radicals, generated in acidic water, may react with transient oxygen radicals formed from water that could potentially decrease the FE (%).
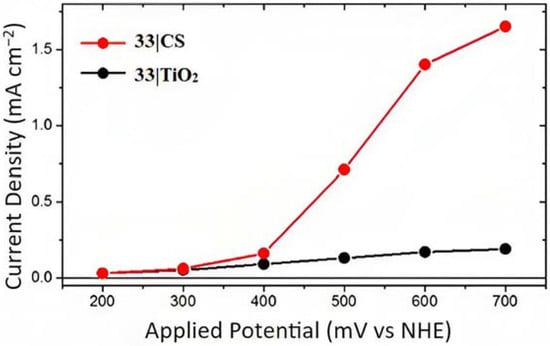
Figure 23.
Photocurrent density–voltage profile for 33|CS (red) and 33|TiO2 (black). Reprinted and adapted with permission of the American Chemical Society from ref. [154].
In 2018, G. Meyer investigated the effect of varying the redox properties of the surface-bound photocatalyst from a new family of Ru(II) polypyridyl complexes towards HBr splitting. These complexes had a general formula [RuII(LL)2P]n+ (where P is 2,2′-bipyridyl-4,4′-diphosphonic acid and LL is 2,2′-bipyridine (bpy), 4,4′-bis(trifluoromethyl)-2,2′-bipyridine (btfmb), 4,4′-bis[(trimethylamino)-methyl]-2,2′-bipyridine (tmam), and 2,2′-bipyrazine (bpz) (4, 33, 34, and 35) [158].
To investigate the injection quantum yield, the charge recombination, and the regeneration efficiency, the photophysical analysis was carried out by means of the nanosecond transient absorption spectroscopy. The formation of the charge separated state (Core/Shell(e-)/[Ru(III)(LL)2P]) occurred within the laser pulse, resulting in a kinj > 108 s−1. was determined 30 ns after the 532 nm laser excitation, employing 4|CS as an actionometric standard and sampling at the ground state/excited state isosbestic point. The values obtained were 1.00 for 4|CS, 0.59 for 33|CS, 0.77 for 34|CS, and 0.09 for 35|CS. Charge recombination was probed at the ground-state/excited-state isosbestic point near 400 nm for each Ru-based complexes loaded onto the core/shell. The average charge recombination rate constants (kcr) were determined by fitting the kinetics by means of the KWW function, resulting in values of 5.2 × 102, 2.8 × 102, and 8.9 × 102 s−1 for 4|CS, 33|CS, and 34|CS (Figure 24a), respectively. A bromide titration was performed to extrapolate the Kreg for the [Ru(III)(LL)2P] complexes in the presence of Br-; in particular, it was computed from the slope of the average rate constant (kKWW) versus bromide concentration for each compound (Figure 24a-insert). These data led to a regeneration efficiency spanning from 1.00 for 35|CS to 0.037 to 4|CS consistent with unfavorable energetics for bromide oxidation exhibited by the latter (E0(4) = 1.35 and E0(Br•/−) = 1.92 V vs. NHE). The determination of the total quantum yield for bromide photo-oxidation, defined as , among the series was on the basis that 34 had the highest of 0.64. Photoelectrochemical experiments were conducted in a standard three-electrode cell by separating the anode and cathode compartments with a PEM membrane, in the presence of 1 M HBr solution. 33 showed the best performance among the series (Figure 24b), with a sustained photocurrent close to 1.5 mA cm−2, an IPCE of 11%, and an FE (%) of 81%. The authors noted that the most efficient compounds were not the most oxidant or photoreductant among the series but, rather, they exhibited a balance between both the properties. 33 displayed sufficient oxidation ability for near-unity regeneration and it was also photoreductant enough to enable a significant electron injection.
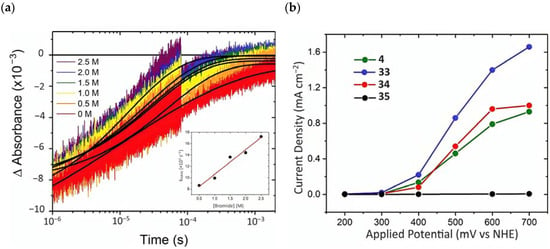
Figure 24.
(a) Kinetics of 405 nm recorded for 34|CS in 1 M HClO4(aq) (argon saturated) with the indicated bromide concentrations. In black is the reported fitting with the KWW function. (b) Photocurrent density–voltage curves for the different complexes (4, 33, 34, and 35) in 1 M HBr under white illumination. Reprinted and adapted with permission of the American Chemical Society from ref. [158].
Other promising results towards the HBr splitting were obtained in the presence of Zinc porphyrin-based sensitizers. Orbelli et al. [75] reported a series of perfluorinated ZnII porphyrins bearing different acceptor linker groups, investigating the electronic and photoelectrochemical properties in the framework of DSPECs, to drive the HBr and water splitting. The authors introduced four electron-withdrawing C6F6 groups to obtain an electron-deficient tetrapyrrolic ring in order to increase the driving force for electron transfer in oxidation reactions. They also introduced a π-conjugated electron acceptor linker equipped with either a carboxylic or a cyanoacrylic group (see the structures of 15, 16, and 17 in Table 1), tasked to bind metal oxides and drive an efficient directionality of the excited state. The absorption spectra in the toluene medium of 15 and 16 reported the typical pattern of β-substituted ZnII porphyrins, showing two well-defined Q bands and a prominent redshift of about 10 nm if compared to the meso-based porphyrin (17). This is a consequence of the more pronounced push–pull character of the formers 15 and 16 dyes (Figure 25a). The nanosecond laser spectroscopy was employed to gain insight into the electron dynamics occurring onto TiO2 or SnO2/TiO2 (active layer of SnO2 with a thin compact TiO2 overlayer) electrodes. The relatively negative dye excited state oxidation potential (Eox* ≈ −0.66 V vs. NHE) should lead to a significant charge injection also onto TiO2 but the authors revealed an excited state residual on the ns–µs timescale. This was ascribed to the introduction of the perfluorinated groups which may significantly increase the inner-sphere reorganization energy (λin) (it is estimated to be close to 0.8–1 eV), affecting the activation energy for electron transfer ΔGET*, according to the Marcus equation ΔGET* = (E*ox − EFB + λ)2/4λ) where λ = λin + λo (λo is the outer-sphere reorganization energy) [159]. Due to the lower flat band potential of the SnO2/TiO2 system (ca. 0.44 V vs. NHE), this resulted in a higher injection quantum yield. SnO2 was first introduced by Bergeron et al. [160] for dyes that are not capable of an efficient charge injection owing to their lower reductant excited state and then, it was introduced in the framework of DSPECs by other authors. In both conditions, the charge-separated state (CS) was found to be long-lived: the recovery of the oxidized chromophore was far from being complete on a 50 µs time scale, allowing sufficient time for intercepting the target molecule of interest (Figure 25b).
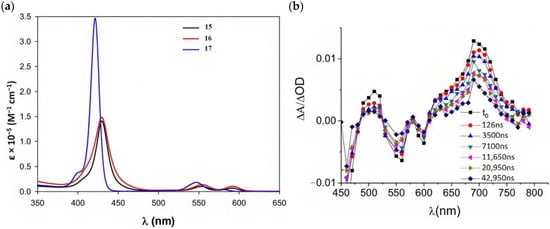
Figure 25.
(a) Absorption spectra recorded for 15, 16, and 17. (b) Transient absorption spectra for 16 loaded on SnO2/TiO2. Thin films were in contact with 0.1 M NaClO4, pH 3. Reprinted and adapted with permission of the American Chemical Society from ref. [75].
When ZnII-based porphyrins were loaded onto SnO2/TiO2, the photocurrent density–voltage curve in 0.1 M HBr underwent a 5- to 10-fold increase in photocurrent density, suggesting an improved charge injection and a slowed recombination, likely attributable to the efficient surface state passivation by TiO2. An optimized electrolyte mixture was found with 0.3 M NaBr and 0.1 M HBr, resulting in the highest photocurrent of 0.4 mA cm−2 for 16 (Figure 26). Under these conditions, the IPCE% was 7% and 2% in correspondence of the B and Q features, respectively. Combining the APCE (=Φinjηreg) data with the regeneration efficiency computed from nanosecond laser kinetics in the presence of Br-, an injection quantum yield of only 14% was obtained.
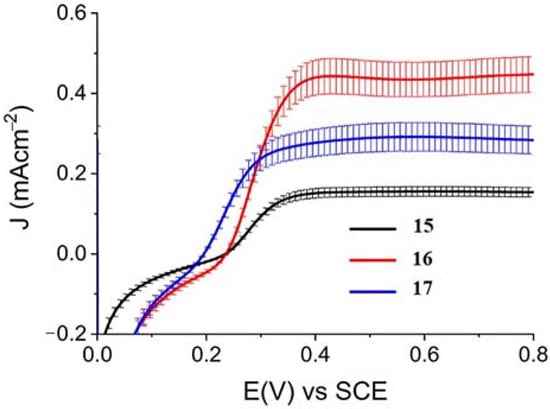
Figure 26.
Photocurrent density–voltage curves for 15, 16, and 17 in contact with 0.1 M HBr/0.3 M NaBr. Reprinted and adapted with permission of the American Chemical Society from ref. [75].
Later, some of the authors reported on a new family of two asymmetric perfluorinated ZnII porphyrins modified with an electron withdrawing branch in either β (36) or meso (37) positions for HBr splitting [76]. Additionally, 36 was further modified by introducing benzothiadiazole (BTD) moieties, aimed at enhancing the absorption at longer wavelengths. DFT and TD-DFT calculations revealed that delocalization of the excited states on the electron withdrawing link was partially successful with the BTD electron acceptor (Figure 27). However, the electronic density on the cyano-acrylic moiety remained comparatively smaller than to that localized in the ring. The photocurrent extrapolated for 36 resulted in higher results than those one of 37, mainly due to the higher excited state directionality. However, the photocurrent associated with the Br− oxidation resulted relatively low, especially if compared to the photocurrent obtained from the oxidation of the ascorbic acid (AA) as a hole scavenger, which reached the value of 300 µA cm−2. Analysis of the transient absorption spectrum by means of the nanosecond laser apparatus indicated that the 10-fold decrease in photocurrent was mainly ascribed to the insufficient driving force for the HBr splitting, paving the way for the design and synthesis of efficient sensitizers.
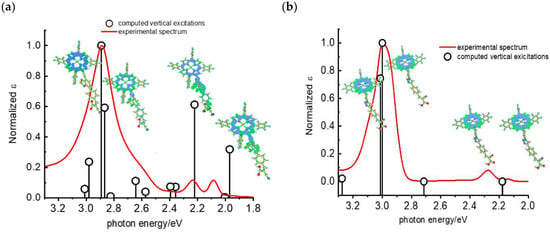
Figure 27.
Computed vertical transitions at the TDDFT 6311 Gd,p level (black vertical bars) overlapped with the experimental spectrum of (a) 36 and (b) 37. Reprinted and adapted with permission of MDPI from ref. [76].
All the photoelectrochemical details such as the type of sensitizer, the photocurrent density, and the applied potential as well as the experimental conditions are summarized in Table 6.
Table 6.
Experimental details for the DSPECs described in the main text for the HBr oxidation.
Table 5.
Structures of the sensitizers reported in the main text to drive HBr splitting.
Table 5.
Structures of the sensitizers reported in the main text to drive HBr splitting.
| Structure | ||||
|---|---|---|---|---|
 | 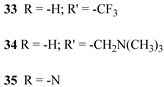 | 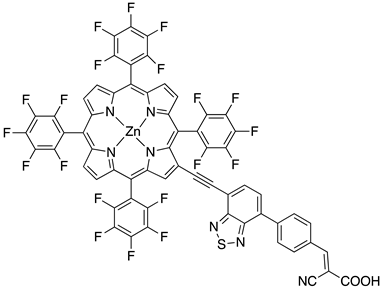 | 36 | |
| S [154,158] | S [76] | |||
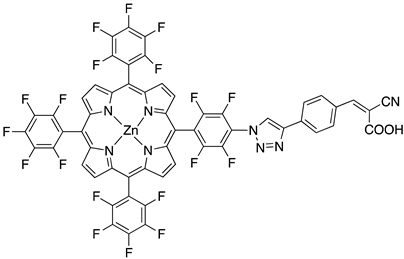 | 37 | |||
| S [76] | ||||
7. Organic Reactions
Nowadays, DSPECs have been extensively explored in the field of organic reactions, aiming to replace expensive noble metal-based catalysts and achieve valuable chemical transformations, such as the generation of new carbonyl compounds or biomass remediation. This approach allows for the efficient replacement of the oxygen evolution reaction (OER) with an alternative oxidative pathway, coupled with the production of storable fuels like hydrogen (H2) or methane (CH4). After preliminary contributions concerning the photodehydrogenation of isopropanol to acetone [161] and to other photocatalytic systems [162,163,164], in 2014, T. Meyer [97] reported the photo-driven oxidation of benzyl alcohol to benzaldehyde in the presence of 7 (see Table 1), which was employed before as a WOC for water oxidation [165]. The catalyst was co-loaded onto a nanoITO/TiO2 core/shell system with the chromophore 4 (see Table 1). Under the presence of 0.1 M of benzyl alcohol, the system was able to generate current densities of 60 and 200 µA cm−2 for TiO2 and the core/shell system, respectively. After other attempts to successfully perform the alcohol oxidation by coupling both a ruthenium chromophore and a Ru-based catalyst, the tendency was to start to replace this expensive and toxic noble metal with fully organic chromophores or catalysts. A comprehensive historical overview of this progress can be found in a recent review [166]. One of the most promising approaches involves the use of the low-cost and readily accessible, 2,2,6,6-tetramethyl-1-piperidine N-oxyl (TEMPO), a well-known organocatalyst for selective and clean electrochemical alcohol oxidation [167,168,169,170]. In this contest, the pristine form of the catalyst TEMPO can be easily restored by reacting with the target substrate, as depicted in the reaction mechanism shown in Figure 28 [171]. TEMPO has been employed in electrocatalysis for selective oxidation reactions [172] and, more recently, it has been applied in photoelectrocatalysis by Choi [173] and Berlinguette [174] to promote the 5-hydroxymethylfurfural (HMF) at a BiVO4 semiconductor. In addition, due to its fast electrochemistry, TEMPO has also been used as a redox mediator for dye-sensitized solar cells (DSSCs) [86,175].
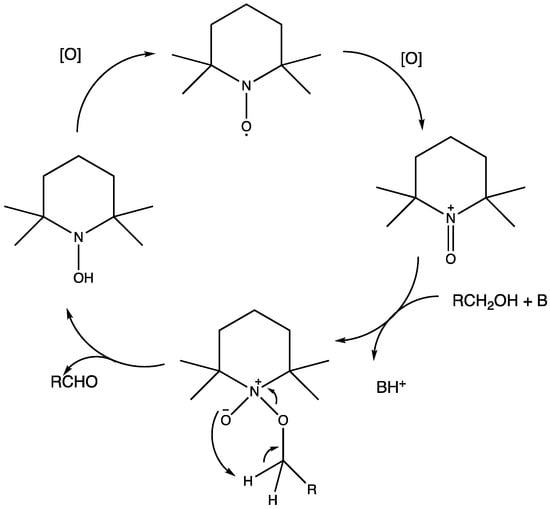
Figure 28.
Structure of TEMPO mediator together with the oxidative cycle involved in the alcohol oxidation in the presence of a base.
The group of Odobel reported an example of a DSPEC based on a triad (NDADI-Ru(bpy)3-TEMPO, where NADI = naphthalenedicarboxyanhydride) (38, see Table 7) was supported onto ITO nanoparticles for para-methoxybenzyl alcohol (Meo-BA) oxidation to para-methoxybenzaldehyde (Figure 29a) [176].
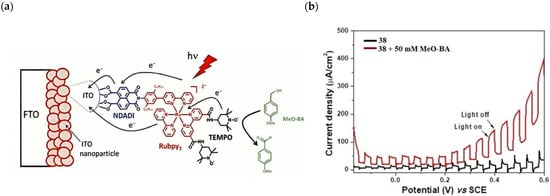
Figure 29.
(a) Schematic representation of the photoanodic compartment for 38 triad supported onto ITO nanoparticles. (b) Photocurrent density–voltage curves under chopped light irradiation of triad 38 in the absence (black trace) and in the presence of 50 mM of MeO-BA (red trace) in carbonate buffer at pH 10. Reprinted and adapted with permission of Wiley from ref. [176].
The dyad NDADI-Ru(bpy)3 was designed to enhance the charge separation, leading to the formation of the ITO(e−)NDADI/Ru(III) CS, as previously reported by Wenger and co-workers [177]. The TEMPO has been incorporated to minimize the spatial separation from the chromophore. After having found suitable conditions for TEMPO-mediated benzyl-alcohol oxidation, the authors employed the supported triad to perform the photo-driven reaction. The chopped photocurrent–density voltage curve (Figure 29b), in the presence of 50 mM of MeO-BA, demonstrated an increased photocurrent response for applied potentials higher than 0.2 V, resulting in a net photocurrent of about 150–180 µA cm−2. Under optimal conditions, the average Faradaic efficiency was on the order of 80%. To explore the replacement of the rare, toxic, and expensive ruthenium-based complexes with zinc porphyrins, to perform the same target reaction, Odobel’s group synthesized two porphyrin dyads covalently linked to TEMPO, bearing a carboxylic acid (39) or hydroxamic acid (40) moiety, respectively. Their structures are displayed in Table 7. The hydroxamic acid anchoring group of 40 was expected to provide higher stability of the TiO2-dye bond over time.
The photocurrent analysis, at pH 8 for 40, highlighted the important role played by the TEMPO catalyst in performing the alcohol oxidation. As depicted in Figure 30, for a voltage larger than −0.06 V vs. NHE, the photocurrent density reached the limit value of 150–200 µA cm−2, significantly higher than the experiment performed without the catalyst (<<50 µA cm−2). The authors also explored the alcohol oxidation in acetonitrile medium because this organic solvent is compatible with proton reduction and is even more interesting for CO2 reduction owing to its 7–8 times higher solubility in organic solvents than in water [178]. In the case of the carbon dioxide reduction, this result is interesting both with respect to the possibility of having a higher substrate concentration at the cathodic counterpart and of avoiding CO2 hydration products, affecting the thermodynamic potentials for generating certain reduction molecules. In these conditions, a base-rich electrolyte is required as the oxidation resulted in proton release (Figure 28). The lower efficiency observed in acetonitrile medium was ascribed to the faster desorption rate of the dyad owing to their higher solubility in organic medium. In all the situations, the Faradaic efficiency for the aldehyde production ranged from 76 to 93%. Comparable experiments have been performed with 39; this device gave a similar photocurrent and FE (%) in water while the faster dye desorption in acetonitrile resulted in impossibility to quantify the produced aldehyde, confirming the higher stability provided by the hydroxamic acid moiety.
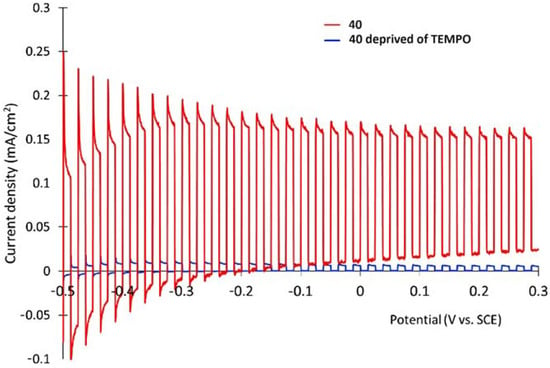
Figure 30.
Chopped light photocurrent–density voltage curves recorded under white light irradiation for 40 and 40 deprived of TEMPO on a TiO2 electrode in contact with a 0.1 M borate buffer at pH 8, containing 50 mM [MeO-Ph-CH2OH] and 0.1 M [NaClO4]. Reprinted and adapted with permission of the American Chemical Society from ref. [28].
DSPECs have recently found applications in exploring biomass valorization via oxidative remediation. For instance, the glycerol oxidation products find application in cosmetics and in the preparation of polyesters and adhesives [179]. Generally, DSPECs are not suitable for this molecular conversion owing to the basic conditions required (pH 8.5), which result in dye desorption [180,181]. In this contest, to drive the glycerol oxidation mediated by TEMPO, Bruggeman et al. reported on a thienopyrroledione-based dye (41, Table 7) encased in an acetonitrile-based redox-gel, which is able to protect the TiO2 photoanode from degradation [182]. The DPSEC was assembled by generating a by-phasic system in which the 41-based photoanode was embedded in a poly (vinylidene fluoride-co-hexafluoropropylene (PVDF-HFP) gel layer and the resulting assembly was put in contact with an aqueous electrolyte. This arrangement allows TEMPO to regenerate the photo-oxidized dye which, in turn, produces the oxammonium TEMPO(+) cation that can diffuse through the gel to reach the gel–aqueous interface where the glycerol oxidation takes place, as depicted in Figure 31a.
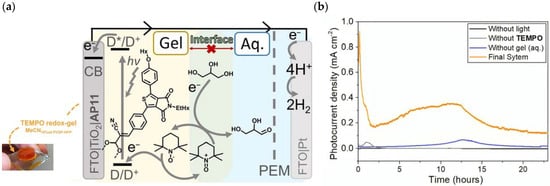
Figure 31.
(a) Schematic representation of an aqueous (blue) biphasic DSPEC with a TEMPO containing redox-gel layer (yellow) for glycerol oxidation. (b) Photocurrent density–time curve in light in the absence of TEMPO, in the full water condition, for the final by-phasic device and in the dark. Reprinted and adapted with permission of Wiley from ref. [182].
The FE (%) computed by analyzing the products accumulated by chronoamperometry experiments (Figure 31b) of the full water- and the TEMPO redox-gel-based systems, resulted in almost quantitative results for both cases. However, the latter device exhibited at least 10-fold higher photocurrent density, thereby leading to faster overall conversion. In addition, improved stability was outlined by devices assembled with the redox-gel system, which preserved its typical orange color throughout the entire 48 h electrolysis. This confirmed how the use of the PVDF-HFP gel layer was effective in preserving not only the chemical stability of the sensitizer but, in general, that of the entire photoanode unit. By contrast, in the same experimental conditions, devices fabricated with the conventional architecture no longer generated photocurrent after 15 h as a consequence of the dye desorption. Thus, albeit the two systems exhibited comparable FE (%) values, in the presence of the redox gel the overall yield of glycerol oxidation was larger if compared to the full water architecture. It is worth noting that just a slight percentage of TEMPO (~6%) was found in the aqueous layer after 48 h, confirming that the mediator is strongly incorporated into the redox gel matrix, which in fact avoids the diffusion of TEMPO outside the film, keeping it in a region close to the semiconductor interface. Moreover, only a slight amount of water (~1%) was found entrapped into the redox-gel after the same period of time (48 h), which is beneficial to give effective protection to the photoanode, thereby preventing the possible hydrolysis of the Ti-Dye bond. The by-phasic architecture of these devices highlights a smart way for stabilizing the dyes towards alkaline environments without the employment of expensive and demanding techniques, such as the aforementioned ALD [59]. Apart from glycerol oxidation, in the framework of biomass remediation, the photo-assisted lignin oxidation, or cleavage, is another promising application in which, to date, only a few examples linked to DSPECs have been reported. Lignin represents the largest noncarbohydrate component in lignocellulosic biomass and its native structure is characterized by interunit linkages including alkyl-aryl-ether, phenylcoumaran, resinol, biphenyl, and other related groups [183]. In 2020, G. Leem reported a photochemical device able to drive the initial oxidation of the secondary benzyl alcohol in lignin model compound (LMC, characterized by the most abundant interunit linkage in natural lignin) and real lignin, as part of a selective two-step process prior to C–O bond cleavage in the lignin backbone [184]. Furthermore, the same group reported a heterogeneous approach to cleave the Caryl-Cα bond in a phenolic LMC by employing a one-dimensional TiO2 nanoroad array (TiO2 NRAs) DSPEC in combination with a Halogen Atom Transfer (HAT) mediator [185]. 1 (Table 1) was employed as a sensitizer, while 4-acetamido-2,2,6,6tetramethyl-1-peperidine-N-oxyl (ACT) was used as the HAT system (Figure 32).
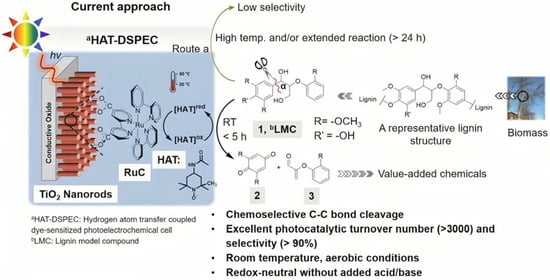
Figure 32.
Illustration of the FTO|TiO2 NRAs|1 in the presence of the HAT and the LMC. Reprinted and adapted with permission of the American Chemical Society from ref. [185].
The photocurrent response of the FTO|TiO2 NRAs|1 has been investigated in acetonitrile by increasing the ACT concentration and by analyzing the chopped photocurrent density–time response. When the concentration of ACT was kept constant, the addition of the LMC led to a significant increase in the response from 190 to 690 μA cm−2, as shown in Figure 33a. The presence of the cleavage products has been investigated by means of both 1H and 13C NMR after 5 h continuous illumination. According to the literature, the author demonstrated no oxidative cleavage of the C-C bond for the nonphenolic lignin model in DSPEC nor by following a thermal catalytic oxidation in the presence of a vanadium catalyst and pyridine at 80 °C for 48 h (Figure 33b). The authors concluded that the presence of the phenoxy group in LMC is an essential moiety to lead to the C-C cleavage.
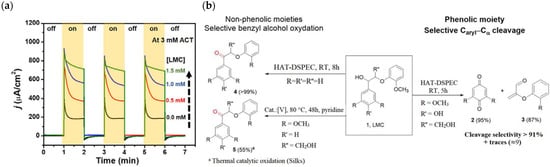
Figure 33.
(a) Photocurrent density-time curves for FTO|TiO2 NRAs|1 electrodes in the presence of 3 mM ACT and increasing LMC concentrations from 0 to 1.5 mM. (b) Different oxidation/cleavage pathways for the LMC in the presence of different substituents and conditions. Reprinted and adapted with permission of the American Chemical Society from ref. [185].
In the near future, several efforts should be taken to explore the possibility of reaching the C-C cleavage in the native lignin compound.
All the photoelectrochemical details such as the type of sensitizer and catalyst, the photocurrent density, and the applied potential as well as the experimental conditions are summarized in Table 8.
Table 8.
Experimental details for the DSPECs described in the main text in the framework of organic reactions.
Table 7.
Structures of the molecules reported in the main text to drive organic reactions. S represents a sensitizer, while A represents an assembly.
Table 7.
Structures of the molecules reported in the main text to drive organic reactions. S represents a sensitizer, while A represents an assembly.
| Structures | ||||
|---|---|---|---|---|
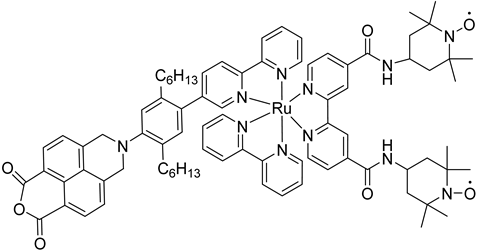 | 38 | |||
| A [176] | ||||
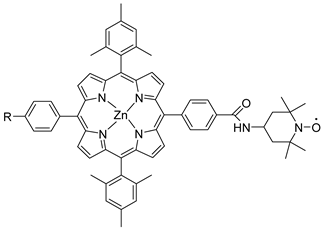 |  | 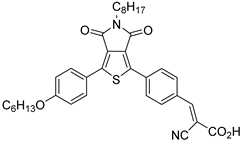 | 41 | |
| A [28] | S [182] | |||
8. Conclusions and Future Outlooks
Since Mallouk’s initial contribution in 2009, DSPECs have emerged as promising alternatives to the classical PEC devices, capable of converting solar light into chemical bonds such as O2, H2, or X2. Several efforts have been devoted to finding strategies to prevent the cleavage of the ester dye-TiO2 bond, especially under alkaline conditions. Several contributions reported higher stability for dye bearing –PO3H2 anchoring groups compared to their -COOH analog. However, both grafting units are precluded under alkaline conditions. Other promising grafting moieties, such as -py groups or diazonium groups, have been identified to enable stability under alkaline conditions. An ALD overlayer of Al2O3 or TiO2 has proven to be an efficient passivation strategy to prevent the hydrolysis of the ester bond. Since the outset of the DSPECs, the splitting of water has been considered the most interesting reaction due to its safety and natural abundance. Numerous sensitizers, catalysts, or assemblies of both have been developed. Alternative oxidative pathways have been explored to overcome limitations arising from using water. HX splitting results in a promising inexpensive storage way for solar energy, with particular attention given to HBr oxidation as a source of Br2 for fuel cells. Recently, organic reactions such as the generation of new carbonyl compounds have garnered attention in the scientific community as a strategy to replace the expensive and toxic noble metal employed until now. Future development should focus on identifying new stabilizing strategies to preserve dye absorption without affecting the electronic dynamics occurring at the semiconductor/dye interface, which could otherwise decrease the overall yield of the device.
Author Contributions
Conceptualization, E.M. and S.C. (Stefano Carli); writing—original draft preparation, E.M. and S.C. (Stefano Carli); writing—review and editing, S.C. (Stefano Caramori). All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Informed Consent Statement
Not applicable.
Data Availability Statement
No datasets were generated or analyzed during the current study.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Lewis, N.S.; Nocera, D.G. Powering the planet: Chemical challenges in solar energy utilization. Proc. Natl. Acad. Sci. USA 2006, 103, 15729–15735. [Google Scholar] [CrossRef] [PubMed]
- Hammarstrom, L.; Hammes-Schiffer, S. Artificial photosynthesis and solar fuels. Acc. Chem. Res. 2009, 42, 1859–1860. [Google Scholar] [CrossRef] [PubMed]
- Maddah, H.A. Modeling the Relation between Carbon Dioxide Emissions and Sea Level Rise for the Determination of Future (2100) Sea Level. Am. J. Environ. Eng. 2016, 6, 52–61. [Google Scholar]
- Marques Lameirinhas, R.A.; Torres, J.P.N.; de Melo Cunha, J.P. A Photovoltaic Technology Review: History, Fundamentals and Applications. Energies 2022, 15, 1823. [Google Scholar] [CrossRef]
- Sudhagar, P.; Roy, N.; Vedarajan, R.; Devadoss, A.; Terashima, C.; Nakata, K.; Fujishima, A. Hydrogen and CO2 Reduction Reactions: Mechanisms and Catalysts. In Photoelectrochemical Solar Fuel Production: From Basic Principles to Advanced Devices; Giménez, S., Bisquert, J., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 105–160. [Google Scholar]
- Newman, J.; Hoertz, P.G.; Bonino, C.A.; Trainham, J.A. Review: An Economic Perspective on Liquid Solar Fuels. J. Electrochem. Soc. 2012, 159, A1722–A1729. [Google Scholar] [CrossRef]
- Li, H.; Tu, W.; Zhou, Y.; Zou, Z. Z-Scheme Photocatalytic Systems for Promoting Photocatalytic Performance: Recent Progress and Future Challenges. Adv. Sci. 2016, 3, 1500389. [Google Scholar] [CrossRef] [PubMed]
- Fujishima, A.; Honda, K. Electrochemical photolysis of water at a semiconductor electrode. Nature 1972, 238, 37–38. [Google Scholar] [CrossRef] [PubMed]
- Kalyanasundaram, K.; Grätzel, M. Cyclic Cleavage of Water into H2 and O2 by Visible Light with Coupled Redox Catalysts. Angew. Chem. Int. Ed. Engl. 1979, 18, 701–702. [Google Scholar] [CrossRef]
- Grätzel, M. Photochemical Methods for the Conversion of Light into Chemical Energy. Berichte Der Bunsenges. Für Phys. Chem. 1980, 84, 981–991. [Google Scholar] [CrossRef]
- Duonghong, D.; Borgarello, E.; Graetzel, M. Dynamics of light-induced water cleavage in colloidal systems. J. Am. Chem. Soc. 2002, 103, 4685–4690. [Google Scholar] [CrossRef]
- Cristino, V.; Caramori, S.; Argazzi, R.; Meda, L.; Marra, G.L.; Bignozzi, C.A. Efficient photoelectrochemical water splitting by anodically grown WO3 electrodes. Langmuir 2011, 27, 7276–7284. [Google Scholar] [CrossRef] [PubMed]
- Bignozzi, C.A.; Caramori, S.; Cristino, V.; Argazzi, R.; Meda, L.; Tacca, A. Nanostructured photoelectrodes based on WO3: Applications to photooxidation of aqueous electrolytes. Chem. Soc. Rev. 2013, 42, 2228–2246. [Google Scholar] [CrossRef] [PubMed]
- Berardi, S.; Cristino, V.; Bignozzi, C.A.; Grandi, S.; Caramori, S. Hematite-based photoelectrochemical interfaces for solar fuel production. Inorg. Chim. Acta 2022, 535, 120862. [Google Scholar] [CrossRef]
- Berardi, S.; Kopula Kesavan, J.; Amidani, L.; Meloni, E.M.; Marelli, M.; Boscherini, F.; Caramori, S.; Pasquini, L. Better Together: Ilmenite/Hematite Junctions for Photoelectrochemical Water Oxidation. ACS Appl. Mater. Interfaces 2020, 12, 47435–47446. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Pendlebury, S.R.; Reynal, A.; Le Formal, F.; Durrant, J.R. Dynamics of photogenerated holes in undoped BiVO4 photoanodes for solar water oxidation. Chem. Sci. 2014, 5, 2964–2973. [Google Scholar] [CrossRef]
- Vecchi, P.; Piccioni, A.; Mazzaro, R.; Mazzanti, M.; Cristino, V.; Caramori, S.; Pasquini, L. Charge Separation Efficiency in WO3/BiVO4 Photoanodes with CoFe Prussian Blue Catalyst Studied by Wavelength-Dependent Intensity-Modulated Photocurrent Spectroscopy. Sol. RRL 2022, 6, 2200108. [Google Scholar] [CrossRef]
- Lhermitte, C.R.; Bartlett, B.M. Advancing the Chemistry of CuWO4 for Photoelectrochemical Water Oxidation. Acc. Chem. Res. 2016, 49, 1121–1129. [Google Scholar] [CrossRef]
- Youngblood, W.J.; Lee, S.-H.A.; Kobayashi, Y.; Hernandez-Pagan, E.A.; Hoertz, P.G.; Moore, T.A.; Moore, A.L.; Gust, D.; Mallouk, T.E. Photoassisted Overall Water Splitting in a Visible Light-Absorbing Dye-Sensitized Photoelectrochemical Cell. J. Am. Chem. Soc. 2009, 131, 926–927. [Google Scholar] [CrossRef]
- Yu, Z.; Li, F.; Sun, L. Recent advances in dye-sensitized photoelectrochemical cells for solar hydrogen production based on molecular components. Energy Environ. Sci. 2015, 8, 760–775. [Google Scholar] [CrossRef]
- Liang, X.; Cao, X.; Sun, W.; Ding, Y. Recent Progress in Visible Light Driven Water Oxidation Using Semiconductors Coupled with Molecular Catalysts. ChemCatChem 2019, 11, 6190–6202. [Google Scholar] [CrossRef]
- Zhang, B.; Sun, L. Artificial photosynthesis: Opportunities and challenges of molecular catalysts. Chem. Soc. Rev. 2019, 48, 2216–2264. [Google Scholar] [CrossRef]
- Nocera, D.G. Chemistry of personalized solar energy. Inorg. Chem. 2009, 48, 10001–10017. [Google Scholar] [CrossRef] [PubMed]
- White, C.; Steeper, R.; Lutz, A. The hydrogen-fueled internal combustion engine: A technical review. Int. J. Hydrogen Energy 2006, 31, 1292–1305. [Google Scholar] [CrossRef]
- Livshits, V.; Ulus, A.; Peled, E. High-power H2/Br2 fuel cell. Electrochem. Commun. 2006, 8, 1358–1362. [Google Scholar] [CrossRef]
- Kreutzer, H.; Yarlagadda, V.; Van Nguyen, T. Performance Evaluation of a Regenerative Hydrogen-Bromine Fuel Cell. J. Electrochem. Soc. 2012, 159, F331–F337. [Google Scholar] [CrossRef]
- Cho, K.T.; Ridgway, P.; Weber, A.Z.; Haussener, S.; Battaglia, V.; Srinivasan, V. High Performance Hydrogen/Bromine Redox Flow Battery for Grid-Scale Energy Storage. J. Electrochem. Soc. 2012, 159, A1806–A1815. [Google Scholar] [CrossRef]
- Nikoloudakis, E.; Pati, P.B.; Charalambidis, G.; Budkina, D.S.; Diring, S.; Planchat, A.; Jacquemin, D.; Vauthey, E.; Coutsolelos, A.G.; Odobel, F. Dye-Sensitized Photoelectrosynthesis Cells for Benzyl Alcohol Oxidation Using a Zinc Porphyrin Sensitizer and TEMPO Catalyst. ACS Catal. 2021, 11, 12075–12086. [Google Scholar] [CrossRef]
- Rahimi, N.; Pax, R.A.; Gray, E.M. Review of functional titanium oxides. I: TiO2 and its modifications. Prog. Solid State Chem. 2016, 44, 86–105. [Google Scholar] [CrossRef]
- Sun, C.; Yang, J.; Xu, M.; Cui, Y.; Ren, W.; Zhang, J.; Zhao, H.; Liang, B. Recent intensification strategies of SnO2-based photocatalysts: A review. Chem. Eng. J. 2022, 427, 131564. [Google Scholar] [CrossRef]
- Ghaffar, S.; Abbas, A.; Naeem-Ul-Hassan, M.; Assad, N.; Sher, M.; Ullah, S.; Alhazmi, H.A.; Najmi, A.; Zoghebi, K.; Al Bratty, M.; et al. Improved Photocatalytic and Antioxidant Activity of Olive Fruit Extract-Mediated ZnO Nanoparticles. Antioxidants 2023, 12, 1201. [Google Scholar] [CrossRef]
- Hagfeldt, A.; Boschloo, G.; Sun, L.; Kloo, L.; Pettersson, H. Dye-sensitized solar cells. Chem. Rev. 2010, 110, 6595–6663. [Google Scholar] [CrossRef]
- Bomben, P.G.; Robson, K.C.D.; Koivisto, B.D.; Berlinguette, C.P. Cyclometalated ruthenium chromophores for the dye-sensitized solar cell. Coord. Chem. Rev. 2012, 256, 1438–1450. [Google Scholar] [CrossRef]
- Kaneko, H.; Minegishi, T.; Nakabayashi, M.; Shibata, N.; Kuang, Y.; Yamada, T.; Domen, K. A Novel Photocathode Material for Sunlight-Driven Overall Water Splitting: Solid Solution of ZnSe and Cu(In,Ga)Se2. Adv. Funct. Mater. 2016, 26, 4570–4577. [Google Scholar] [CrossRef]
- Hammarstrom, L. Accumulative charge separation for solar fuels production: Coupling light-induced single electron transfer to multielectron catalysis. Acc. Chem. Res. 2015, 48, 840–850. [Google Scholar] [CrossRef] [PubMed]
- Nozik, A.J. Photoelectrolysis of water using semiconducting TiO2 crystals. Nature 1975, 257, 383–386. [Google Scholar] [CrossRef]
- Nayak, P.K.; Garcia-Belmonte, G.; Kahn, A.; Bisquert, J.; Cahen, D. Photovoltaic efficiency limits and material disorder. Energy Environ. Sci. 2012, 5, 6022–6039. [Google Scholar] [CrossRef]
- Yun, S.; Vlachopoulos, N.; Qurashi, A.; Ahmad, S.; Hagfeldt, A. Dye sensitized photoelectrolysis cells. Chem. Soc. Rev. 2019, 48, 3705–3722. [Google Scholar] [CrossRef] [PubMed]
- Galoppini, E. Linkers for anchoring sensitizers to semiconductor nanoparticles. Coord. Chem. Rev. 2004, 248, 1283–1297. [Google Scholar] [CrossRef]
- Bignozzi, C.A.; Argazzi, R.; Boaretto, R.; Busatto, E.; Carli, S.; Ronconi, F.; Caramori, S. The role of transition metal complexes in dye sensitized solar devices. Coord. Chem. Rev. 2013, 257, 1472–1492. [Google Scholar] [CrossRef]
- Polo, A.S.; Itokazu, M.K.; Murakami Iha, N.Y. Metal complex sensitizers in dye-sensitized solar cells. Coord. Chem. Rev. 2004, 248, 1343–1361. [Google Scholar] [CrossRef]
- Park, H.; Bae, E.; Lee, J.J.; Park, J.; Choi, W. Effect of the anchoring group in Ru-bipyridyl sensitizers on the photoelectrochemical behavior of dye-sensitized TiO2 electrodes: Carboxylate versus phosphonate linkages. J. Phys. Chem. B 2006, 110, 8740–8749. [Google Scholar] [CrossRef] [PubMed]
- Bae, E.; Choi, W. Effect of the anchoring group (carboxylate vs phosphonate) in Ru-complex-sensitized TiO2 on hydrogen production under visible light. J. Phys. Chem. B 2006, 110, 14792–14799. [Google Scholar] [CrossRef] [PubMed]
- Hanson, K.; Brennaman, M.K.; Luo, H.; Glasson, C.R.; Concepcion, J.J.; Song, W.; Meyer, T.J. Photostability of phosphonate-derivatized, Ru(II) polypyridyl complexes on metal oxide surfaces. ACS Appl. Mater. Interfaces 2012, 4, 1462–1469. [Google Scholar] [CrossRef] [PubMed]
- Hyde, J.T.; Hanson, K.; Vannucci, A.K.; Lapides, A.M.; Alibabaei, L.; Norris, M.R.; Meyer, T.J.; Harrison, D.P. Electrochemical Instability of Phosphonate-Derivatized, Ruthenium(III) Polypyridyl Complexes on Metal Oxide Surfaces. ACS Appl. Mater. Interfaces 2015, 7, 9554–9562. [Google Scholar] [CrossRef] [PubMed]
- Raber, M.M.; Brady, M.D.; Troian-Gautier, L.; Dickenson, J.C.; Marquard, S.L.; Hyde, J.T.; Lopez, S.J.; Meyer, G.J.; Meyer, T.J.; Harrison, D.P. Fundamental Factors Impacting the Stability of Phosphonate-Derivatized Ruthenium Polypyridyl Sensitizers Adsorbed on Metal Oxide Surfaces. ACS Appl. Mater. Interfaces 2018, 10, 22821–22833. [Google Scholar] [CrossRef] [PubMed]
- Gillaizeau-Gauthier, I.; Odobel, F.; Alebbi, M.; Argazzi, R.; Costa, E.; Bignozzi, C.A.; Qu, P.; Meyer, G.J. Phosphonate-based bipyridine dyes for stable photovoltaic devices. Inorg. Chem. 2001, 40, 6073–6079. [Google Scholar] [CrossRef] [PubMed]
- Wee, K.R.; Brennaman, M.K.; Alibabaei, L.; Farnum, B.H.; Sherman, B.; Lapides, A.M.; Meyer, T.J. Stabilization of ruthenium(II) polypyridyl chromophores on nanoparticle metal-oxide electrodes in water by hydrophobic PMMA overlayers. J. Am. Chem. Soc. 2014, 136, 13514–13517. [Google Scholar] [CrossRef]
- Lapides, A.M.; Ashford, D.L.; Hanson, K.; Torelli, D.A.; Templeton, J.L.; Meyer, T.J. Stabilization of a ruthenium(II) polypyridyl dye on nanocrystalline TiO2 by an electropolymerized overlayer. J. Am. Chem. Soc. 2013, 135, 15450–15458. [Google Scholar] [CrossRef]
- Ashford, D.L.; Lapides, A.M.; Vannucci, A.K.; Hanson, K.; Torelli, D.A.; Harrison, D.P.; Templeton, J.L.; Meyer, T.J. Water oxidation by an electropolymerized catalyst on derivatized mesoporous metal oxide electrodes. J. Am. Chem. Soc. 2014, 136, 6578–6581. [Google Scholar] [CrossRef]
- Ashford, D.L.; Sherman, B.D.; Binstead, R.A.; Templeton, J.L.; Meyer, T.J. Electro-assembly of a chromophore-catalyst bilayer for water oxidation and photocatalytic water splitting. Angew. Chem. Int. Ed. Engl. 2015, 54, 4778–4781. [Google Scholar] [CrossRef]
- Moss, J.A.; Yang, J.C.; Stipkala, J.M.; Wen, X.; Bignozzi, C.A.; Meyer, G.J.; Meyer, T.J. Sensitization and stabilization of TiO2 photoanodes with electropolymerized overlayer films of ruthenium and zinc polypyridyl complexes: A stable aqueous photoelectrochemical cell. Inorg. Chem. 2004, 43, 1784–1792. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.G.; Schauer, P.A.; Borau-Garcia, J.; Fancy, B.R.; Berlinguette, C.P. Stabilization of ruthenium sensitizers to TiO2 surfaces through cooperative anchoring groups. J. Am. Chem. Soc. 2013, 135, 1692–1695. [Google Scholar] [CrossRef] [PubMed]
- Szpakolski, K.; Latham, K.; Rix, C.; Rani, R.A.; Kalantar-zadeh, K. Silane: A new linker for chromophores in dye-sensitised solar cells. Polyhedron 2013, 52, 719–732. [Google Scholar] [CrossRef]
- Materna, K.L.; Rudshteyn, B.; Brennan, B.J.; Kane, M.H.; Bloomfield, A.J.; Huang, D.L.; Shopov, D.Y.; Batista, V.S.; Crabtree, R.H.; Brudvig, G.W. Heterogenized Iridium Water-Oxidation Catalyst from a Silatrane Precursor. ACS Catal. 2016, 6, 5371–5377. [Google Scholar] [CrossRef]
- Materna, K.L.; Brennan, B.J.; Brudvig, G.W. Silatranes for binding inorganic complexes to metal oxide surfaces. Dalton Trans. 2015, 44, 20312–20315. [Google Scholar] [CrossRef]
- Brennan, B.J.; Llansola Portoles, M.J.; Liddell, P.A.; Moore, T.A.; Moore, A.L.; Gust, D. Comparison of silatrane, phosphonic acid, and carboxylic acid functional groups for attachment of porphyrin sensitizers to TiO2 in photoelectrochemical cells. Phys. Chem. Chem. Phys. 2013, 15, 16605–16614. [Google Scholar] [CrossRef] [PubMed]
- Zakeeruddin, S.M.; Nazeeruddin, M.K.; Humphry-Baker, R.; Péchy, P.; Quagliotto, P.; Barolo, C.; Viscardi, G.; Grätzel, M. Design, Synthesis, and Application of Amphiphilic Ruthenium Polypyridyl Photosensitizers in Solar Cells Based on Nanocrystalline TiO2 Films. Langmuir 2002, 18, 952–954. [Google Scholar] [CrossRef]
- Vannucci, A.K.; Alibabaei, L.; Losego, M.D.; Concepcion, J.J.; Kalanyan, B.; Parsons, G.N.; Meyer, T.J. Crossing the divide between homogeneous and heterogeneous catalysis in water oxidation. Proc. Natl. Acad. Sci. USA 2013, 110, 20918–20922. [Google Scholar] [CrossRef]
- Lapides, A.M.; Sherman, B.D.; Brennaman, M.K.; Dares, C.J.; Skinner, K.R.; Templeton, J.L.; Meyer, T.J. Synthesis, characterization, and water oxidation by a molecular chromophore-catalyst assembly prepared by atomic layer deposition. The “mummy” strategy. Chem. Sci. 2015, 6, 6398–6406. [Google Scholar] [CrossRef]
- Takijiri, K.; Morita, K.; Nakazono, T.; Sakai, K.; Ozawa, H. Highly stable chemisorption of dyes with pyridyl anchors over TiO2: Application in dye-sensitized photoelectrochemical water reduction in aqueous media. Chem. Commun. 2017, 53, 3042–3045. [Google Scholar] [CrossRef]
- Ooyama, Y.; Inoue, S.; Nagano, T.; Kushimoto, K.; Ohshita, J.; Imae, I.; Komaguchi, K.; Harima, Y. Dye-Sensitized Solar Cells Based On Donor-Acceptor π-Conjugated Fluorescent Dyes with a Pyridine Ring as an Electron-Withdrawing Anchoring Group. Angew. Chem. 2011, 123, 7567–7571. [Google Scholar] [CrossRef]
- Ooyama, Y.; Nagano, T.; Inoue, S.; Imae, I.; Komaguchi, K.; Ohshita, J.; Harima, Y. Dye-sensitized solar cells based on donor-pi-acceptor fluorescent dyes with a pyridine ring as an electron-withdrawing-injecting anchoring group. Chemistry 2011, 17, 14837–14843. [Google Scholar] [CrossRef]
- Shibayama, N.; Ozawa, H.; Abe, M.; Ooyama, Y.; Arakawa, H. A new cosensitization method using the Lewis acid sites of a TiO2 photoelectrode for dye-sensitized solar cells. Chem. Commun. 2014, 50, 6398–6401. [Google Scholar] [CrossRef] [PubMed]
- Gray, C.L.; Xu, P.; Rothenberger, A.J.; Koehler, S.J.; Elacqua, E.; Milosavljevic, B.H.; Mallouk, T.E. Oligomeric Ruthenium Polypyridyl Dye for Improved Stability of Aqueous Photoelectrochemical Cells. J. Phys. Chem. C 2020, 124, 3542–3550. [Google Scholar] [CrossRef]
- Swierk, J.R.; McCool, N.S.; Saunders, T.P.; Barber, G.D.; Mallouk, T.E. Effects of electron trapping and protonation on the efficiency of water-splitting dye-sensitized solar cells. J. Am. Chem. Soc. 2014, 136, 10974–10982. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.Y.; Ardo, S. Direct observation of sequential oxidations of a titania-bound molecular proxy catalyst generated through illumination of molecular sensitizers. Nat. Chem. 2018, 10, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Ardo, S.; Meyer, G.J. Direct observation of photodriven intermolecular hole transfer across TiO2 nanocrystallites: Lateral self-exchange reactions and catalyst oxidation. J. Am. Chem. Soc. 2010, 132, 9283–9285. [Google Scholar] [CrossRef]
- Bangle, R.; Sampaio, R.N.; Troian-Gautier, L.; Meyer, G.J. Surface Grafting of Ru(II) Diazonium-Based Sensitizers on Metal Oxides Enhances Alkaline Stability for Solar Energy Conversion. ACS Appl. Mater. Interfaces 2018, 10, 3121–3132. [Google Scholar] [CrossRef]
- Linfoot, C.L.; Richardson, P.; McCall, K.L.; Durrant, J.R.; Morandeira, A.; Robertson, N. A nickel-complex sensitiser for dye-sensitised solar cells. Sol. Energy 2011, 85, 1195–1203. [Google Scholar] [CrossRef]
- Ma, Y.; Wang, K.; Zhang, H.; Liu, H.; Tian, Y.; Wang, Y.; Zhong, C. Sensitizers of Metal Complexes with Sulfur Coordination Achieving a Power Conversion Efficiency of 12.89. ACS Appl. Mater. Interfaces 2023, 15, 35251–35260. [Google Scholar] [CrossRef]
- Conradie, J. Polypyridyl copper complexes as dye sensitizer and redox mediator for dye-sensitized solar cells. Electrochem. Commun. 2022, 134, 107182. [Google Scholar] [CrossRef]
- Marri, A.R.; Marchini, E.; Cabanes, V.D.; Argazzi, R.; Pastore, M.; Caramori, S.; Bignozzi, C.A.; Gros, P.C. A Series of Iron(II)-NHC Sensitizers with Remarkable Power Conversion Efficiency in Photoelectrochemical Cells. Chemistry 2021, 27, 16260–16269. [Google Scholar] [CrossRef]
- Tichnell, C.R.; Miller, J.N.; Liu, C.; Mukherjee, S.; Jakubikova, E.; McCusker, J.K. Influence of Electrolyte Composition on Ultrafast Interfacial Electron Transfer in Fe-Sensitized TiO2-Based Solar Cells. J. Phys. Chem. C 2020, 124, 1794–1811. [Google Scholar] [CrossRef]
- Orbelli Biroli, A.; Tessore, F.; Di Carlo, G.; Pizzotti, M.; Benazzi, E.; Gentile, F.; Berardi, S.; Bignozzi, C.A.; Argazzi, R.; Natali, M.; et al. Fluorinated Zn(II) Porphyrins for Dye-Sensitized Aqueous Photoelectrosynthetic Cells. ACS Appl. Mater. Interfaces 2019, 11, 32895–32908. [Google Scholar] [CrossRef] [PubMed]
- Berardi, S.; Caramori, S.; Benazzi, E.; Zabini, N.; Niorettini, A.; Orbelli Biroli, A.; Pizzotti, M.; Tessore, F.; Di Carlo, G. Electronic Properties of Electron-Deficient Zn(II) Porphyrins for HBr Splitting. Appl. Sci. 2019, 9, 2739. [Google Scholar] [CrossRef]
- Mussini, P.R.; Orbelli Biroli, A.; Tessore, F.; Pizzotti, M.; Biaggi, C.; Di Carlo, G.; Lobello, M.G.; De Angelis, F. Modulating the electronic properties of asymmetric push–pull and symmetric Zn(II)-diarylporphyrinates with para substituted phenylethynyl moieties in 5,15 meso positions: A combined electrochemical and spectroscopic investigation. Electrochim. Acta 2012, 85, 509–523. [Google Scholar] [CrossRef]
- Moore, G.F.; Blakemore, J.D.; Milot, R.L.; Hull, J.F.; Song, H.-e.; Cai, L.; Schmuttenmaer, C.A.; Crabtree, R.H.; Brudvig, G.W. A visible light water-splitting cell with a photoanode formed by codeposition of a high-potential porphyrin and an iridium water-oxidation catalyst. Energy Environ. Sci. 2011, 4, 2389–2392. [Google Scholar] [CrossRef]
- Nayak, A.; Roy, S.; Sherman, B.D.; Alibabaei, L.; Lapides, A.M.; Brennaman, M.K.; Wee, K.R.; Meyer, T.J. Phosphonate-Derivatized Porphyrins for Photoelectrochemical Applications. ACS Appl. Mater. Interfaces 2016, 8, 3853–3860. [Google Scholar] [CrossRef]
- Akamine, K.; Morita, K.; Sakai, K.; Ozawa, H. A Molecular-Based Water Electrolyzer Consisting of Two Mesoporous TiO2 Electrodes Modified with Metalloporphyrin Molecular Catalysts Showing a Quantitative Faradaic Efficiency. ACS Appl. Energy Mater. 2020, 3, 4860–4866. [Google Scholar] [CrossRef]
- Urbani, M.; Ragoussi, M.-E.; Nazeeruddin, M.K.; Torres, T. Phthalocyanines for dye-sensitized solar cells. Coord. Chem. Rev. 2019, 381, 1–64. [Google Scholar] [CrossRef]
- Atta, N.F.; Amin, H.M.; Khalil, M.W.; Galal, A. Nanotube arrays as photoanodes for dye sensitized solar cells using metal phthalocyanine dyes. Int. J. Electrochem. Sci. 2011, 6, 3. [Google Scholar] [CrossRef]
- Nada, F.; Galal, A.; Amin, H.M. Synthesis and photoelectrochemical behavior of a hybrid electrode composed of polyaniline encapsulated in highly ordered TiO2 nanotubes array. Int. J. Electrochem. Sci. 2012, 7, 3610–3626. [Google Scholar] [CrossRef]
- Orbelli Biroli, A.; Tessore, F.; Pizzotti, M.; Biaggi, C.; Ugo, R.; Caramori, S.; Aliprandi, A.; Bignozzi, C.A.; De Angelis, F.; Giorgi, G.; et al. A Multitechnique Physicochemical Investigation of Various Factors Controlling the Photoaction Spectra and of Some Aspects of the Electron Transfer for a Series of Push–Pull Zn(II) Porphyrins Acting as Dyes in DSSCs. J. Phys. Chem. C 2011, 115, 23170–23182. [Google Scholar] [CrossRef]
- Bessho, T.; Zakeeruddin, S.M.; Yeh, C.Y.; Diau, E.W.; Gratzel, M. Highly efficient mesoscopic dye-sensitized solar cells based on donor-acceptor-substituted porphyrins. Angew. Chem. Int. Ed. Engl. 2010, 49, 6646–6649. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Vlachopoulos, N.; Hao, Y.; Hagfeldt, A.; Boschloo, G. Efficient dye regeneration at low driving force achieved in triphenylamine dye LEG4 and TEMPO redox mediator based dye-sensitized solar cells. Phys. Chem. Chem. Phys. 2015, 17, 15868–15875. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Fan, K.; Xu, B.; Gabrielsson, E.; Daniel, Q.; Li, L.; Sun, L. Organic Dye-Sensitized Tandem Photoelectrochemical Cell for Light Driven Total Water Splitting. J. Am. Chem. Soc. 2015, 137, 9153–9159. [Google Scholar] [CrossRef]
- Loudet, A.; Burgess, K. BODIPY dyes and their derivatives: Syntheses and spectroscopic properties. Chem. Rev. 2007, 107, 4891–4932. [Google Scholar] [CrossRef]
- Ulrich, G.; Ziessel, R.; Harriman, A. The chemistry of fluorescent bodipy dyes: Versatility unsurpassed. Angew. Chem. Int. Ed. Engl. 2008, 47, 1184–1201. [Google Scholar] [CrossRef]
- Kubo, Y.; Minowa, Y.; Shoda, T.; Takeshita, K. Synthesis of a new type of dibenzopyrromethene–boron complex with near-infrared absorption property. Tetrahedron Lett. 2010, 51, 1600–1602. [Google Scholar] [CrossRef]
- Kubo, Y.; Eguchi, D.; Matsumoto, A.; Nishiyabu, R.; Yakushiji, H.; Shigaki, K.; Kaneko, M. Boron–dibenzopyrromethene-based organic dyes for application in dye-sensitized solar cells. J. Mater. Chem. A 2014, 2, 5204–5211. [Google Scholar] [CrossRef]
- Kubo, Y.; Watanabe, K.; Nishiyabu, R.; Hata, R.; Murakami, A.; Shoda, T.; Ota, H. Near-infrared absorbing boron-dibenzopyrromethenes that serve as light-harvesting sensitizers for polymeric solar cells. Org. Lett. 2011, 13, 4574–4577. [Google Scholar] [CrossRef] [PubMed]
- Suryani, O.; Higashino, Y.; Mulyana, J.Y.; Kaneko, M.; Hoshi, T.; Shigaki, K.; Kubo, Y. A near-infrared organic photosensitizer for use in dye-sensitized photoelectrochemical water splitting. Chem. Commun. 2017, 53, 6784–6787. [Google Scholar] [CrossRef] [PubMed]
- Badgurjar, D.; Shan, B.; Nayak, A.; Wu, L.; Chitta, R.; Meyer, T.J. Electron-Withdrawing Boron Dipyrromethene Dyes As Visible Light Absorber/Sensitizers on Semiconductor Oxide Surfaces. ACS Appl. Mater. Interfaces 2020, 12, 7768–7776. [Google Scholar] [CrossRef] [PubMed]
- Brimblecombe, R.; Koo, A.; Dismukes, G.C.; Swiegers, G.F.; Spiccia, L. Solar driven water oxidation by a bioinspired manganese molecular catalyst. J. Am. Chem. Soc. 2010, 132, 2892–2894. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Duan, L.; Xu, Y.; Gorlov, M.; Hagfeldt, A.; Sun, L. A photoelectrochemical device for visible light driven water splitting by a molecular ruthenium catalyst assembled on dye-sensitized nanostructured TiO2. Chem. Commun. 2010, 46, 7307–7309. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Vannucci, A.K.; Farnum, B.H.; Lapides, A.M.; Brennaman, M.K.; Kalanyan, B.; Alibabaei, L.; Concepcion, J.J.; Losego, M.D.; Parsons, G.N.; et al. Visible light driven benzyl alcohol dehydrogenation in a dye-sensitized photoelectrosynthesis cell. J. Am. Chem. Soc. 2014, 136, 9773–9779. [Google Scholar] [CrossRef] [PubMed]
- Michaux, K.E.; Gambardella, A.A.; Alibabaei, L.; Ashford, D.L.; Sherman, B.D.; Binstead, R.A.; Meyer, T.J.; Murray, R.W. Visible Photoelectrochemical Water Splitting Based on a Ru(II) Polypyridyl Chromophore and Iridium Oxide Nanoparticle Catalyst. J. Phys. Chem. C 2015, 119, 17023–17027. [Google Scholar] [CrossRef]
- Zhu, Y.; Wang, D.; Huang, Q.; Du, J.; Sun, L.; Li, F.; Meyer, T.J. Stabilization of a molecular water oxidation catalyst on a dye-sensitized photoanode by a pyridyl anchor. Nat. Commun. 2020, 11, 4610. [Google Scholar] [CrossRef]
- Walter, M.G.; Warren, E.L.; McKone, J.R.; Boettcher, S.W.; Mi, Q.; Santori, E.A.; Lewis, N.S. Solar water splitting cells. Chem. Rev. 2010, 110, 6446–6473. [Google Scholar] [CrossRef]
- Cook, T.R.; Dogutan, D.K.; Reece, S.Y.; Surendranath, Y.; Teets, T.S.; Nocera, D.G. Solar energy supply and storage for the legacy and nonlegacy worlds. Chem. Rev. 2010, 110, 6474–6502. [Google Scholar] [CrossRef]
- Kirner, J.T.; Finke, R.G. Water-oxidation photoanodes using organic light-harvesting materials: A review. J. Mater. Chem. A 2017, 5, 19560–19592. [Google Scholar] [CrossRef]
- Murphy, A.; Barnes, P.; Randeniya, L.; Plumb, I.; Grey, I.; Horne, M.; Glasscock, J. Efficiency of solar water splitting using semiconductor electrodes. Int. J. Hydrogen Energy 2006, 31, 1999–2017. [Google Scholar] [CrossRef]
- Bolton, J.R.; Strickler, S.J.; Connolly, J.S. Limiting and realizable efficiencies of solar photolysis of water. Nature 1985, 316, 495–500. [Google Scholar] [CrossRef]
- Gersten, S.W.; Samuels, G.J.; Meyer, T.J. Catalytic oxidation of water by an oxo-bridged ruthenium dimer. J. Am. Chem. Soc. 1982, 104, 4029–4030. [Google Scholar] [CrossRef]
- Gilbert, J.A.; Eggleston, D.S.; Murphy, W.R.; Geselowitz, D.A.; Gersten, S.W.; Hodgson, D.J.; Meyer, T.J. Structure and redox properties of the water-oxidation catalyst [(bpy)2(OH2)RuORu(OH2)(bpy)2]4+. J. Am. Chem. Soc. 1985, 107, 3855–3864. [Google Scholar] [CrossRef]
- Karkas, M.D.; Verho, O.; Johnston, E.V.; Akermark, B. Artificial photosynthesis: Molecular systems for catalytic water oxidation. Chem. Rev. 2014, 114, 11863–12001. [Google Scholar] [CrossRef]
- Sens, C.; Romero, I.; Rodriguez, M.; Llobet, A.; Parella, T.; Benet-Buchholz, J. A new Ru complex capable of catalytically oxidizing water to molecular dioxygen. J. Am. Chem. Soc. 2004, 126, 7798–7799. [Google Scholar] [CrossRef] [PubMed]
- Zong, R.; Thummel, R.P. A new family of Ru complexes for water oxidation. J. Am. Chem. Soc. 2005, 127, 12802–12803. [Google Scholar] [CrossRef]
- Concepcion, J.J.; Jurss, J.W.; Templeton, J.L.; Meyer, T.J. One site is enough. Catalytic water oxidation by [Ru(tpy)(bpm)(OH2)]2+ and [Ru(tpy)(bpz)(OH2)]2+. J. Am. Chem. Soc. 2008, 130, 16462–16463. [Google Scholar] [CrossRef]
- Concepcion, J.J.; Jurss, J.W.; Brennaman, M.K.; Hoertz, P.G.; Patrocinio, A.O.; Murakami Iha, N.Y.; Templeton, J.L.; Meyer, T.J. Making oxygen with ruthenium complexes. Acc. Chem. Res. 2009, 42, 1954–1965. [Google Scholar] [CrossRef]
- Xu, Y.; Akermark, T.; Gyollai, V.; Zou, D.; Eriksson, L.; Duan, L.; Zhang, R.; Akermark, B.; Sun, L. A new dinuclear ruthenium complex as an efficient water oxidation catalyst. Inorg. Chem. 2009, 48, 2717–2719. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Fischer, A.; Duan, L.; Tong, L.; Gabrielsson, E.; Åkermark, B.; Sun, L. Chemical and Light-Driven Oxidation of Water Catalyzed by an Efficient Dinuclear Ruthenium Complex. Angew. Chem. 2010, 122, 9118–9121. [Google Scholar] [CrossRef]
- Duan, L.; Fischer, A.; Xu, Y.; Sun, L. Isolated seven-coordinate Ru(IV) dimer complex with [HOHOH](-) bridging ligand as an intermediate for catalytic water oxidation. J. Am. Chem. Soc. 2009, 131, 10397–10399. [Google Scholar] [CrossRef] [PubMed]
- Blakemore, J.D.; Crabtree, R.H.; Brudvig, G.W. Molecular Catalysts for Water Oxidation. Chem. Rev. 2015, 115, 12974–13005. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Sun, L. Ru-bda: Unique Molecular Water-Oxidation Catalysts with Distortion Induced Open Site and Negatively Charged Ligands. J. Am. Chem. Soc. 2019, 141, 5565–5580. [Google Scholar] [CrossRef] [PubMed]
- Harriman, A.; Pickering, I.J.; Thomas, J.M.; Christensen, P.A. Metal oxides as heterogeneous catalysts for oxygen evolution under photochemical conditions. J. Chem. Soc. Faraday Trans. 1 Phys. Chem. Condens. Phases 1988, 84, 2795–2806. [Google Scholar] [CrossRef]
- Ruttinger, W.; Dismukes, G.C. Synthetic Water-Oxidation Catalysts for Artificial Photosynthetic Water Oxidation. Chem. Rev. 1997, 97, 1–24. [Google Scholar] [CrossRef]
- Ruettinger, W.F.; Campana, C.; Dismukes, G.C. Synthesis and Characterization of Mn4O4L6Complexes with Cubane-like Core Structure: A New Class of Models of the Active Site of the Photosynthetic Water Oxidase. J. Am. Chem. Soc. 1997, 119, 6670–6671. [Google Scholar] [CrossRef]
- Brimblecombe, R.; Swiegers, G.F.; Dismukes, G.C.; Spiccia, L. Sustained water oxidation photocatalysis by a bioinspired manganese cluster. Angew. Chem. Int. Ed. Engl. 2008, 47, 7335–7338. [Google Scholar] [CrossRef]
- Duan, L.; Xu, Y.; Zhang, P.; Wang, M.; Sun, L. Visible light-driven water oxidation by a molecular ruthenium catalyst in homogeneous system. Inorg. Chem. 2010, 49, 209–215. [Google Scholar] [CrossRef]
- Hoogeveen, D.A.; Fournier, M.; Bonke, S.A.; Nattestad, A.; Mishra, A.; Bäuerle, P.; Spiccia, L.; Mozer, A.J.; Simonov, A.N. Origin of Photoelectrochemical Generation of Dihydrogen by a Dye-Sensitized Photocathode without an Intentionally Introduced Catalyst. J. Phys. Chem. C 2017, 121, 25836–25846. [Google Scholar] [CrossRef]
- Kamire, R.J.; Materna, K.L.; Hoffeditz, W.L.; Phelan, B.T.; Thomsen, J.M.; Farha, O.K.; Hupp, J.T.; Brudvig, G.W.; Wasielewski, M.R. Photodriven Oxidation of Surface-Bound Iridium-Based Molecular Water-Oxidation Catalysts on Perylene-3,4-dicarboximide-Sensitized TiO2 Electrodes Protected by an Al2O3 Layer. J. Phys. Chem. C 2017, 121, 3752–3764. [Google Scholar] [CrossRef]
- Xu, P.; Gray, C.L.; Xiao, L.; Mallouk, T.E. Charge Recombination with Fractional Reaction Orders in Water-Splitting Dye-Sensitized Photoelectrochemical Cells. J. Am. Chem. Soc. 2018, 140, 11647–11654. [Google Scholar] [CrossRef] [PubMed]
- Hisatomi, T.; Kubota, J.; Domen, K. Recent advances in semiconductors for photocatalytic and photoelectrochemical water splitting. Chem. Soc. Rev. 2014, 43, 7520–7535. [Google Scholar] [CrossRef] [PubMed]
- Tian, Z.; Zhang, P.; Qin, P.; Sun, D.; Zhang, S.; Guo, X.; Zhao, W.; Zhao, D.; Huang, F. Novel Black BiVO4/TiO2−x Photoanode with Enhanced Photon Absorption and Charge Separation for Efficient and Stable Solar Water Splitting. Adv. Energy Mater. 2019, 9, 1901287. [Google Scholar] [CrossRef]
- Concepcion, J.J.; Zhong, D.K.; Szalda, D.J.; Muckerman, J.T.; Fujita, E. Mechanism of water oxidation by [Ru(bda)(L)2]: The return of the “blue dimer”. Chem. Commun. 2015, 51, 4105–4108. [Google Scholar] [CrossRef]
- Wang, D.; Sampaio, R.N.; Troian-Gautier, L.; Marquard, S.L.; Farnum, B.H.; Sherman, B.D.; Sheridan, M.V.; Dares, C.J.; Meyer, G.J.; Meyer, T.J. Molecular Photoelectrode for Water Oxidation Inspired by Photosystem II. J. Am. Chem. Soc. 2019, 141, 7926–7933. [Google Scholar] [CrossRef] [PubMed]
- Ashford, D.L.; Gish, M.K.; Vannucci, A.K.; Brennaman, M.K.; Templeton, J.L.; Papanikolas, J.M.; Meyer, T.J. Molecular Chromophore-Catalyst Assemblies for Solar Fuel Applications. Chem. Rev. 2015, 115, 13006–13049. [Google Scholar] [CrossRef]
- Alibabaei, L.; Brennaman, M.K.; Norris, M.R.; Kalanyan, B.; Song, W.; Losego, M.D.; Concepcion, J.J.; Binstead, R.A.; Parsons, G.N.; Meyer, T.J. Solar water splitting in a molecular photoelectrochemical cell. Proc. Natl. Acad. Sci. USA 2013, 110, 20008–20013. [Google Scholar] [CrossRef]
- Alibabaei, L.; Sherman, B.D.; Norris, M.R.; Brennaman, M.K.; Meyer, T.J. Visible photoelectrochemical water splitting into H2 and O2 in a dye-sensitized photoelectrosynthesis cell. Proc. Natl. Acad. Sci. USA 2015, 112, 5899–5902. [Google Scholar] [CrossRef]
- Lee, H.; Kepley, L.J.; Hong, H.G.; Akhter, S.; Mallouk, T.E. Adsorption of ordered zirconium phosphonate multilayer films on silicon and gold surfaces. J. Phys. Chem. 2002, 92, 2597–2601. [Google Scholar] [CrossRef]
- Lee, H.; Kepley, L.J.; Hong, H.G.; Mallouk, T.E. Inorganic analogs of Langmuir-Blodgett films: Adsorption of ordered zirconium 1,10-decanebisphosphonate multilayers on silicon surfaces. J. Am. Chem. Soc. 2002, 110, 618–620. [Google Scholar] [CrossRef]
- Ishida, T.; Terada, K.-i.; Hasegawa, K.; Kuwahata, H.; Kusama, K.; Sato, R.; Nakano, M.; Naitoh, Y.; Haga, M.-a. Self-assembled monolayer and multilayer formation using redox-active Ru complex with phosphonic acids on silicon oxide surface. Appl. Surf. Sci. 2009, 255, 8824–8830. [Google Scholar] [CrossRef]
- Terada, K.; Kobayashi, K.; Hikita, J.; Haga, M.-a. Electric Conduction Properties of Self-assembled Monolayer Films of Ru Complexes with Disulfide/Phosphonate Anchors in a Au–(Molecular Ensemble)–(Au Nanoparticle) Junction. Chem. Lett. 2009, 38, 416–417. [Google Scholar] [CrossRef]
- Hanson, K.; Torelli, D.A.; Vannucci, A.K.; Brennaman, M.K.; Luo, H.; Alibabaei, L.; Song, W.; Ashford, D.L.; Norris, M.R.; Glasson, C.R.K.; et al. Self-Assembled Bilayer Films of Ruthenium(II)/Polypyridyl Complexes through Layer-by-Layer Deposition on Nanostructured Metal Oxides. Angew. Chem. 2012, 124, 12954–12957. [Google Scholar] [CrossRef]
- Ding, X.; Gao, Y.; Zhang, L.; Yu, Z.; Liu, J.; Sun, L. Visible Light-Driven Water Splitting in Photoelectrochemical Cells with Supramolecular Catalysts on Photoanodes. ACS Catal. 2014, 4, 2347–2350. [Google Scholar] [CrossRef]
- Sheridan, M.V.; Sherman, B.D.; Coppo, R.L.; Wang, D.; Marquard, S.L.; Wee, K.-R.; Murakami Iha, N.Y.; Meyer, T.J. Evaluation of Chromophore and Assembly Design in Light-Driven Water Splitting with a Molecular Water Oxidation Catalyst. ACS Energy Lett. 2016, 1, 231–236. [Google Scholar] [CrossRef]
- Wang, D.; Xu, Z.; Sheridan, M.V.; Concepcion, J.J.; Li, F.; Lian, T.; Meyer, T.J. Photodriven water oxidation initiated by a surface bound chromophore-donor-catalyst assembly. Chem. Sci. 2021, 12, 14441–14450. [Google Scholar] [CrossRef]
- Yeo, R.S.; Chin, D.T. A Hydrogen-Bromine Cell for Energy Storage Applications. J. Electrochem. Soc. 2019, 127, 549–555. [Google Scholar] [CrossRef]
- Savinell, R.F.; Fritts, S.D. Theoretical performance of a hydrogen-bromine rechargeable SPE fuel cell. J. Power Sources 1988, 22, 423–440. [Google Scholar] [CrossRef]
- Park, J.W.; Wycisk, R.; Pintauro, P.N.; Yarlagadda, V.; Van Nguyen, T. Electrospun Nafion((R))/Polyphenylsulfone Composite Membranes for Regenerative Hydrogen Bromine Fuel Cells. Materials 2016, 9, 143. [Google Scholar] [CrossRef] [PubMed]
- Michalowski, T. Calculation of pH and Potential E for Bromine Aqueous Solution. J. Chem. Educ. 1994, 71, 560–561. [Google Scholar] [CrossRef]
- Sheridan, M.V.; Wang, Y.; Wang, D.; Troian-Gautier, L.; Dares, C.J.; Sherman, B.D.; Meyer, T.J. Light-Driven Water Splitting Mediated by Photogenerated Bromine. Angew. Chem. 2018, 130, 3507–3511. [Google Scholar] [CrossRef]
- Cettou, P.; Robertson, P.M.; Ibl, N. On the electrolysis of aqueous bromide solutions to bromate. Electrochim. Acta 1984, 29, 875–885. [Google Scholar] [CrossRef]
- Mastragostino, M.; Gramellini, C. Kinetic study of the electrochemical processes of the bromine/bromine aqueous system on vitreous carbon electrodes. Electrochim. Acta 1985, 30, 373–380. [Google Scholar] [CrossRef]
- Duan, L.; Bozoglian, F.; Mandal, S.; Stewart, B.; Privalov, T.; Llobet, A.; Sun, L. A molecular ruthenium catalyst with water-oxidation activity comparable to that of photosystem II. Nat. Chem. 2012, 4, 418–423. [Google Scholar] [CrossRef] [PubMed]
- Song, N.; Concepcion, J.J.; Binstead, R.A.; Rudd, J.A.; Vannucci, A.K.; Dares, C.J.; Coggins, M.K.; Meyer, T.J. Base-enhanced catalytic water oxidation by a carboxylate-bipyridine Ru(II) complex. Proc. Natl. Acad. Sci. USA 2015, 112, 4935–4940. [Google Scholar] [CrossRef]
- Li, G.; Ward, W.M.; Meyer, G.J. Visible Light Driven Nanosecond Bromide Oxidation by a Ru Complex with Subsequent Br-Br Bond Formation. J. Am. Chem. Soc. 2015, 137, 8321–8323. [Google Scholar] [CrossRef]
- Li, G.; Swords, W.B.; Meyer, G.J. Bromide Photo-oxidation Sensitized to Visible Light in Consecutive Ion Pairs. J. Am. Chem. Soc. 2017, 139, 14983–14991. [Google Scholar] [CrossRef]
- Li, G.; Brady, M.D.; Meyer, G.J. Visible Light Driven Bromide Oxidation and Ligand Substitution Photochemistry of a Ru Diimine Complex. J. Am. Chem. Soc. 2018, 140, 5447–5456. [Google Scholar] [CrossRef]
- Tsai, K.Y.; Chang, I.J. Photocatalytic Oxidation of Bromide to Bromine. Inorg. Chem. 2017, 56, 693–696. [Google Scholar] [CrossRef]
- Tsai, K.Y.; Chang, I.J. Oxidation of Bromide to Bromine by Ruthenium(II) Bipyridine-Type Complexes Using the Flash-Quench Technique. Inorg. Chem. 2017, 56, 8497–8503. [Google Scholar] [CrossRef] [PubMed]
- Brady, M.D.; Sampaio, R.N.; Wang, D.; Meyer, T.J.; Meyer, G.J. Dye-Sensitized Hydrobromic Acid Splitting for Hydrogen Solar Fuel Production. J. Am. Chem. Soc. 2017, 139, 15612–15615. [Google Scholar] [CrossRef] [PubMed]
- Muller, A.V.; de Oliveira, K.T.; Meyer, G.J.; Polo, A.S. Inhibiting Charge Recombination in cis-Ru(NCS)(2) Diimine Sensitizers with Aromatic Substituents. ACS Appl. Mater. Interfaces 2019, 11, 43223–43234. [Google Scholar] [CrossRef] [PubMed]
- Reddy-Marri, A.; Marchini, E.; Cabanes, V.D.; Argazzi, R.; Pastore, M.; Caramori, S.; Gros, P.C. Panchromatic light harvesting and record power conversion efficiency for carboxylic/cyanoacrylic Fe(ii) NHC co-sensitized FeSSCs. Chem. Sci. 2023, 14, 4288–4301. [Google Scholar] [CrossRef] [PubMed]
- Slama-Schwok, A.; Gershuni, S.; Rabani, J.; Cohen, H.; Meyerstein, D. An iridium-bipyridine complex as a photosensitizer for the bromide oxidation to bromine by oxygen. J. Phys. Chem. 2002, 89, 2460–2464. [Google Scholar] [CrossRef]
- Brady, M.D.; Troian-Gautier, L.; Sampaio, R.N.; Motley, T.C.; Meyer, G.J. Optimization of Photocatalyst Excited- and Ground-State Reduction Potentials for Dye-Sensitized HBr Splitting. ACS Appl. Mater. Interfaces 2018, 10, 31312–31323. [Google Scholar] [CrossRef]
- Wilkinson, G.; Gillard, R.D.; McCleverty, J.A. Comprehensive Coordination Chemistry. The Synthesis, Reactions, Properties and Applications of Coordination Compounds. V. 3. Main Group and Early Transition Elements; Pergamon Press: Oxford, UK, 1987. [Google Scholar]
- Bergeron, B.V.; Marton, A.; Oskam, G.; Meyer, G.J. Dye-sensitized SnO2 electrodes with iodide and pseudohalide redox mediators. J. Phys. Chem. B 2005, 109, 937–943. [Google Scholar] [CrossRef]
- Treadway, J.A.; Moss, J.A.; Meyer, T.J. Visible Region Photooxidation on TiO2 with a Chromophore-Catalyst Molecular Assembly. Inorg. Chem. 1999, 38, 4386–4387. [Google Scholar] [CrossRef]
- Ruberu, T.P.A.; Nelson, N.C.; Slowing, I.I.; Vela, J. Selective Alcohol Dehydrogenation and Hydrogenolysis with Semiconductor-Metal Photocatalysts: Toward Solar-to-Chemical Energy Conversion of Biomass-Relevant Substrates. J. Phys. Chem. Lett. 2012, 3, 2798–2802. [Google Scholar] [CrossRef]
- Shimizu, K.; Sugino, K.; Sawabe, K.; Satsuma, A. Oxidant-free dehydrogenation of alcohols heterogeneously catalyzed by cooperation of silver clusters and acid-base sites on alumina. Chemistry 2009, 15, 2341–2351. [Google Scholar] [CrossRef] [PubMed]
- Angelici, R.J. Hydrogen Storage and Energy Recovery Using Aldehydes and Ketones: A Key Role for Catalysis. ACS Catal. 2011, 1, 772–776. [Google Scholar] [CrossRef]
- Song, W.; Ito, A.; Binstead, R.A.; Hanson, K.; Luo, H.; Brennaman, M.K.; Concepcion, J.J.; Meyer, T.J. Accumulation of multiple oxidative equivalents at a single site by cross-surface electron transfer on TiO2. J. Am. Chem. Soc. 2013, 135, 11587–11594. [Google Scholar] [CrossRef] [PubMed]
- Natali, M.; Sartorel, A.; Ruggi, A. Beyond Water Oxidation: Hybrid, Molecular-Based Photoanodes for the Production of Value-Added Organics. Front. Chem. 2022, 10, 907510. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Stahl, S.S. Electrochemical Oxidation of Organic Molecules at Lower Overpotential: Accessing Broader Functional Group Compatibility with Electron-Proton Transfer Mediators. Acc. Chem. Res. 2020, 53, 561–574. [Google Scholar] [CrossRef] [PubMed]
- Nutting, J.E.; Rafiee, M.; Stahl, S.S. Tetramethylpiperidine N-Oxyl (TEMPO), Phthalimide N-Oxyl (PINO), and Related N-Oxyl Species: Electrochemical Properties and Their Use in Electrocatalytic Reactions. Chem. Rev. 2018, 118, 4834–4885. [Google Scholar] [CrossRef]
- Ciriminna, R.; Palmisano, G.; Pagliaro, M. Electrodes Functionalized with the 2,2,6,6-Tetramethylpiperidinyloxy Radical for the Waste-Free Oxidation of Alcohols. ChemCatChem 2015, 7, 552–558. [Google Scholar] [CrossRef]
- Ciriminna, R.; Ghahremani, M.; Karimi, B.; Pagliaro, M. Electrochemical Alcohol Oxidation Mediated by TEMPO-like Nitroxyl Radicals. ChemistryOpen 2017, 6, 5–10. [Google Scholar] [CrossRef]
- Heeres, A.; van Doren, H.A.; Bleeker, I.P.; Gotlieb, K.F. Method for Recovering or Recirculating Stable Nitroxide Radicals. WO Patent 9636621, 14 May 1996. [Google Scholar]
- Schnatbaum, K.; Schäfer, H.J. Electroorganic Synthesis 66: Selective Anodic Oxidation of Carbohydrates Mediated by TEMPO. Synthesis 1999, 1999, 864–872. [Google Scholar] [CrossRef]
- Cha, H.G.; Choi, K.S. Combined biomass valorization and hydrogen production in a photoelectrochemical cell. Nat. Chem. 2015, 7, 328–333. [Google Scholar] [CrossRef]
- Li, T.; Kasahara, T.; He, J.; Dettelbach, K.E.; Sammis, G.M.; Berlinguette, C.P. Photoelectrochemical oxidation of organic substrates in organic media. Nat. Commun. 2017, 8, 390. [Google Scholar] [CrossRef] [PubMed]
- Kato, F.; Kikuchi, A.; Okuyama, T.; Oyaizu, K.; Nishide, H. Nitroxide Radicals as Highly Reactive Redox Mediators in Dye-Sensitized Solar Cells. Angew. Chem. 2012, 124, 10324–10327. [Google Scholar] [CrossRef]
- Pati, P.B.; Abdellah, M.; Diring, S.; Hammarstrom, L.; Odobel, F. Molecular Triad Containing a TEMPO Catalyst Grafted on Mesoporous Indium Tin Oxide as a Photoelectrocatalytic Anode for Visible Light-Driven Alcohol Oxidation. ChemSusChem 2021, 14, 2902–2913. [Google Scholar] [CrossRef] [PubMed]
- Skaisgirski, M.; Guo, X.; Wenger, O.S. Electron Accumulation on Naphthalene Diimide Photosensitized by [Ru(2,2′-Bipyridine)3]2+. Inorg. Chem. 2017, 56, 2432–2439. [Google Scholar] [CrossRef] [PubMed]
- Kumar, B.; Llorente, M.; Froehlich, J.; Dang, T.; Sathrum, A.; Kubiak, C.P. Photochemical and photoelectrochemical reduction of CO2. Annu. Rev. Phys. Chem. 2012, 63, 541–569. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Liu, J.C.; Cai, W.; Ma, J.; Yang, H.B.; Xiao, H.; Li, J.; Xiong, Y.; Huang, Y.; Liu, B. Selective photoelectrochemical oxidation of glycerol to high value-added dihydroxyacetone. Nat. Commun. 2019, 10, 1779. [Google Scholar] [CrossRef]
- Katryniok, B.; Kimura, H.; Skrzyńska, E.; Girardon, J.-S.; Fongarland, P.; Capron, M.; Ducoulombier, R.; Mimura, N.; Paul, S.; Dumeignil, F. Selective catalytic oxidation of glycerol: Perspectives for high value chemicals. Green Chem. 2011, 13, 1960–1979. [Google Scholar] [CrossRef]
- Bobbitt, J.M.; BrüCkner, C.; Merbouh, N. Oxoammonium- and Nitroxide-Catalyzed Oxidations of Alcohols. Organomet. React. 2010, 103–424. [Google Scholar] [CrossRef]
- Bruggeman, D.F.; Laporte, A.A.H.; Detz, R.J.; Mathew, S.; Reek, J.N.H. Aqueous Biphasic Dye-Sensitized Photosynthesis Cells for TEMPO-Based Oxidation of Glycerol. Angew. Chem. Int. Ed. Engl. 2022, 61, e202200175. [Google Scholar] [CrossRef]
- Yoo, C.G.; Yang, Y.; Pu, Y.; Meng, X.; Muchero, W.; Yee, K.L.; Thompson, O.A.; Rodriguez, M.; Bali, G.; Engle, N.L.; et al. Insights of biomass recalcitrance in natural Populus trichocarpa variants for biomass conversion. Green Chem. 2017, 19, 5467–5478. [Google Scholar] [CrossRef]
- Galkin, M.V.; Samec, J.S. Lignin Valorization through Catalytic Lignocellulose Fractionation: A Fundamental Platform for the Future Biorefinery. ChemSusChem 2016, 9, 1544–1558. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Kim, S.; Davis, A.H.; Zhuang, J.; Shuler, E.W.; Willinger, D.; Lee, J.-J.; Zheng, W.; Sherman, B.D.; Yoo, C.G.; et al. Photocatalytic Chemoselective C–C Bond Cleavage at Room Temperature in Dye-Sensitized Photoelectrochemical Cells. ACS Catal. 2021, 11, 3771–3781. [Google Scholar] [CrossRef]
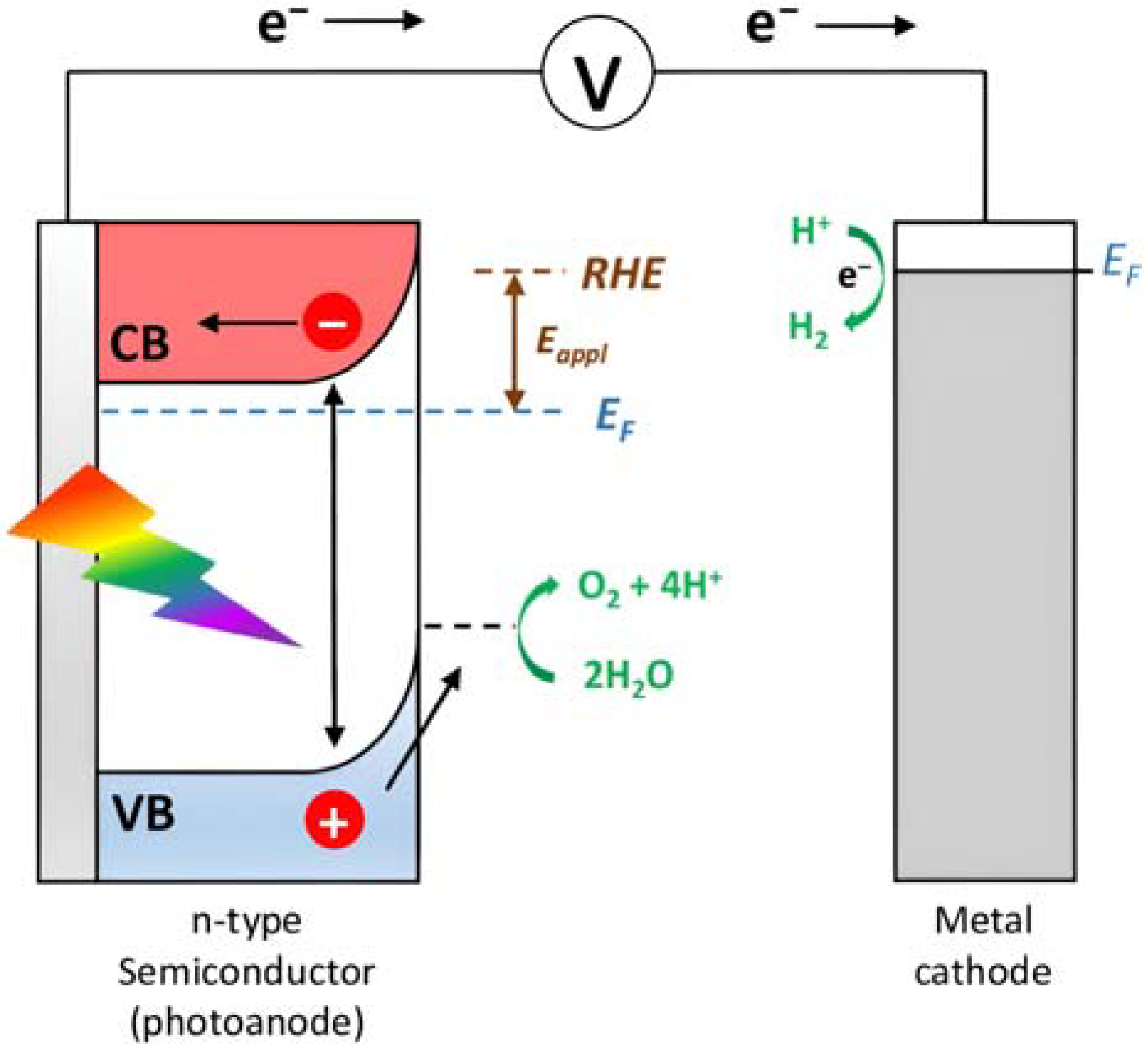
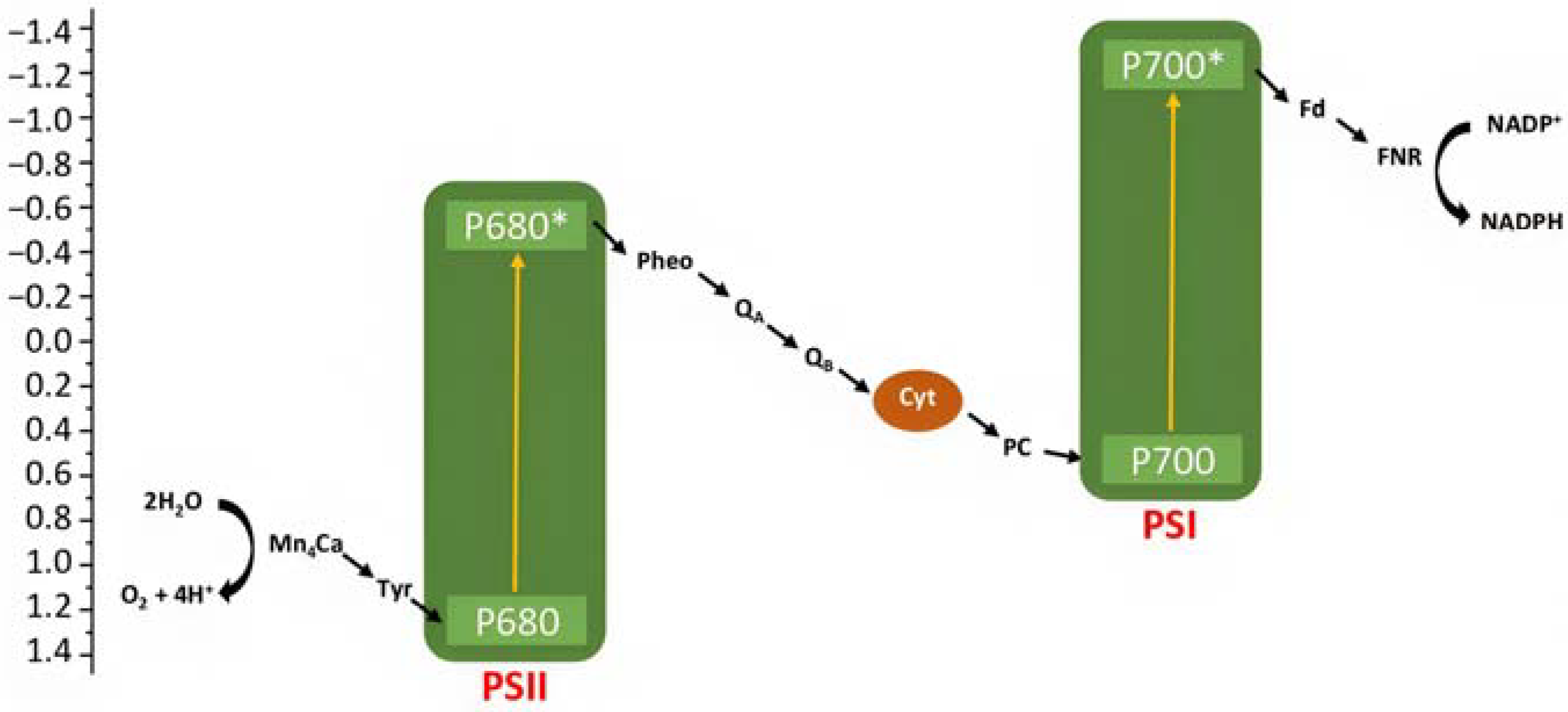
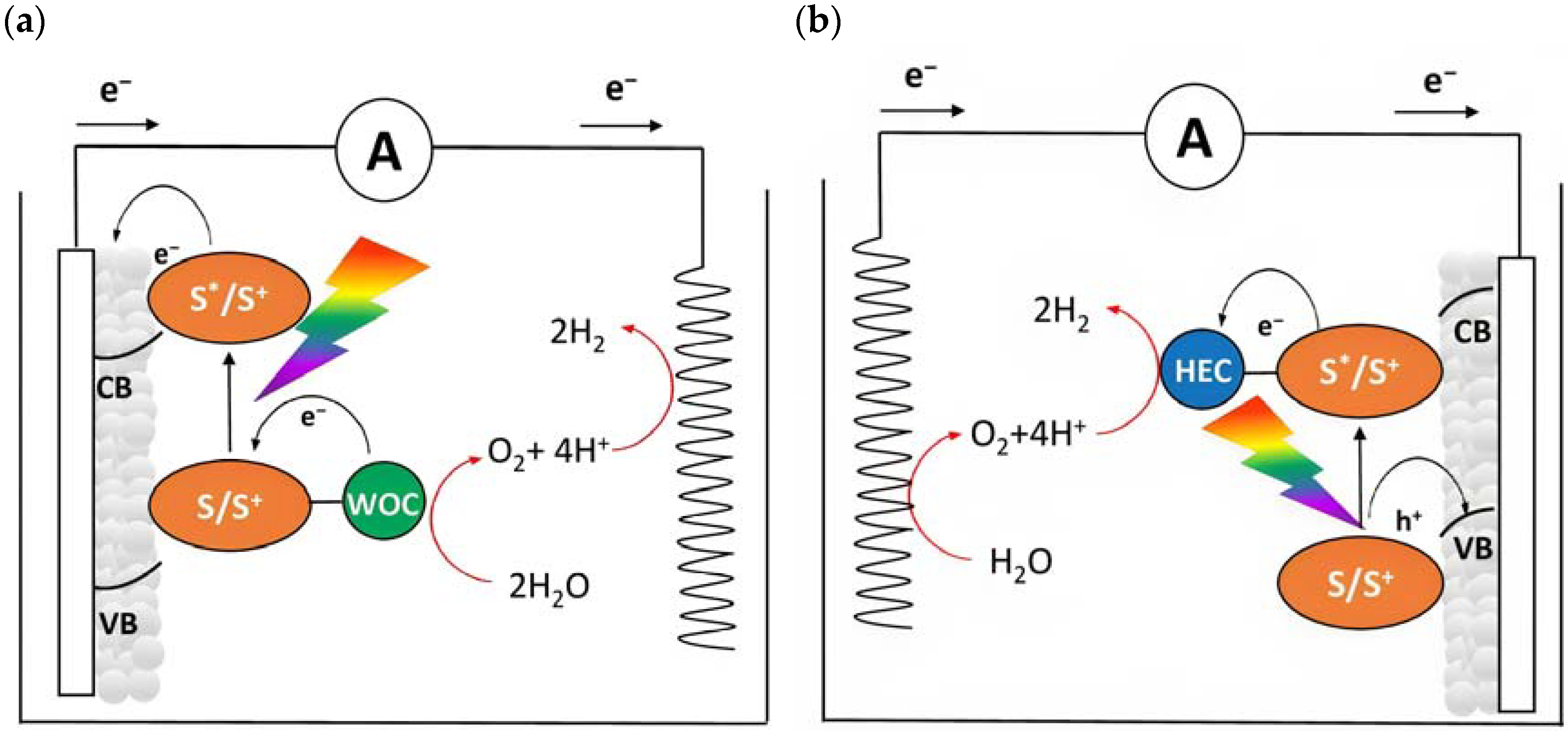
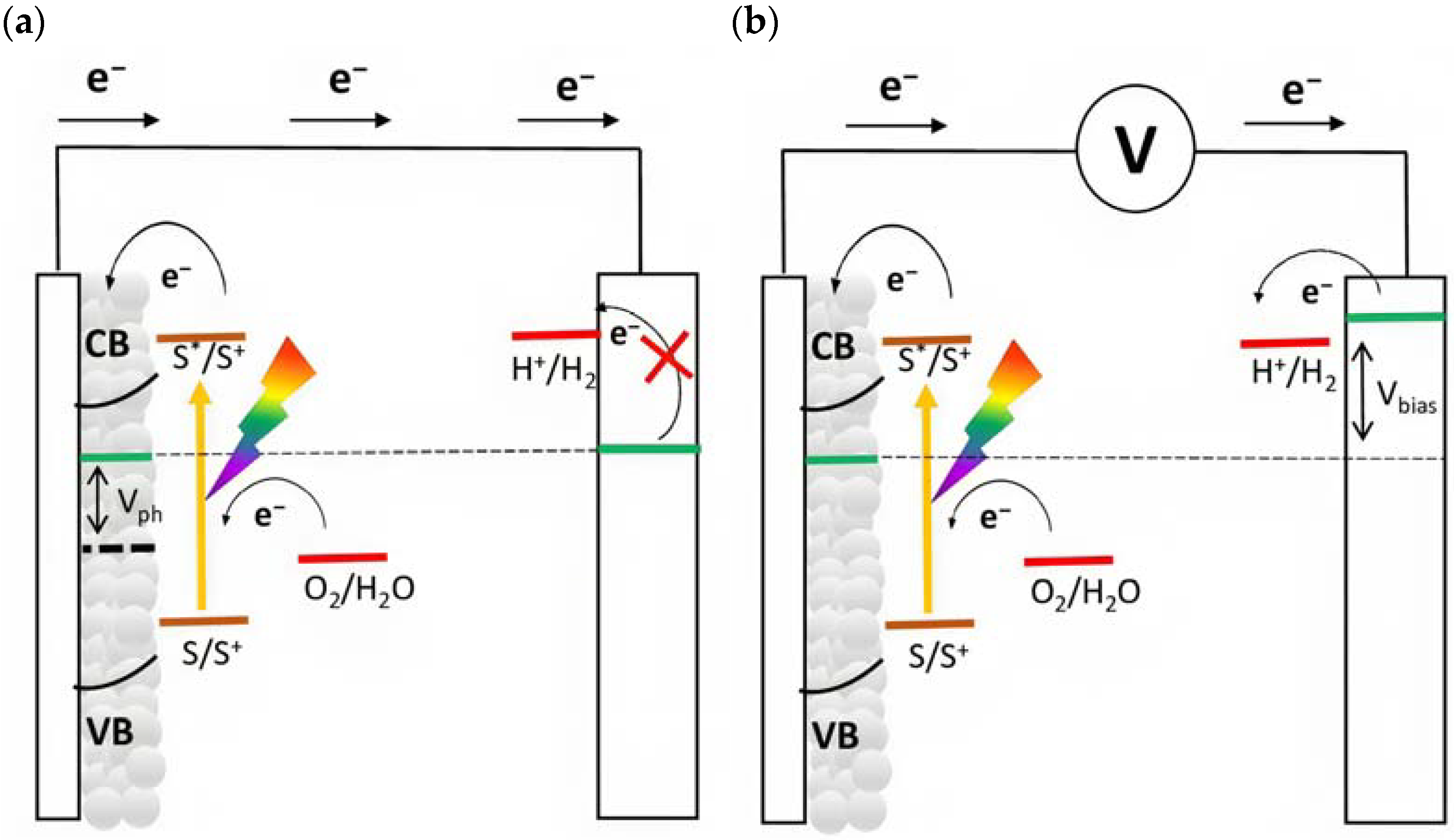
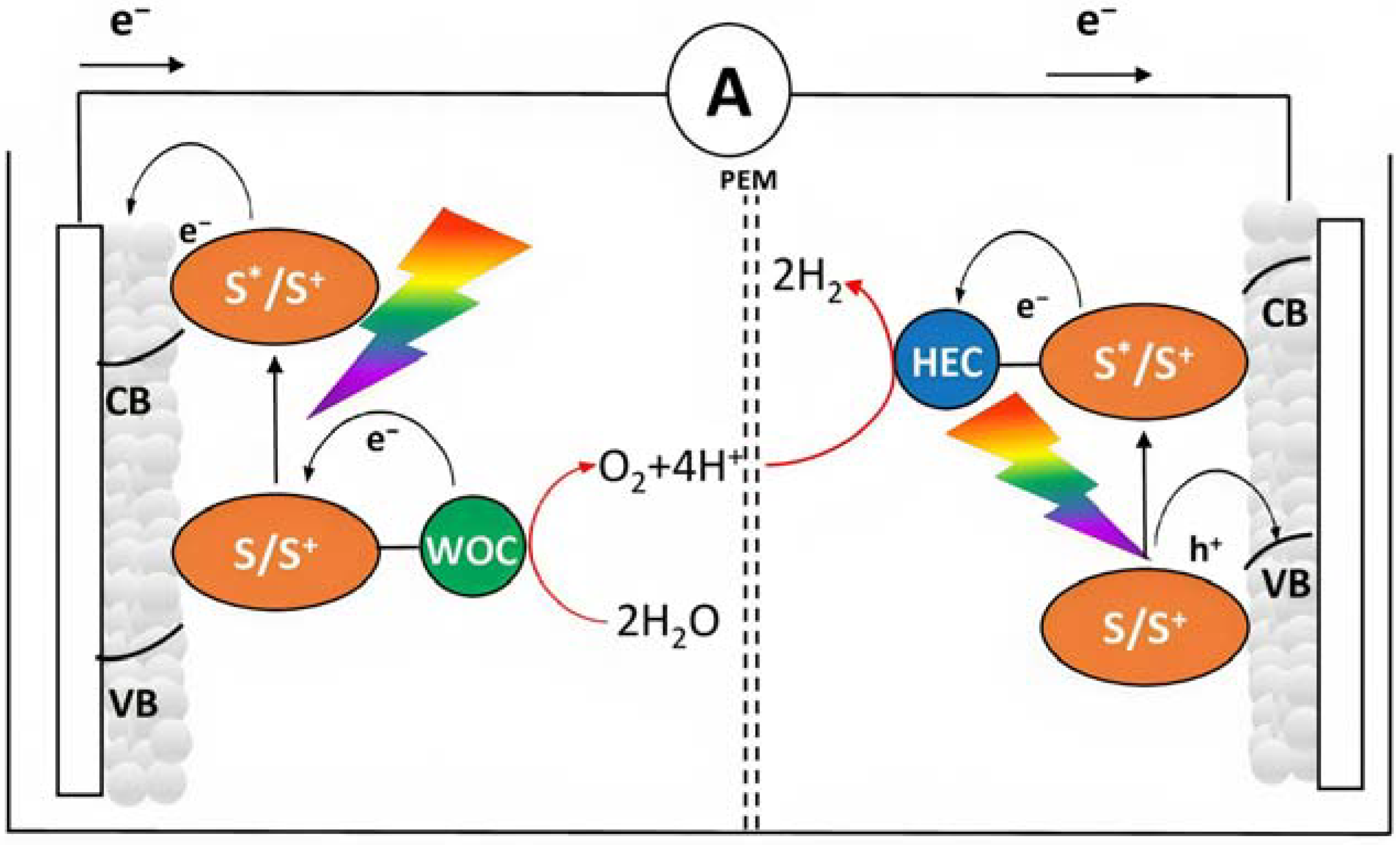
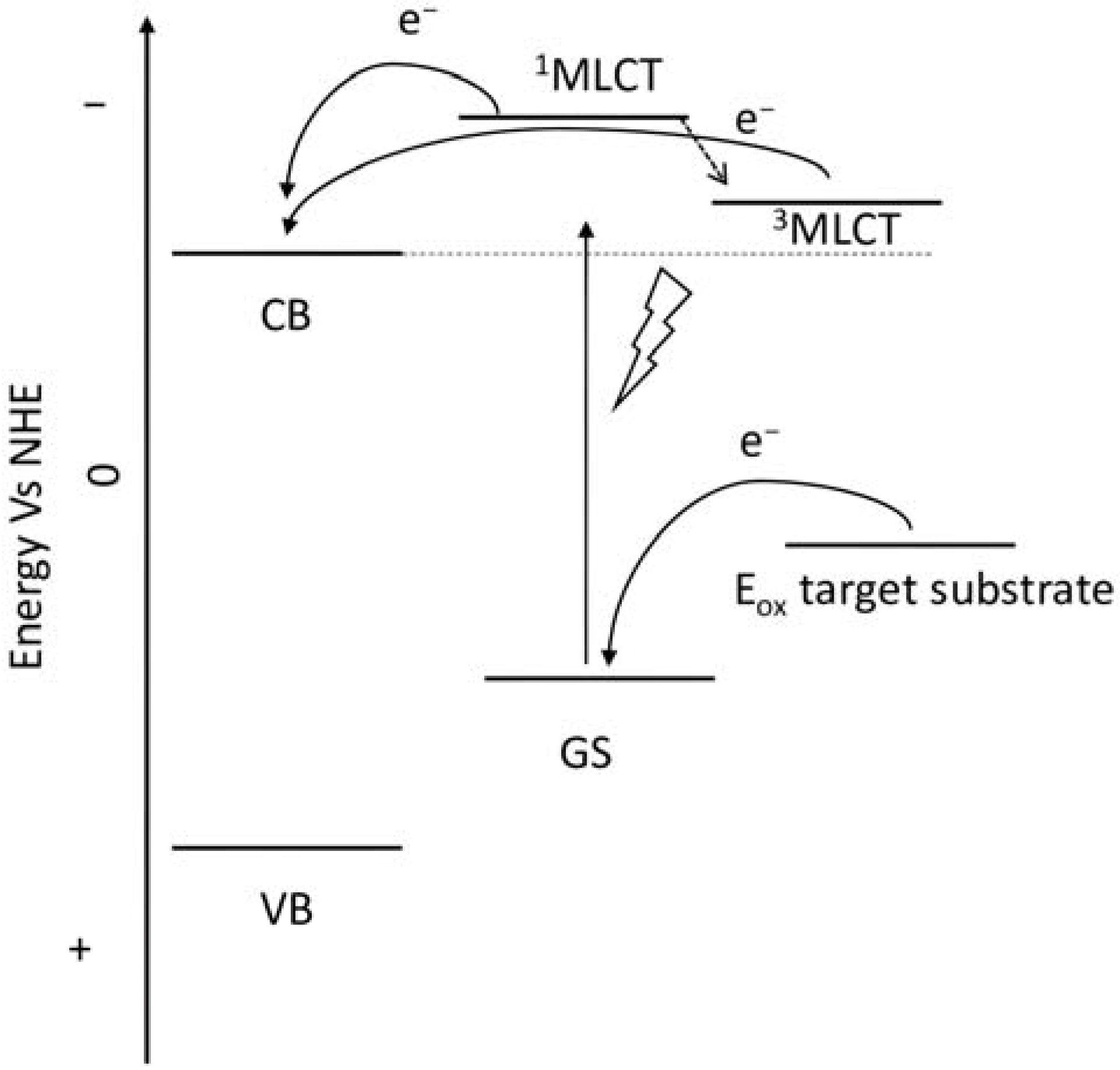
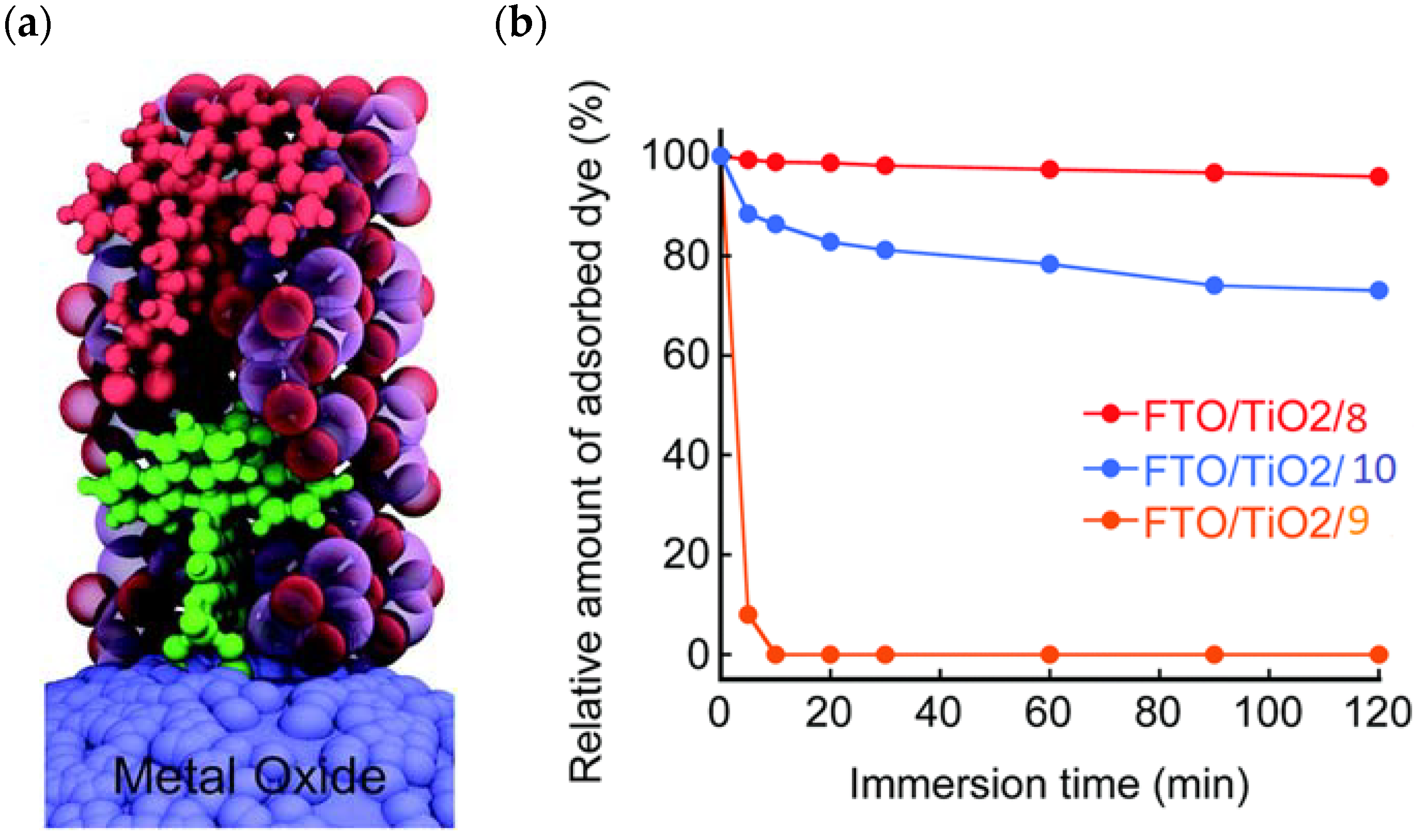
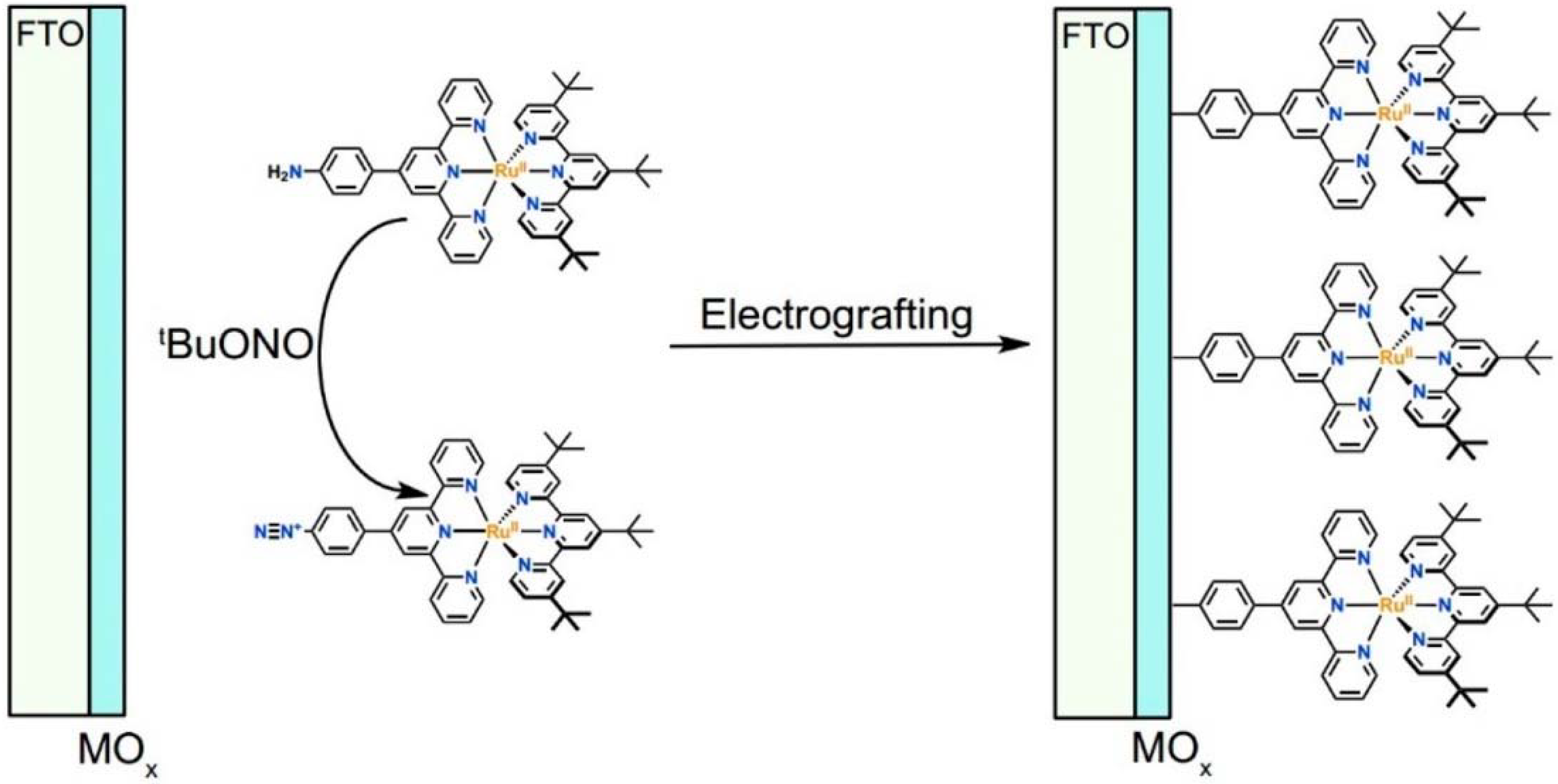
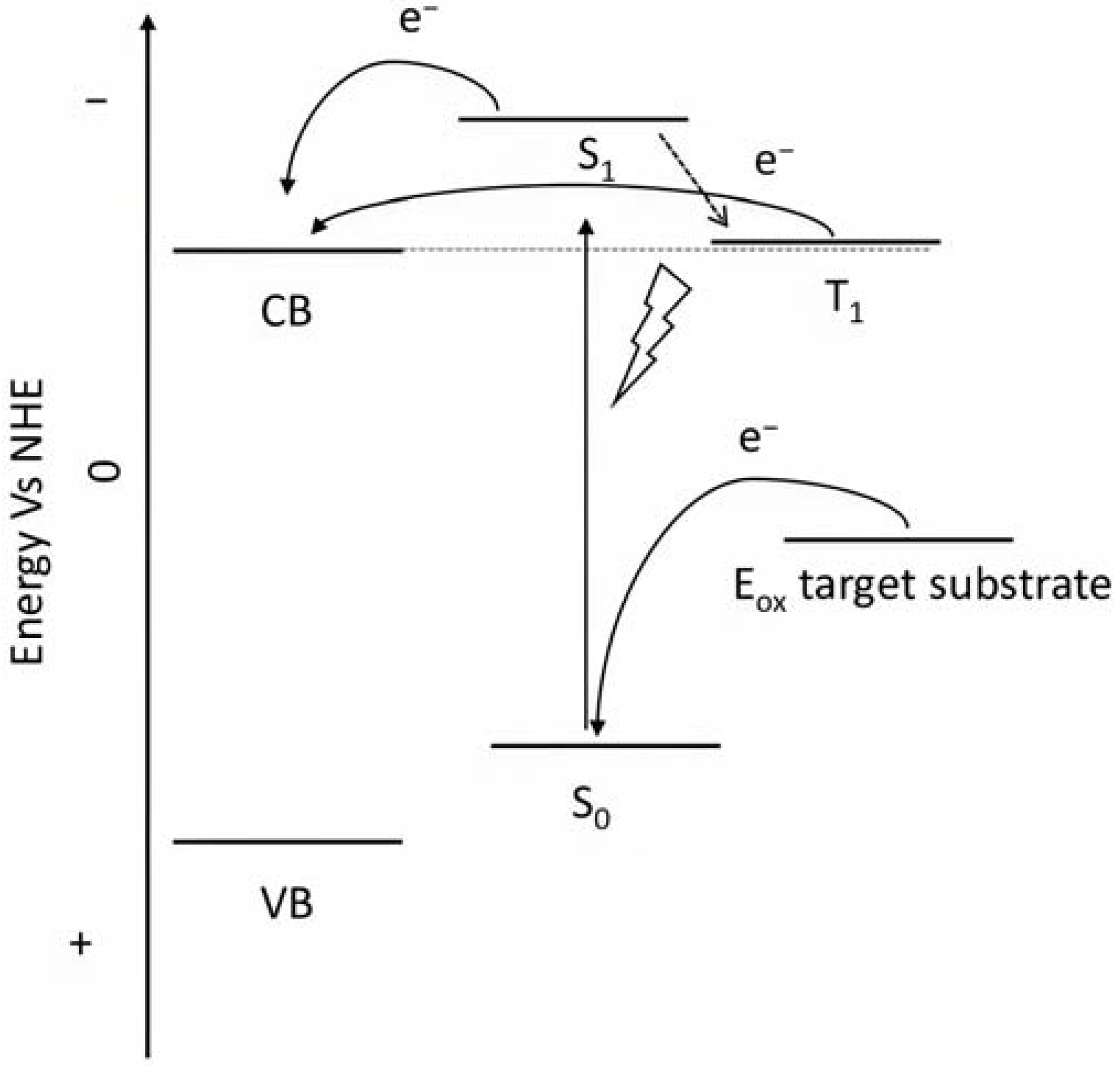
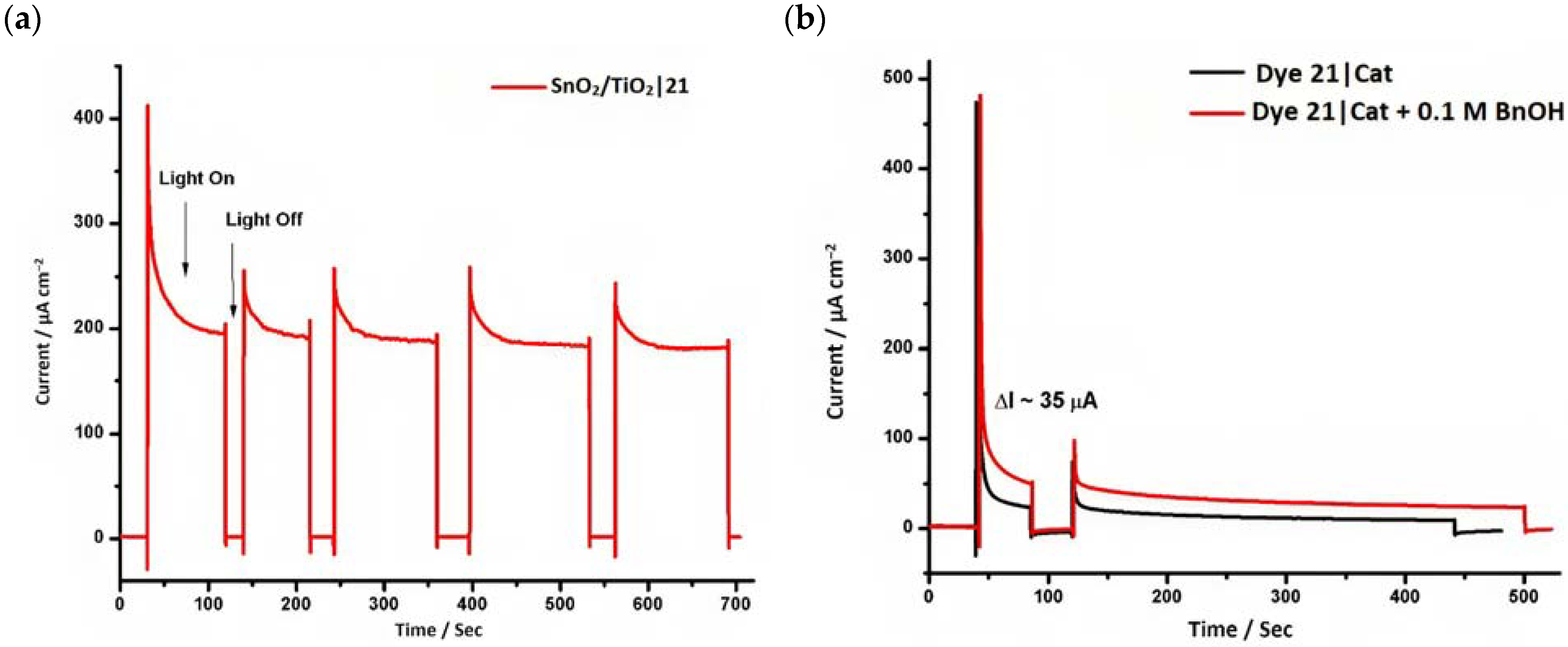
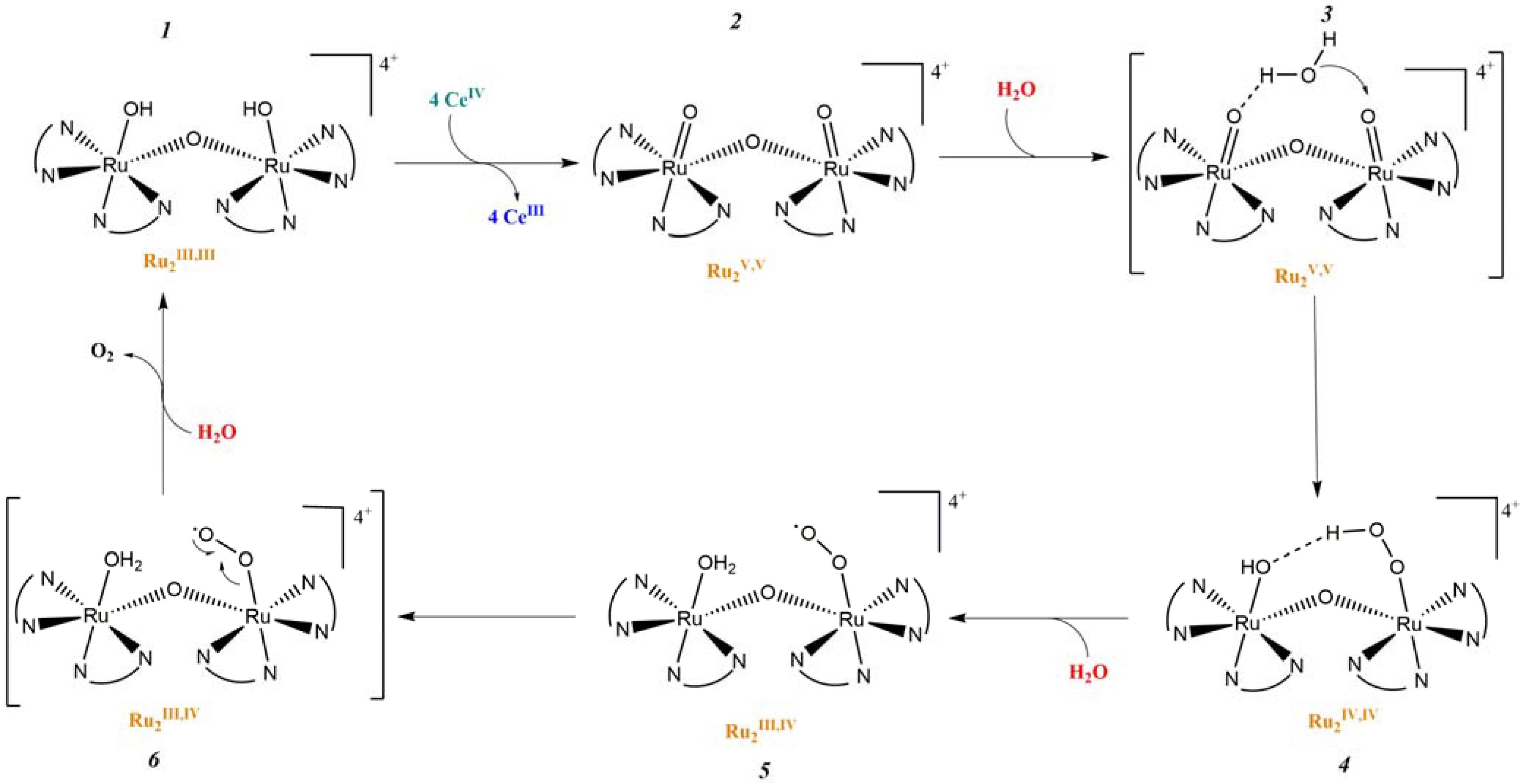
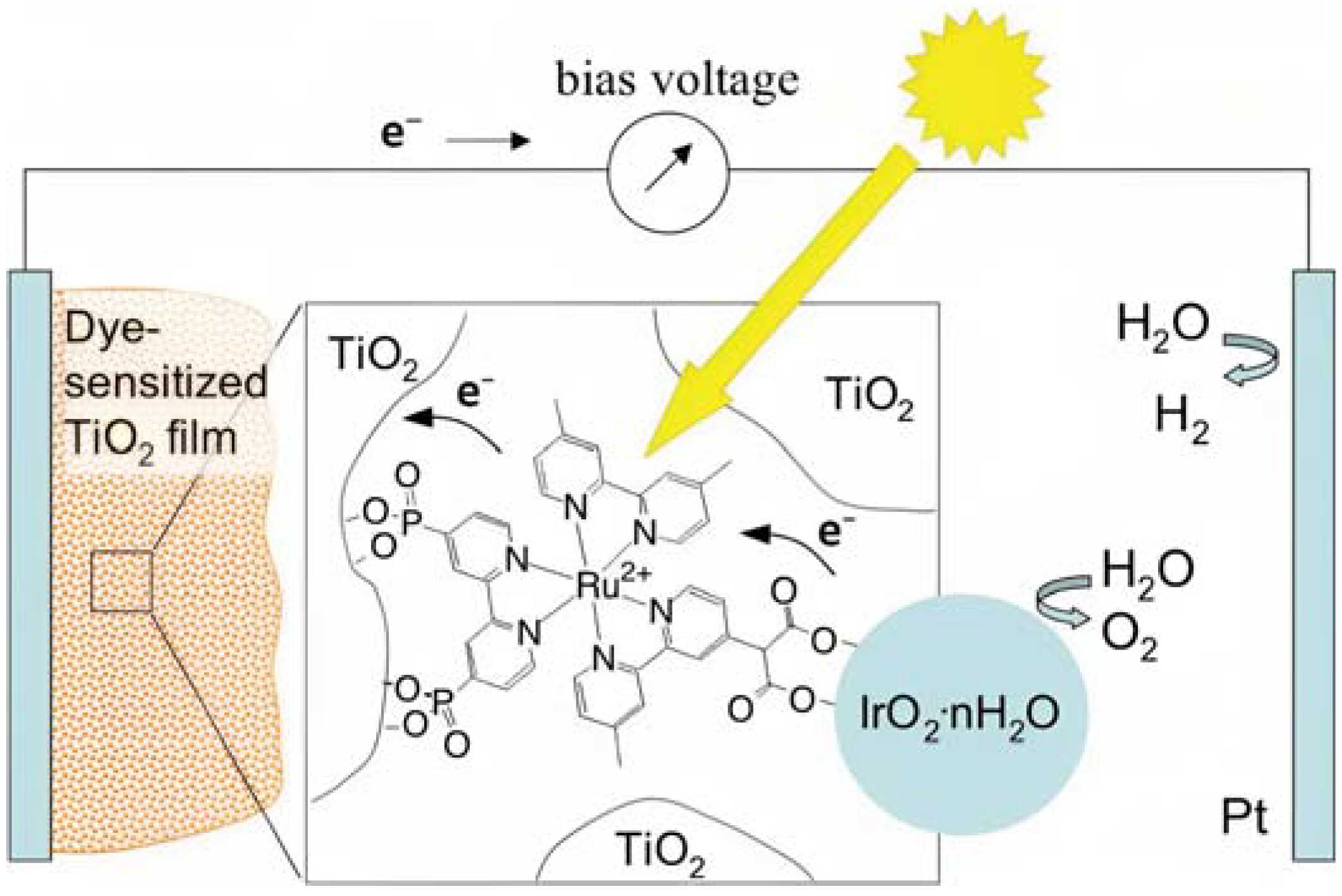
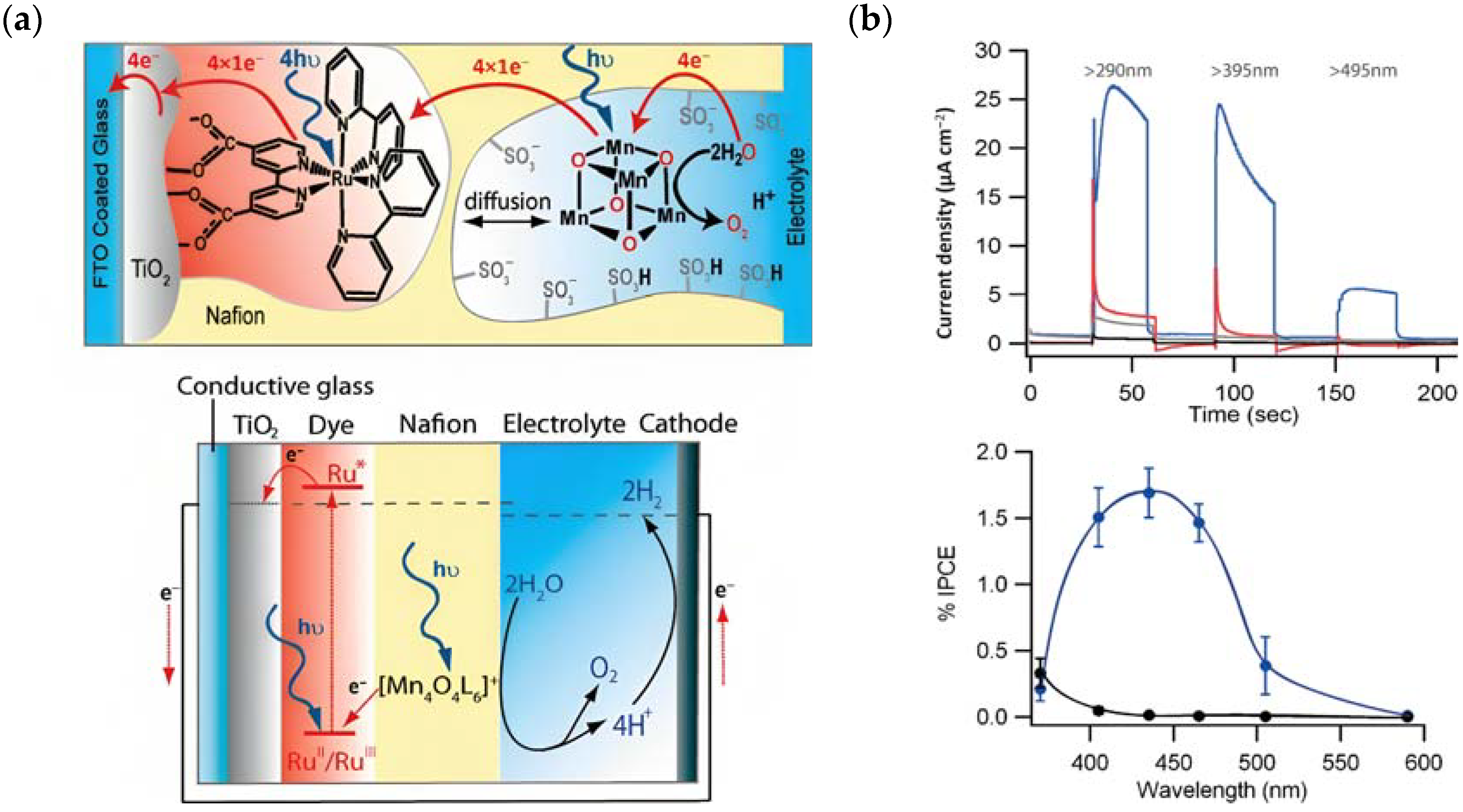
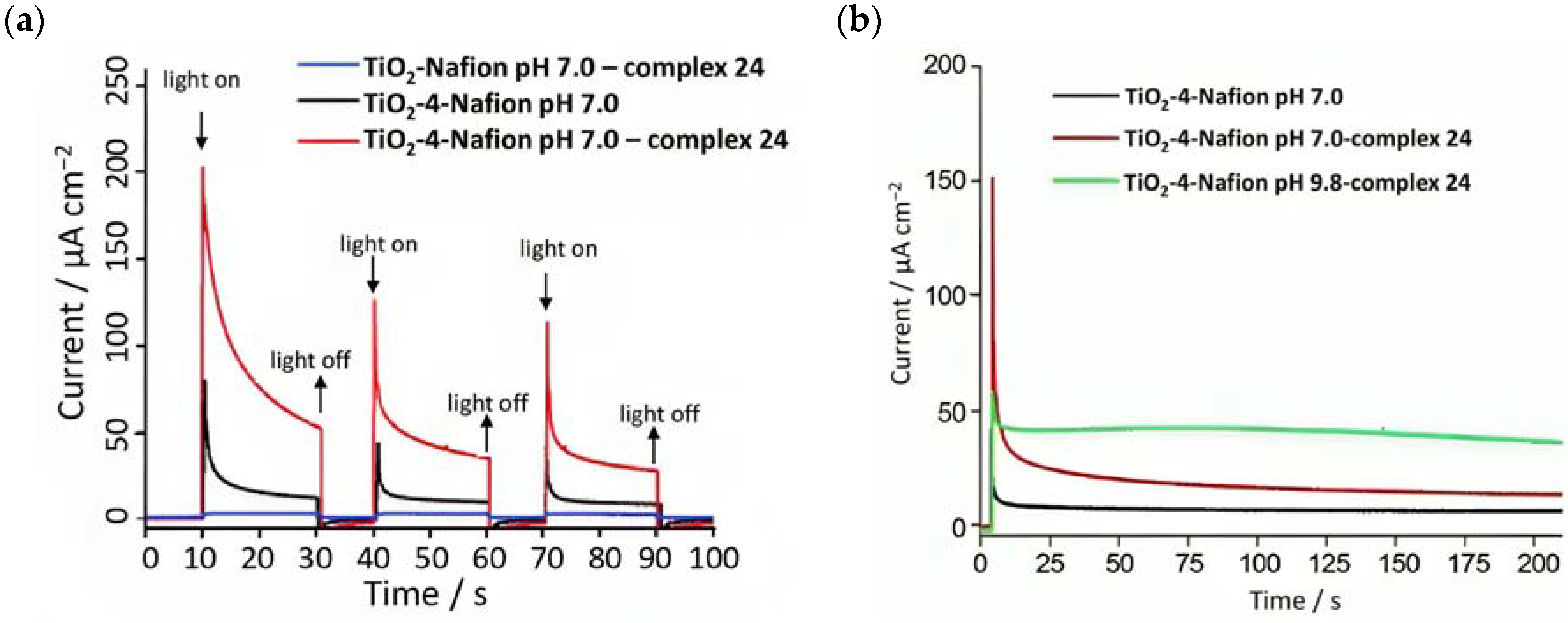
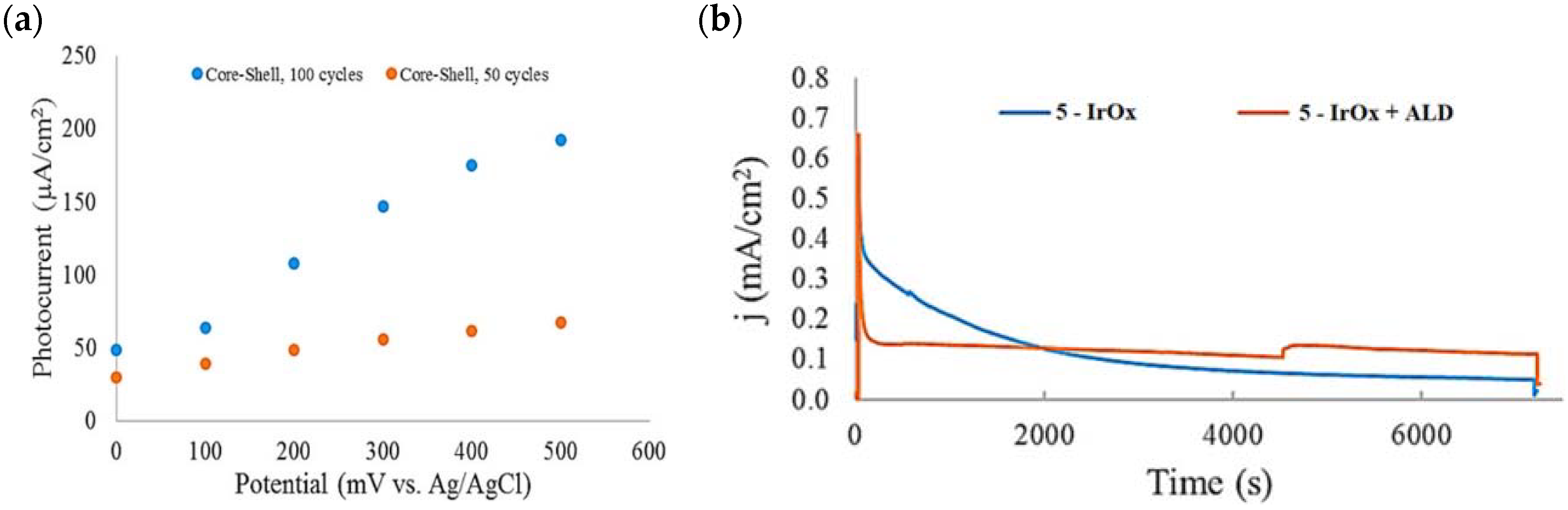
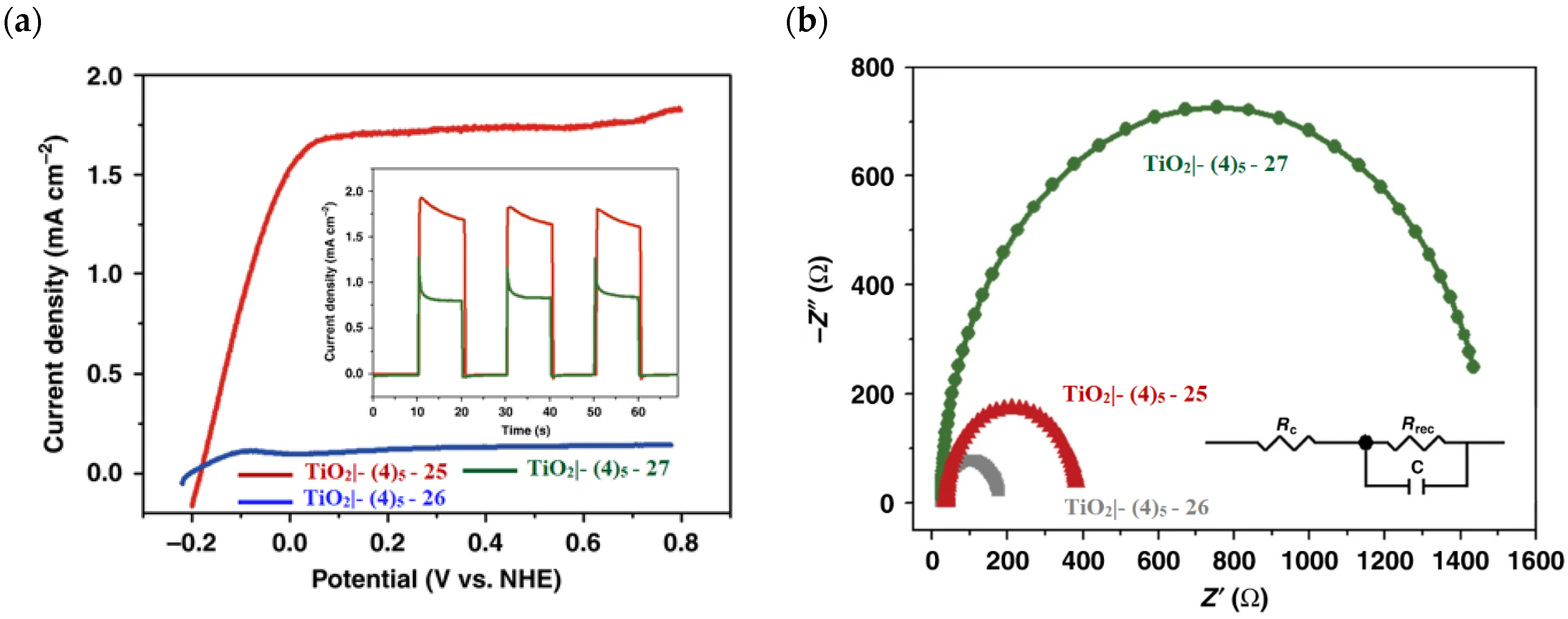
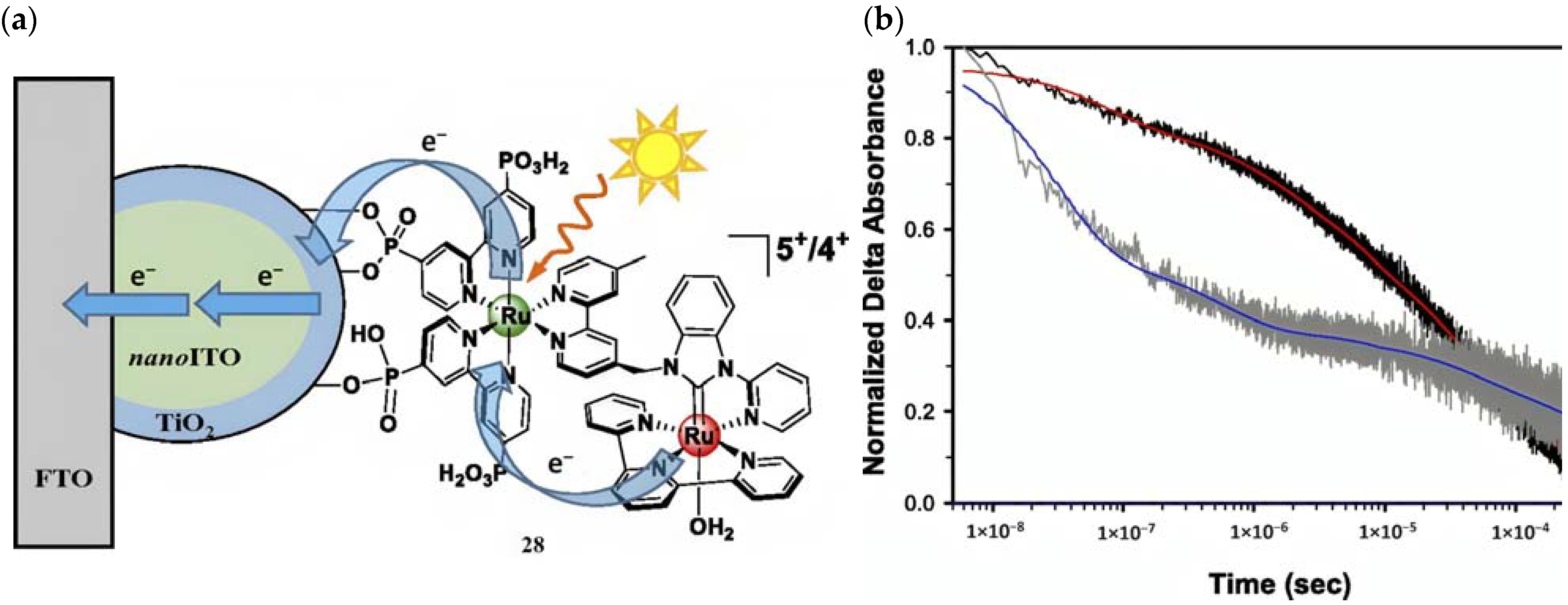
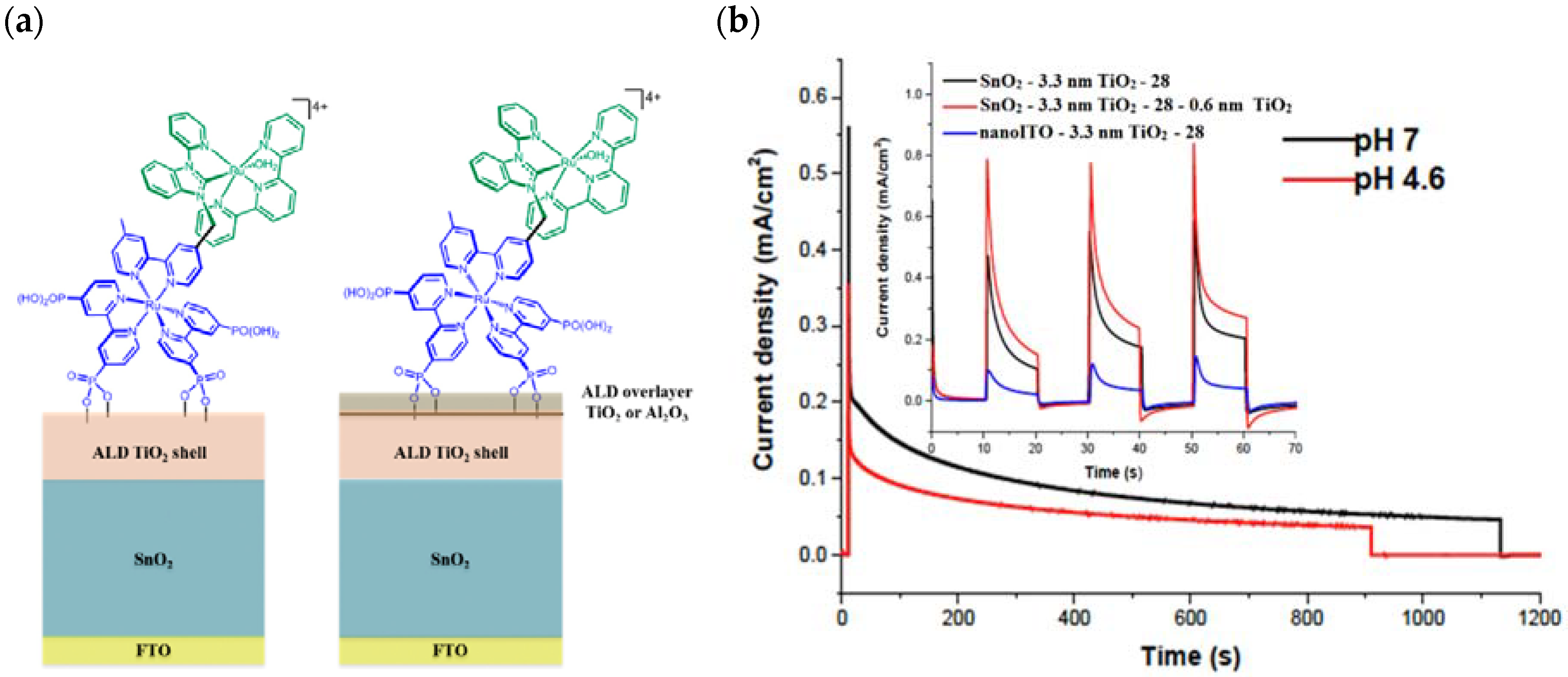
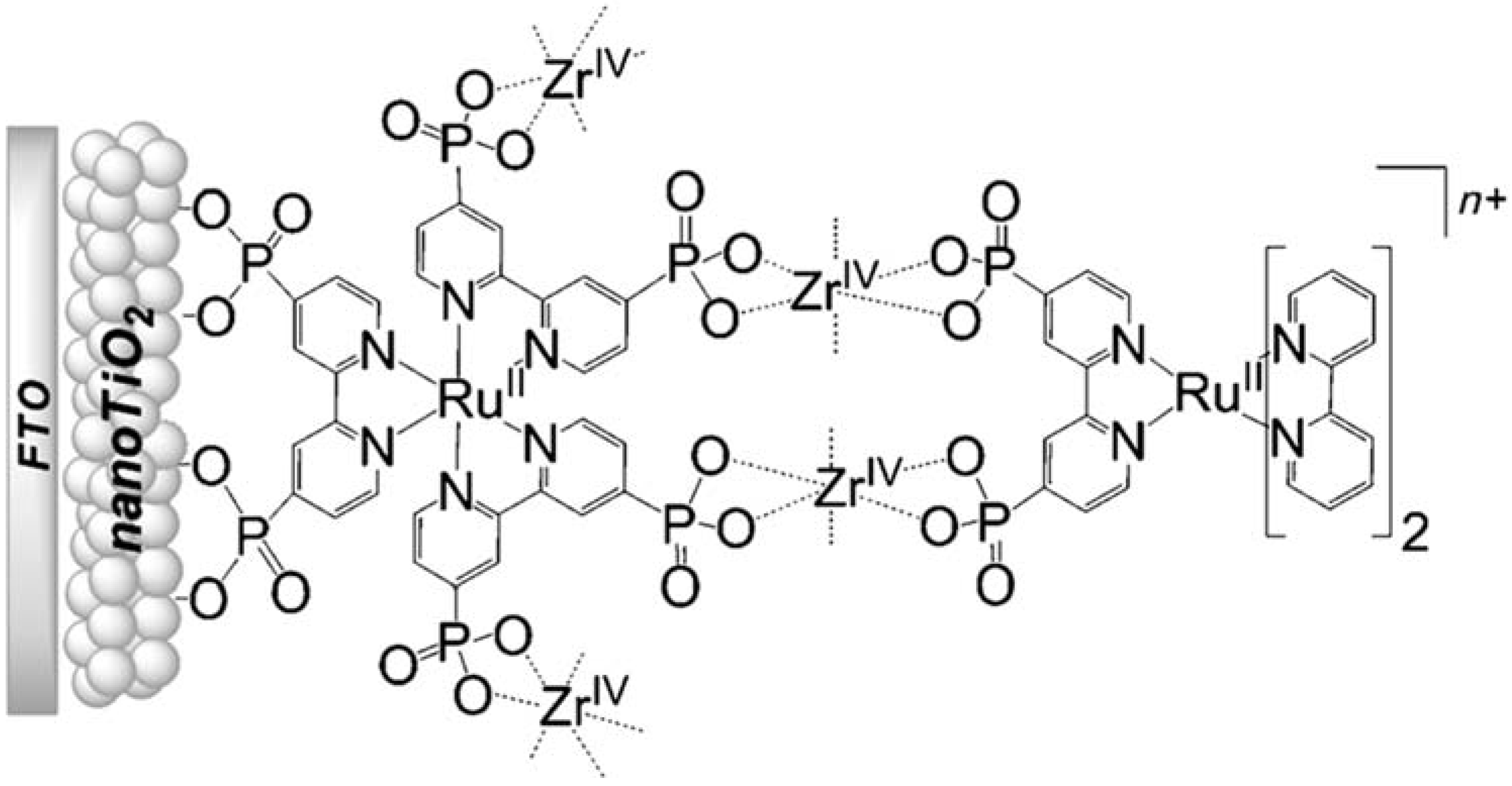
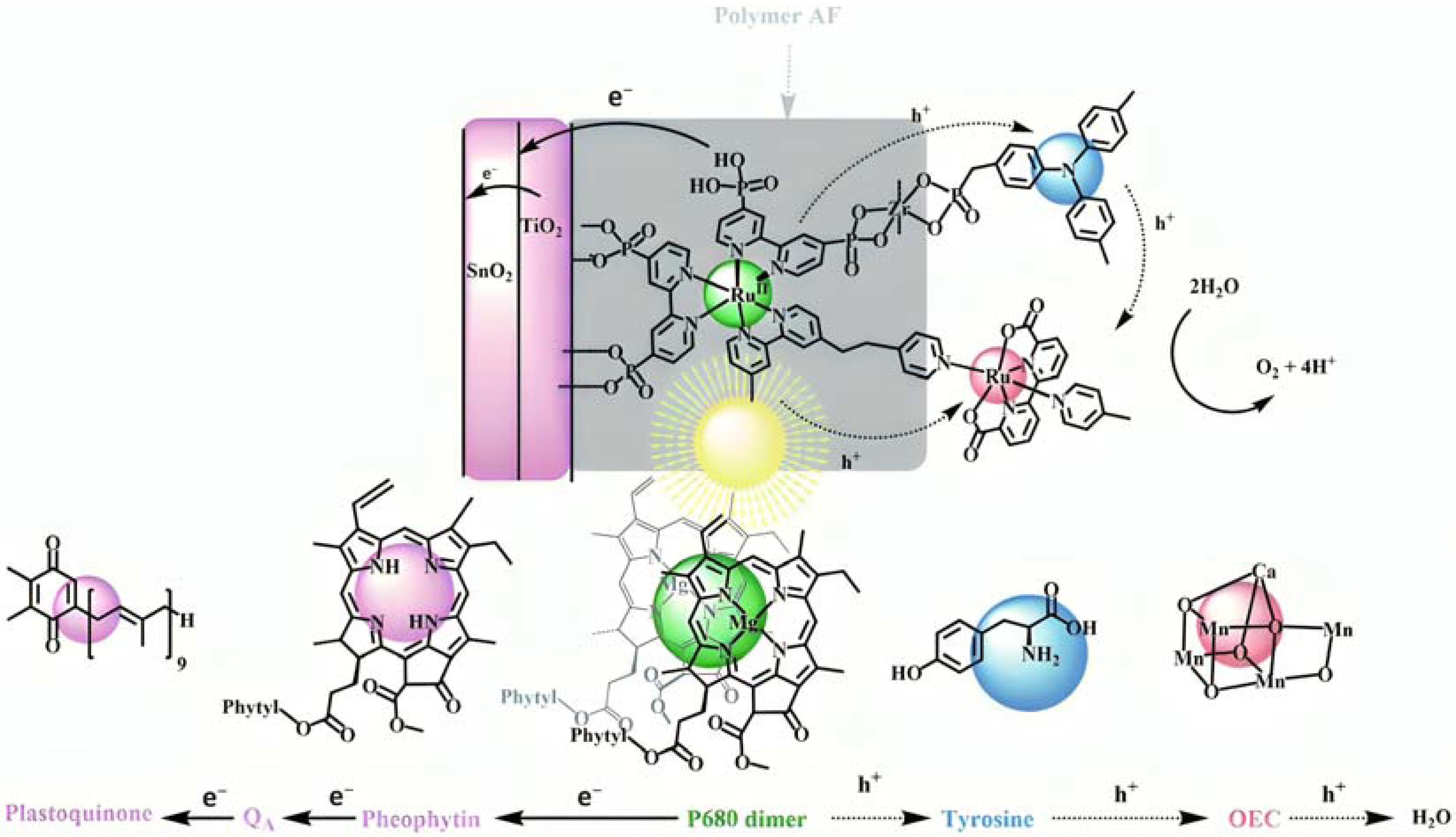
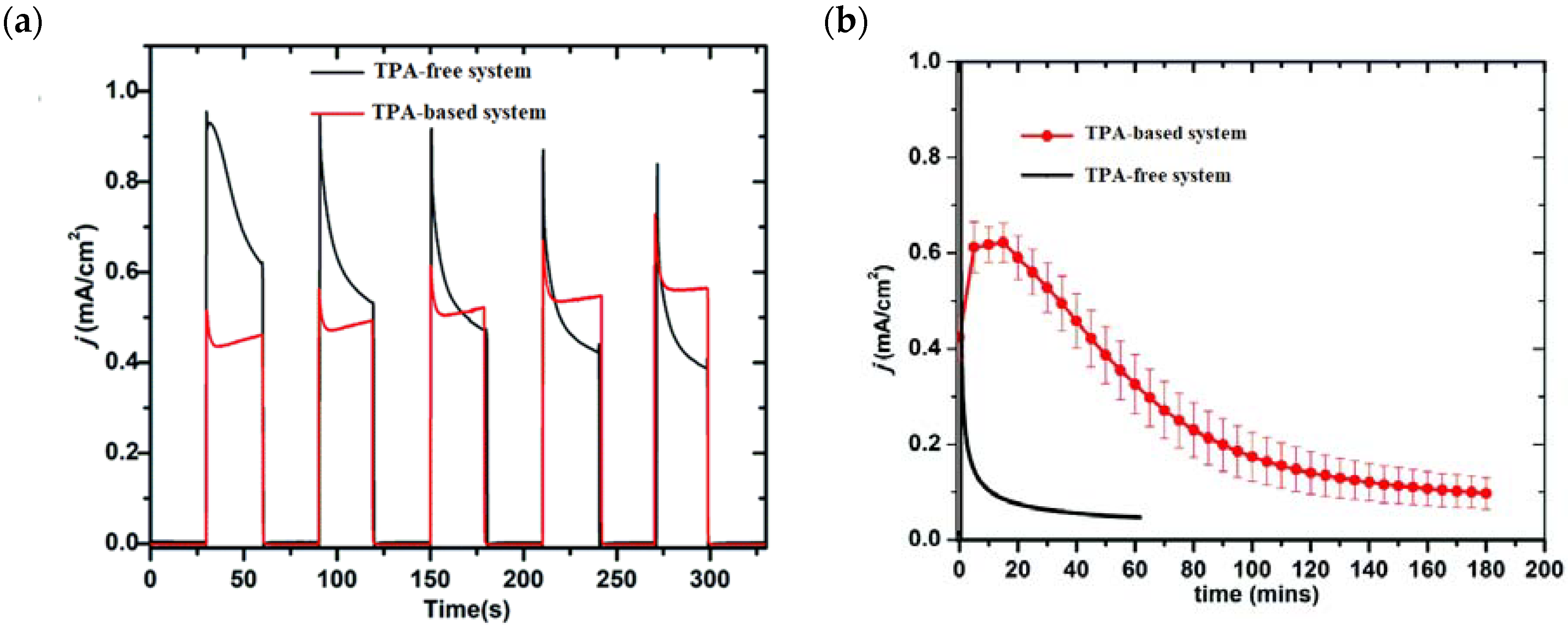
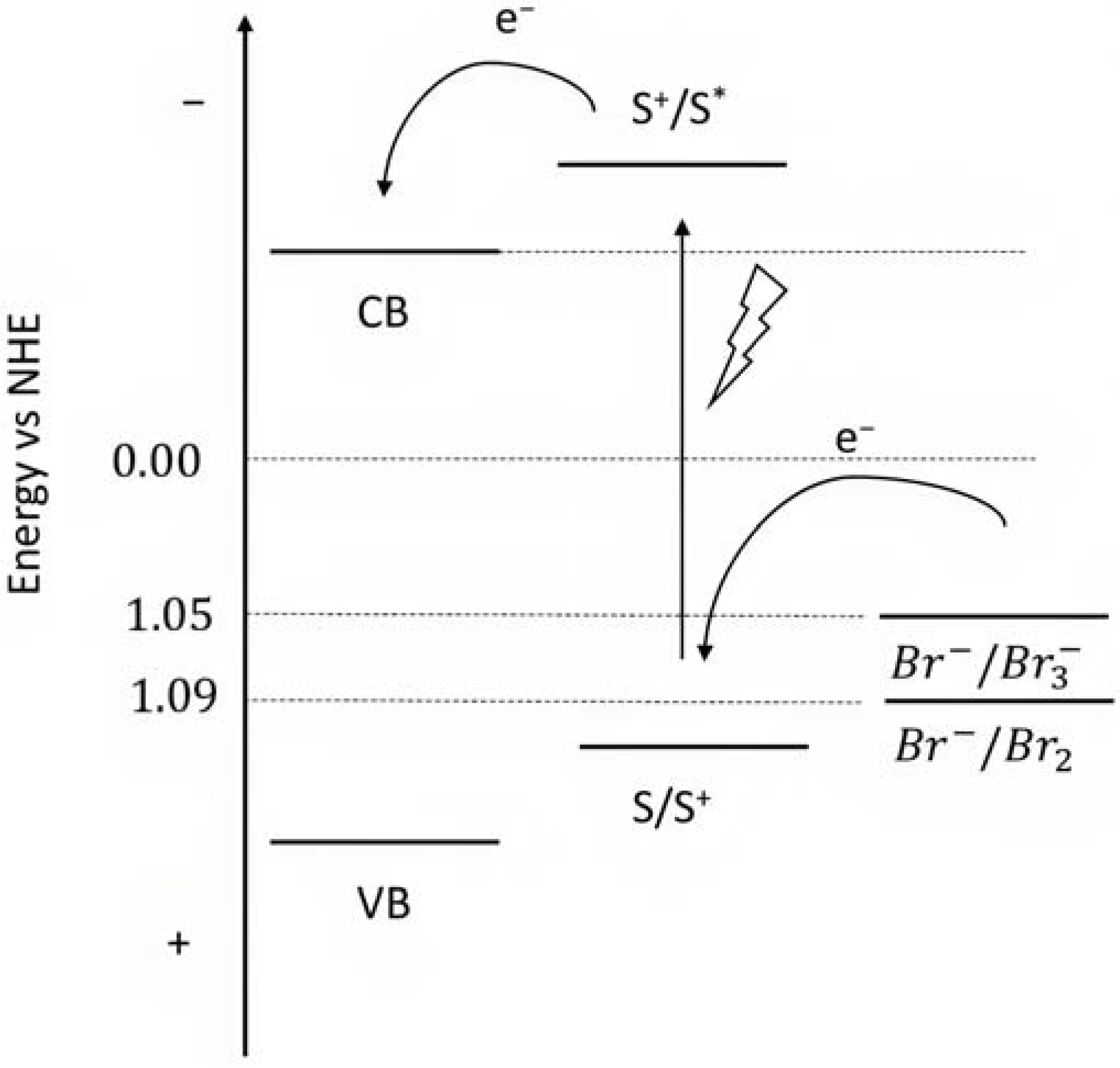
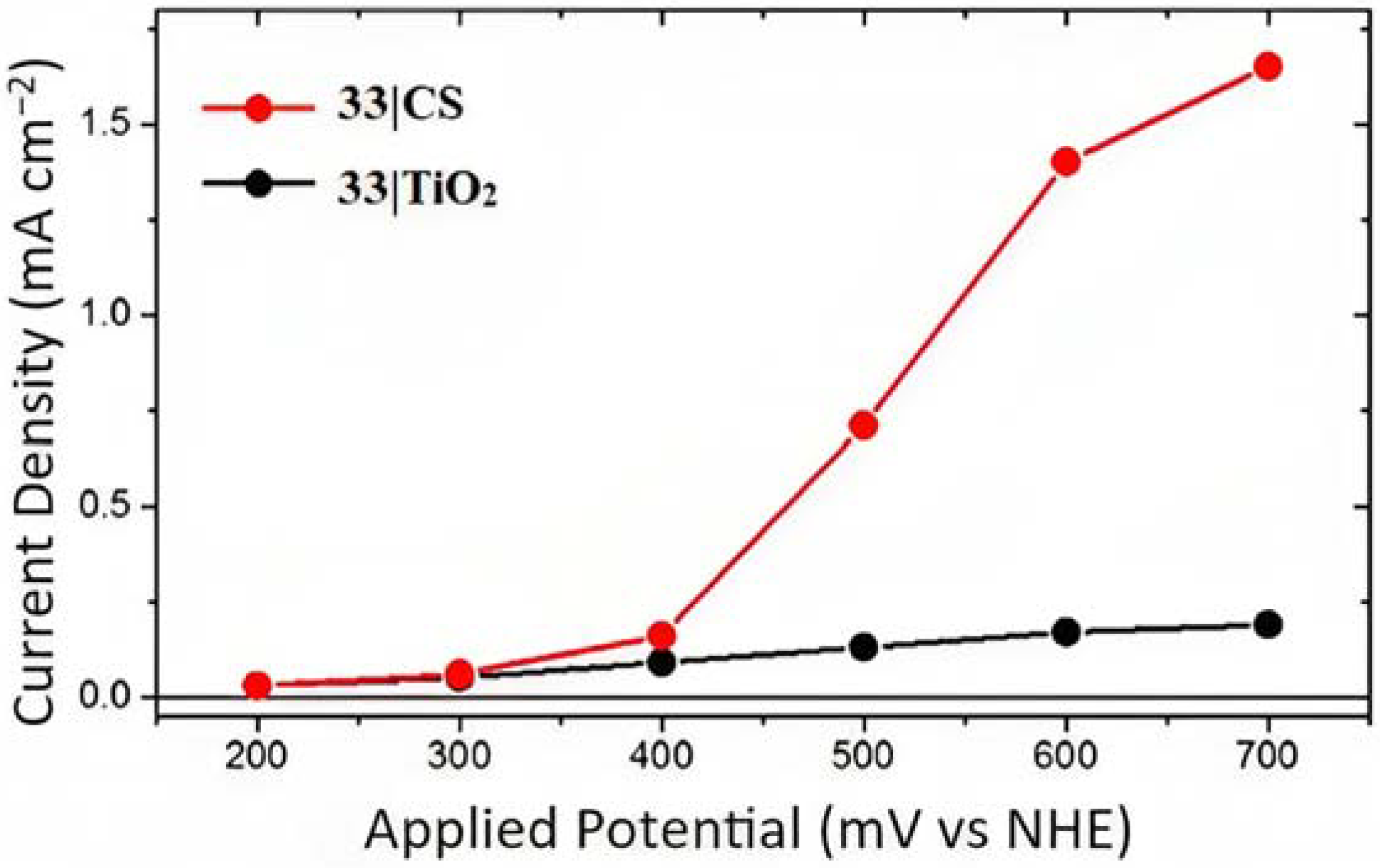
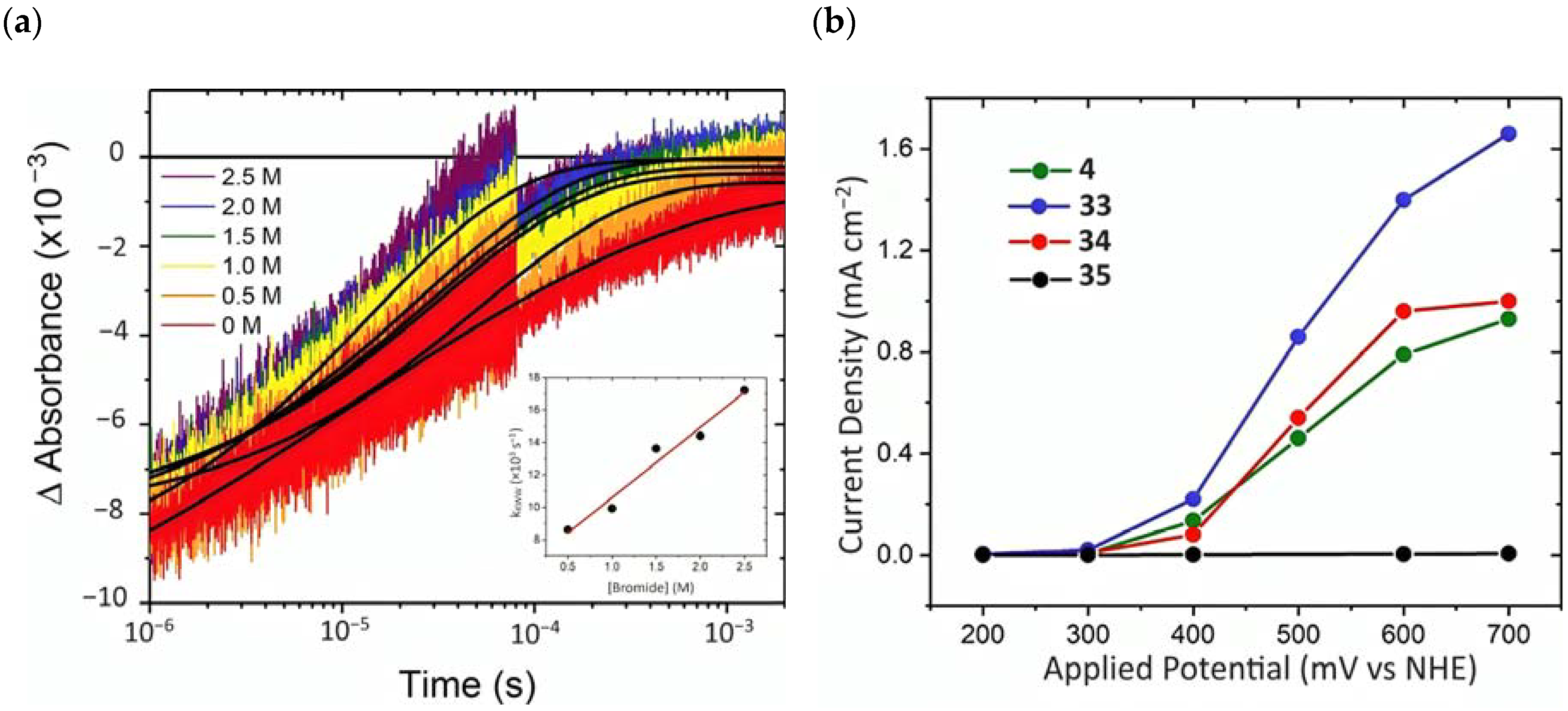
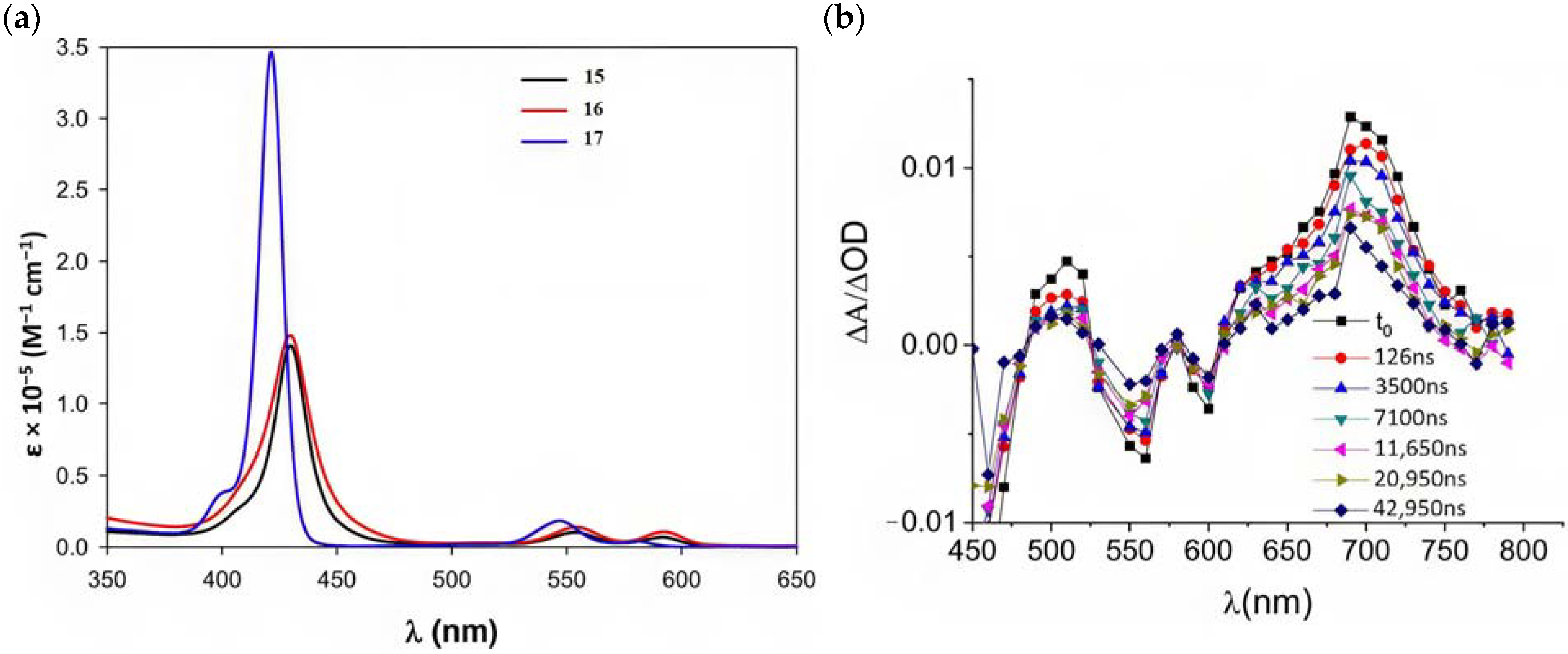
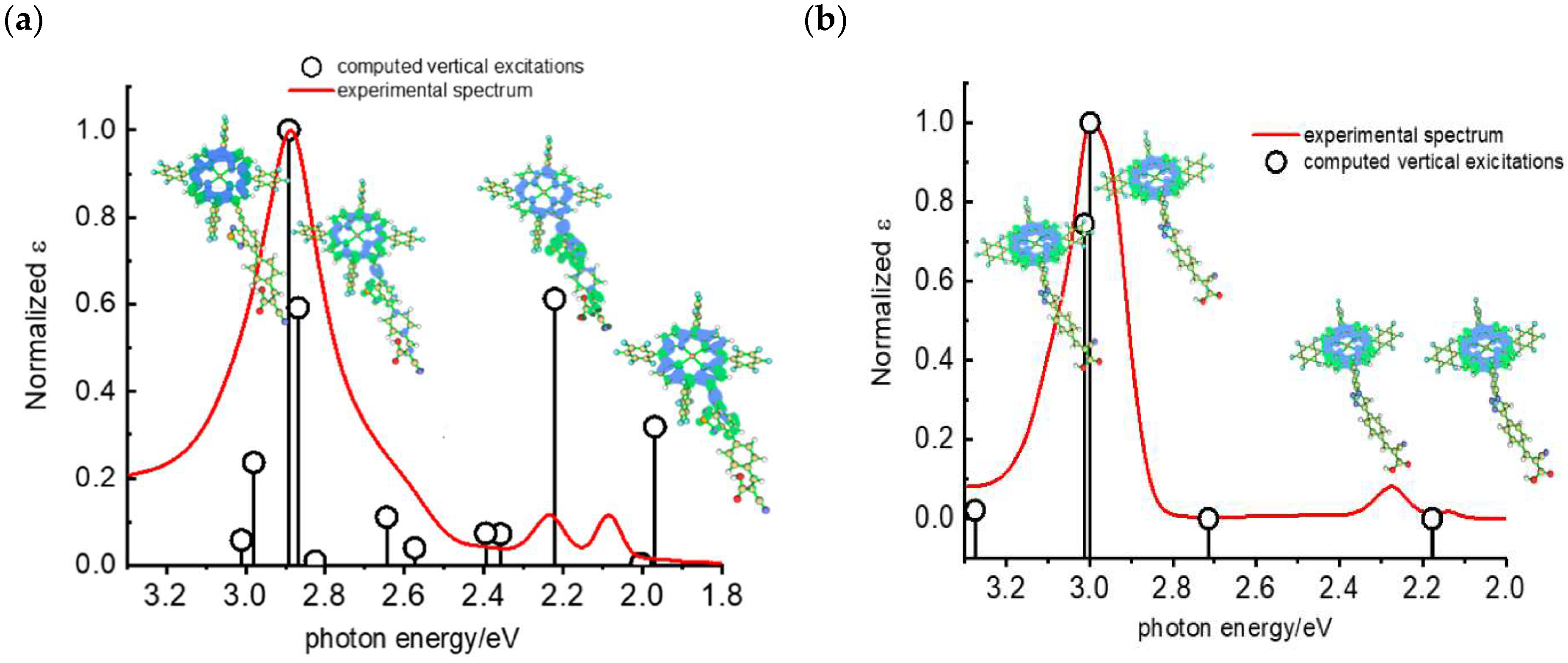
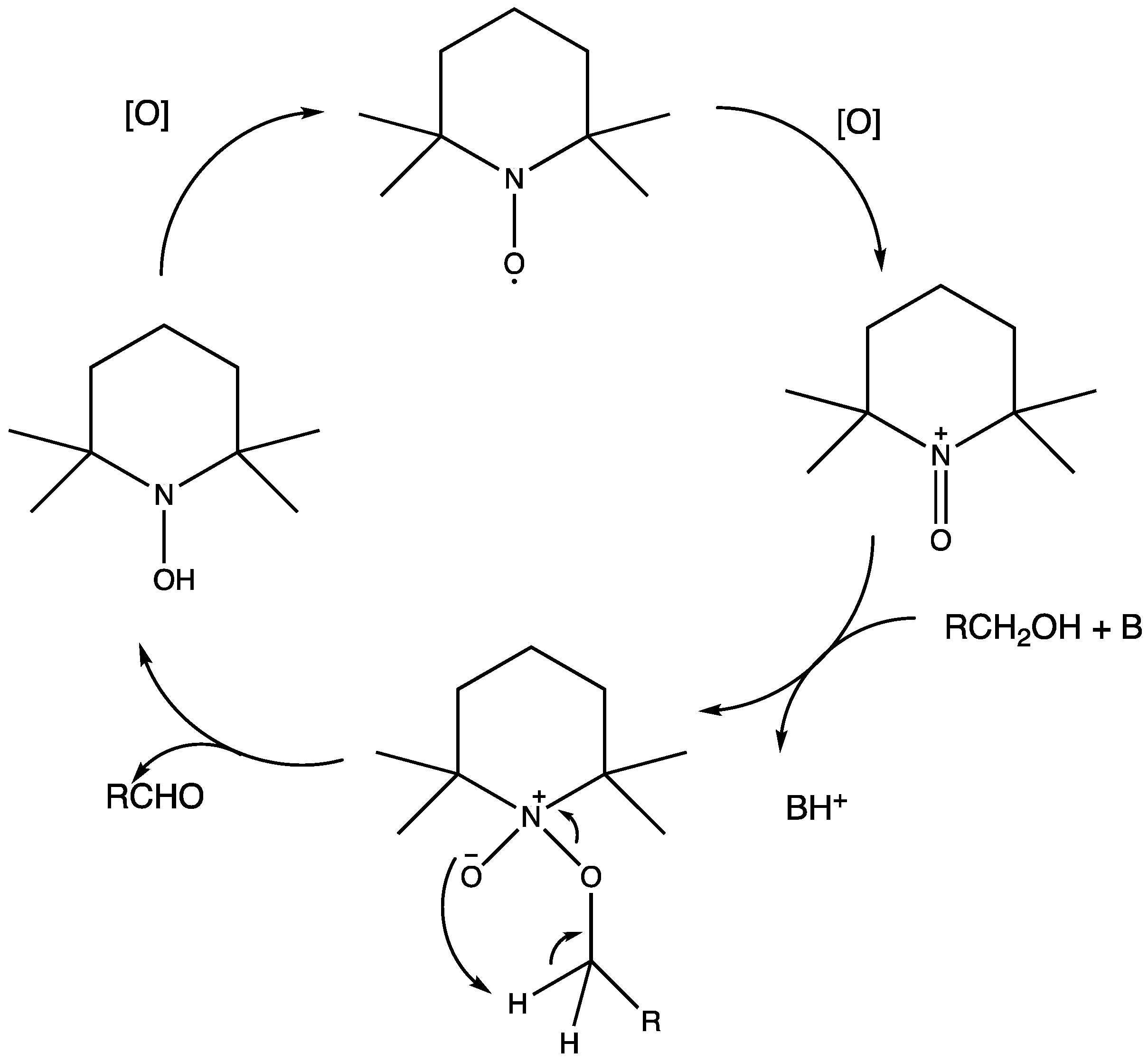
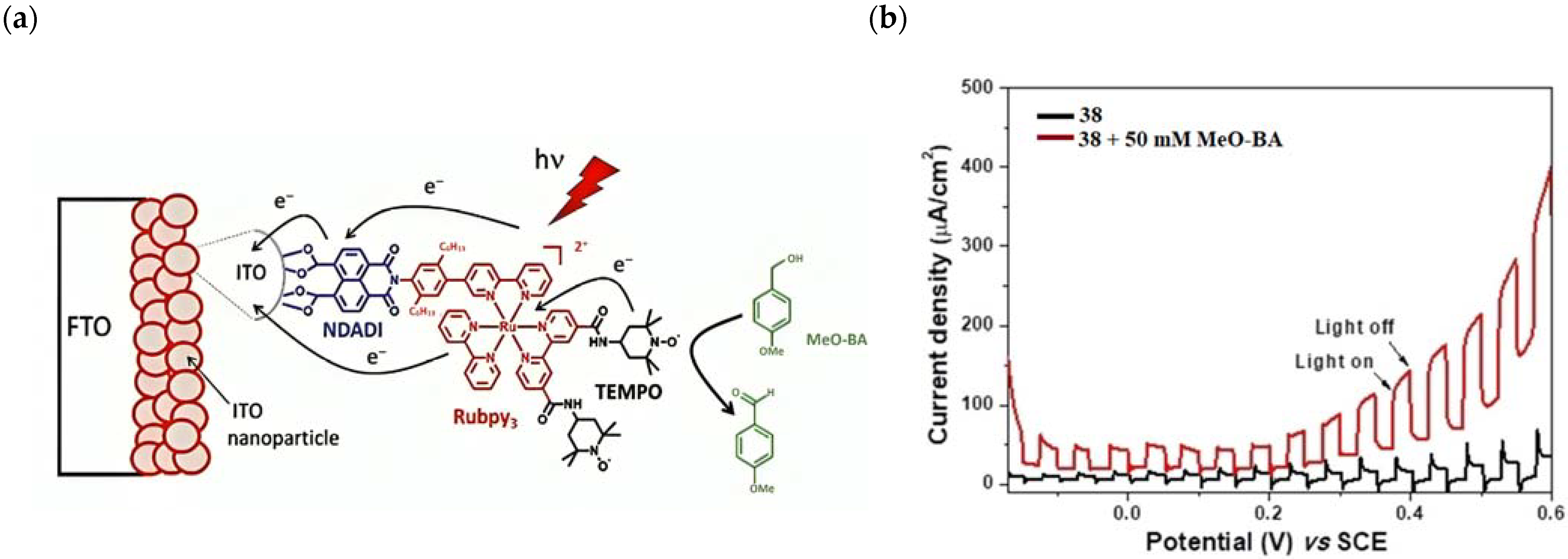
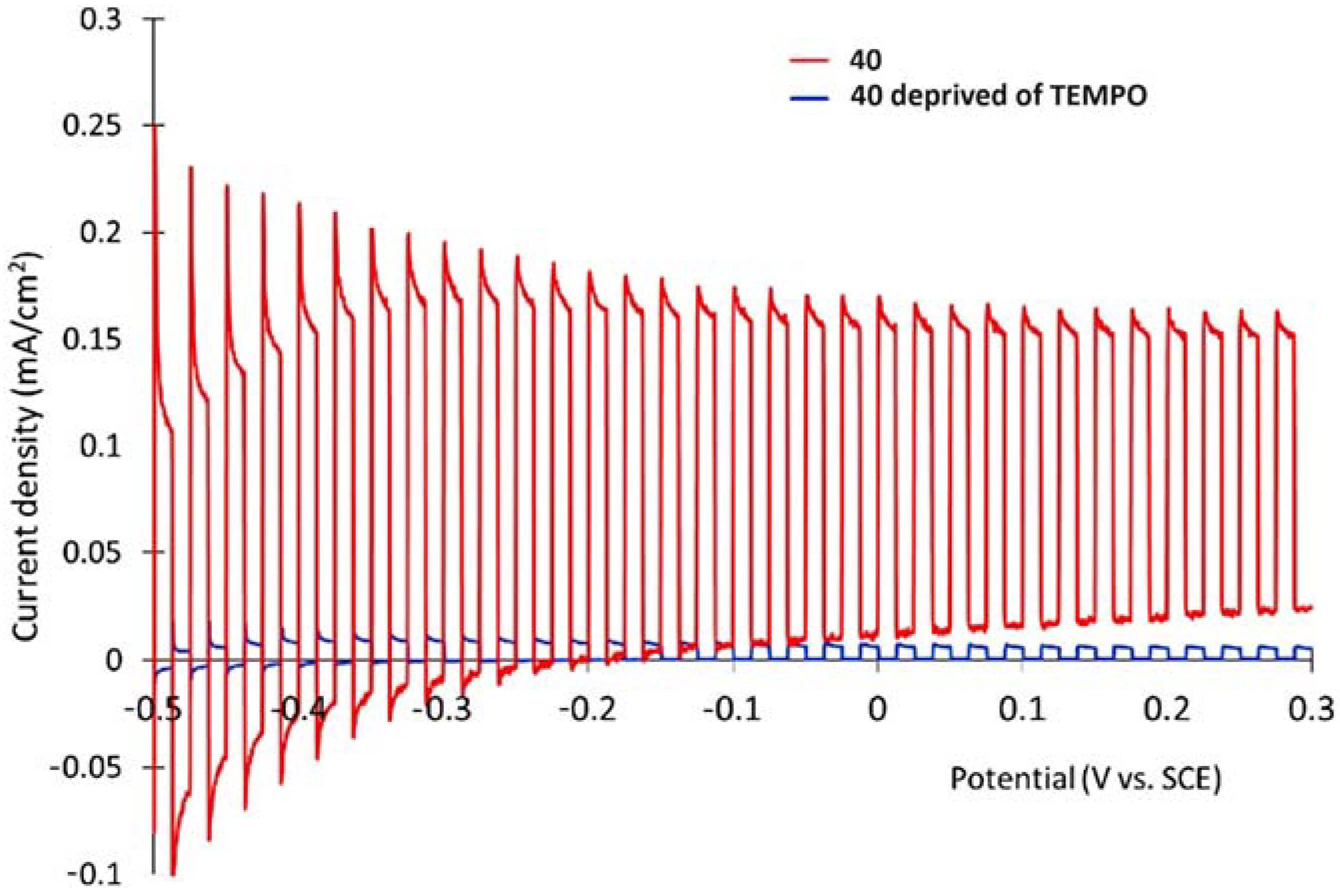
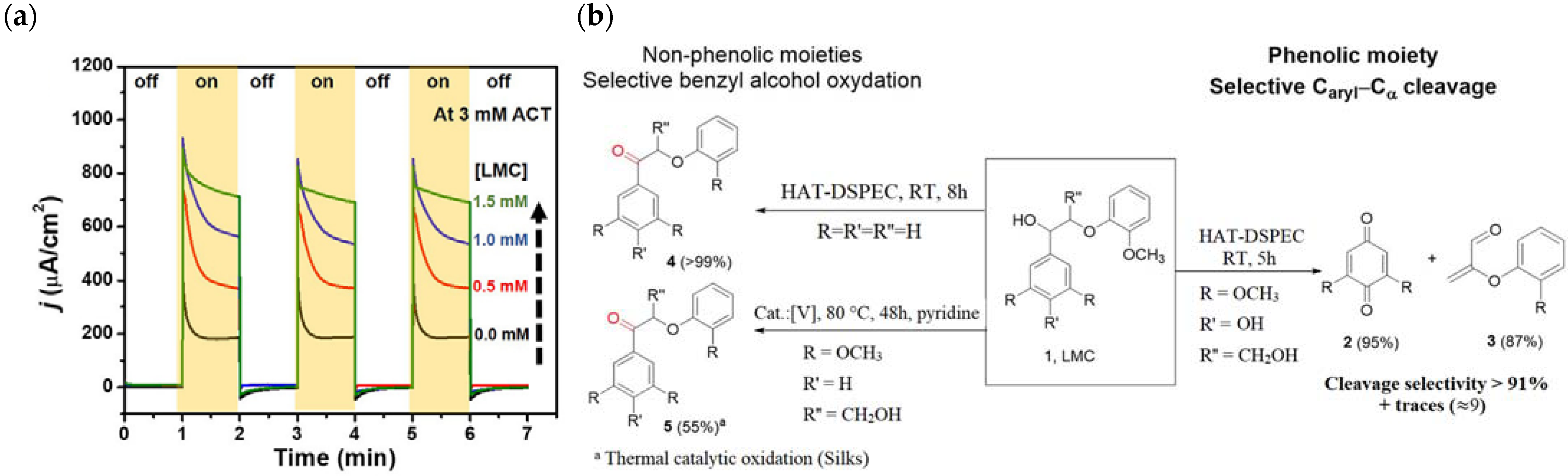
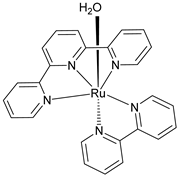
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).