

Exploration of Specific Fluoroquinolone Interaction with SARS-CoV-2 Main Protease (Mpro) to Battle COVID-19: DFT, Molecular Docking, ADME and Cardiotoxicity Studies




Abstract
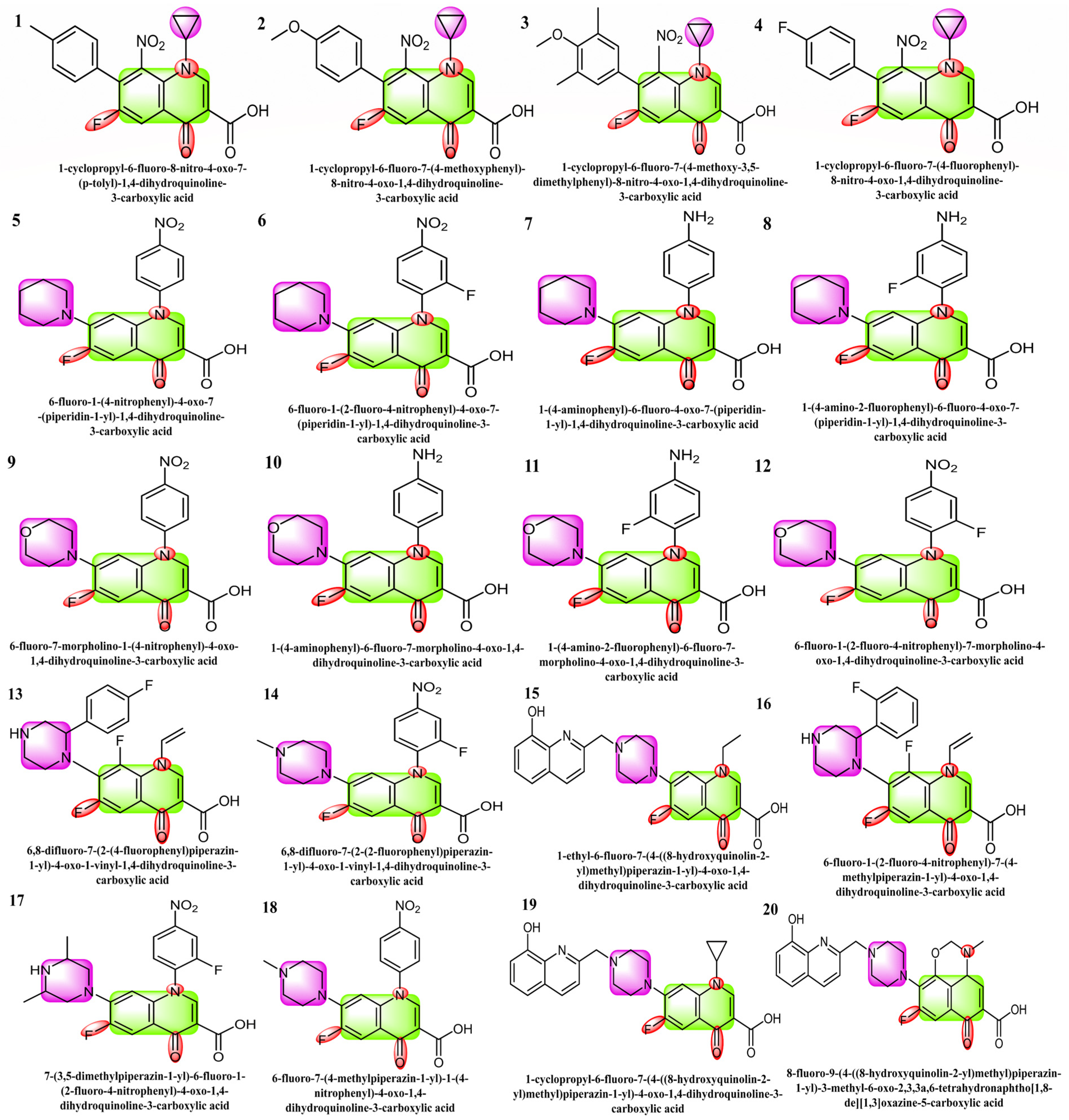
1. Introduction
2. Results and Discussions
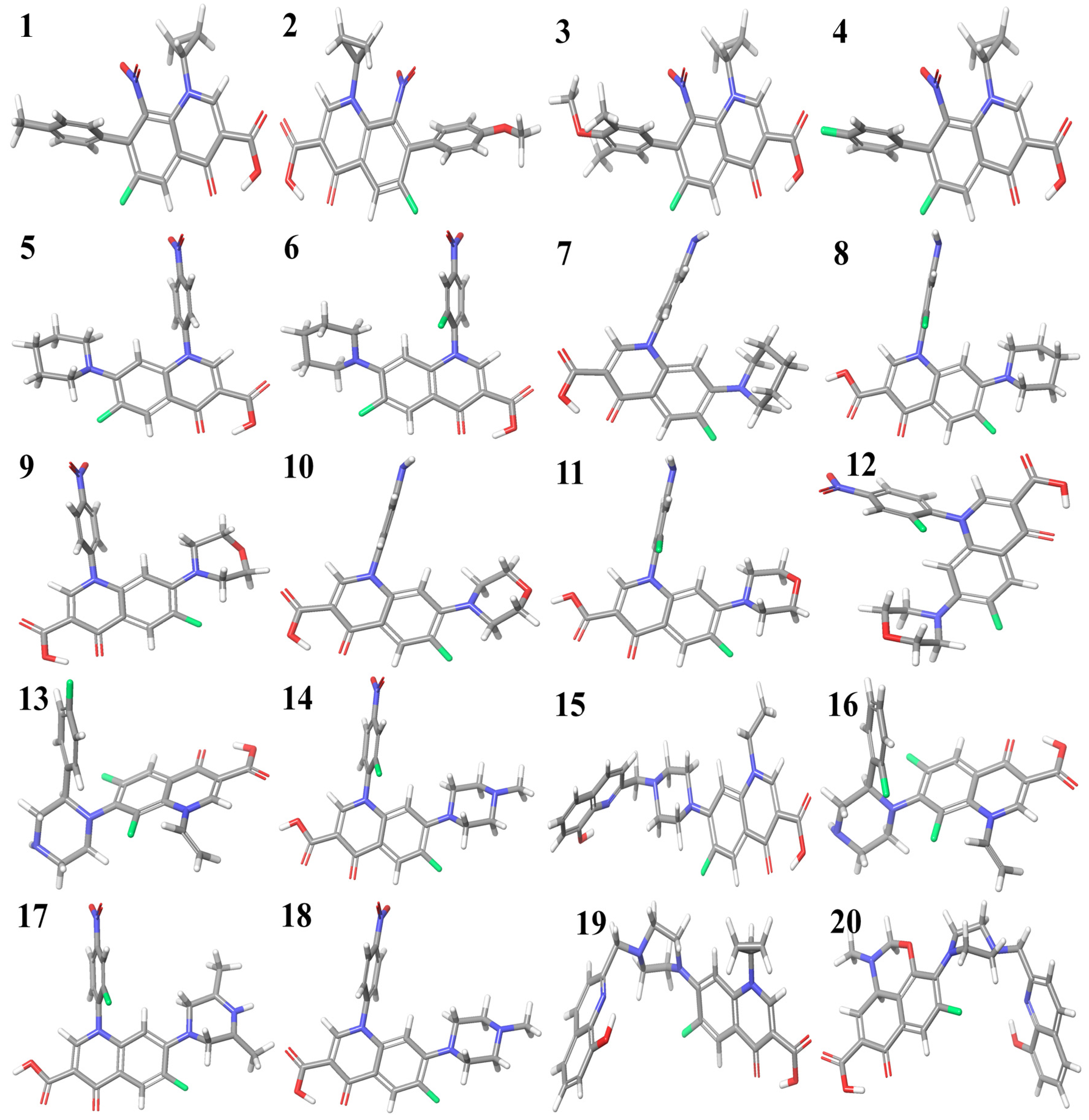
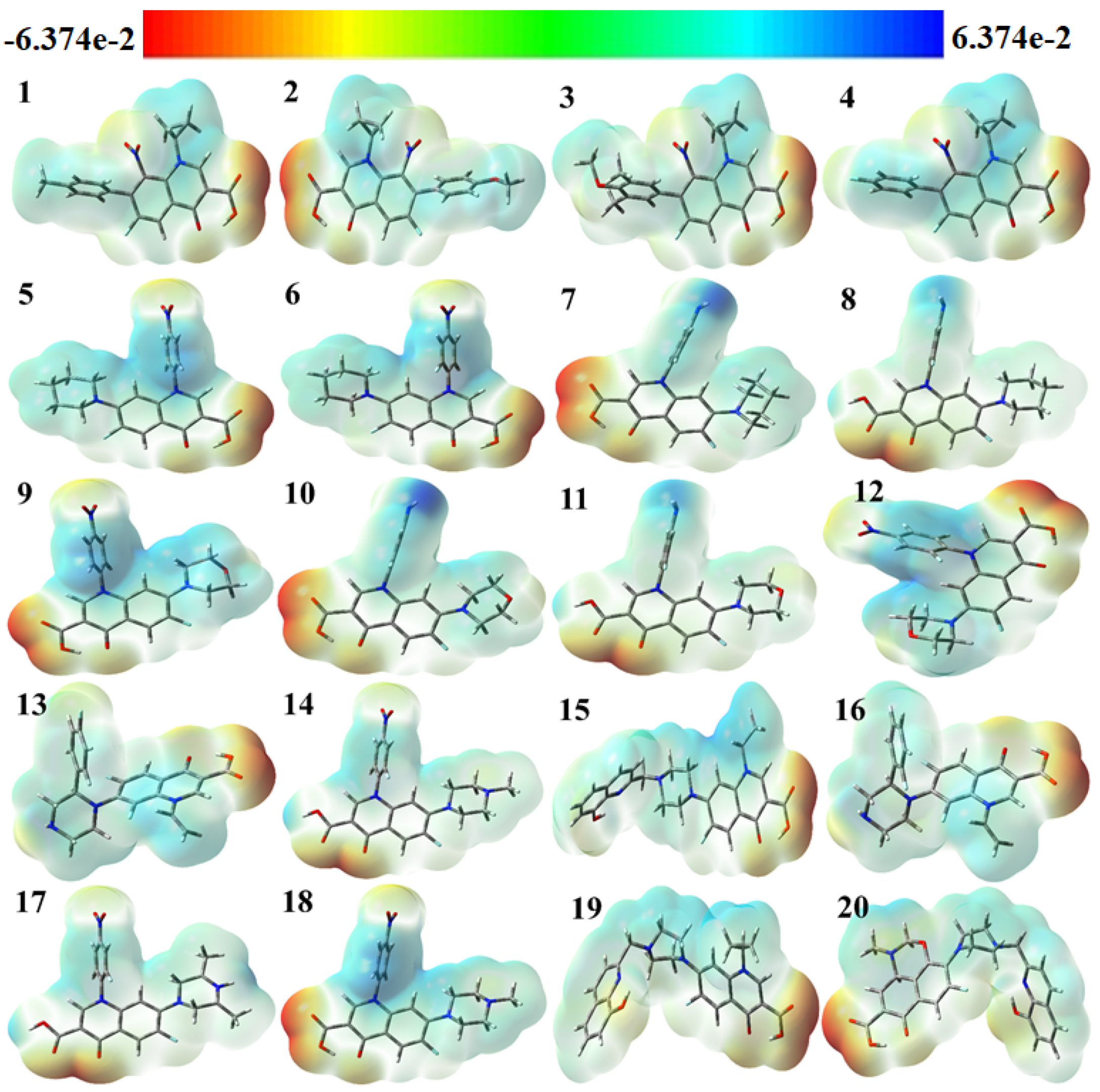
2.1. DFT Study
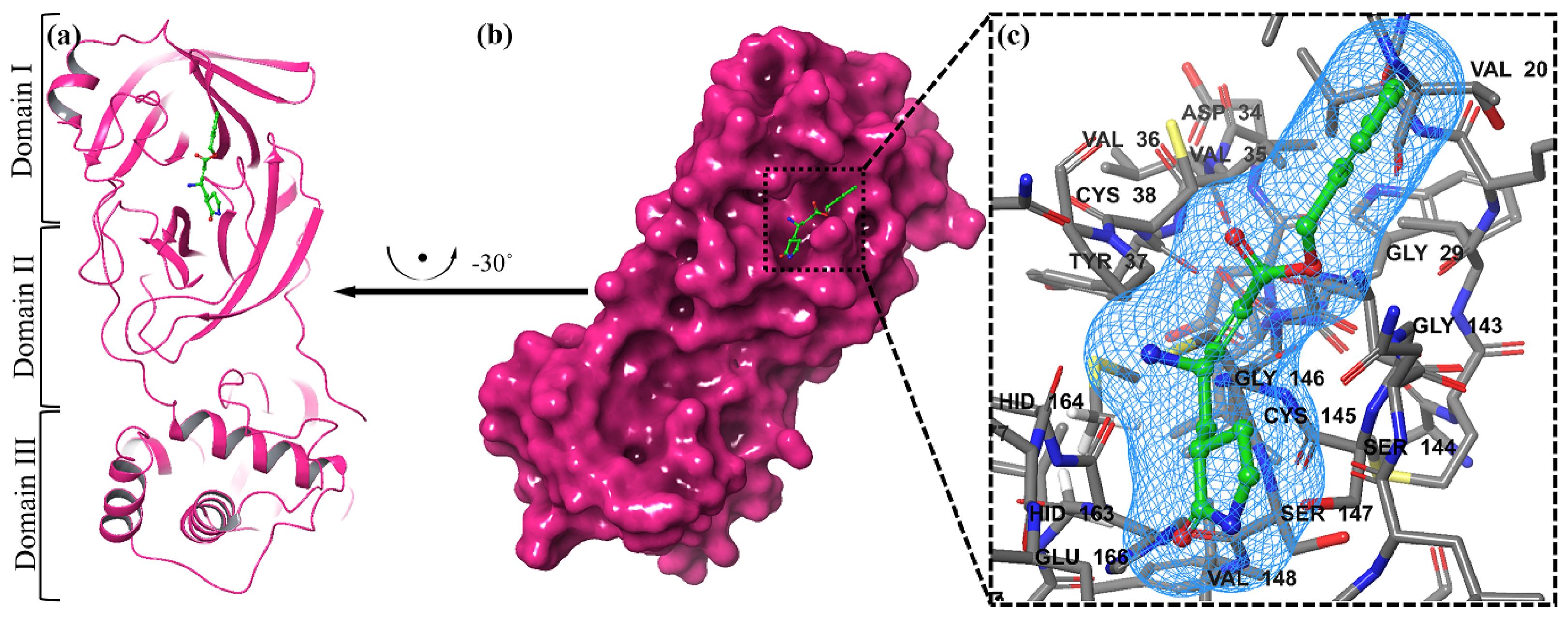
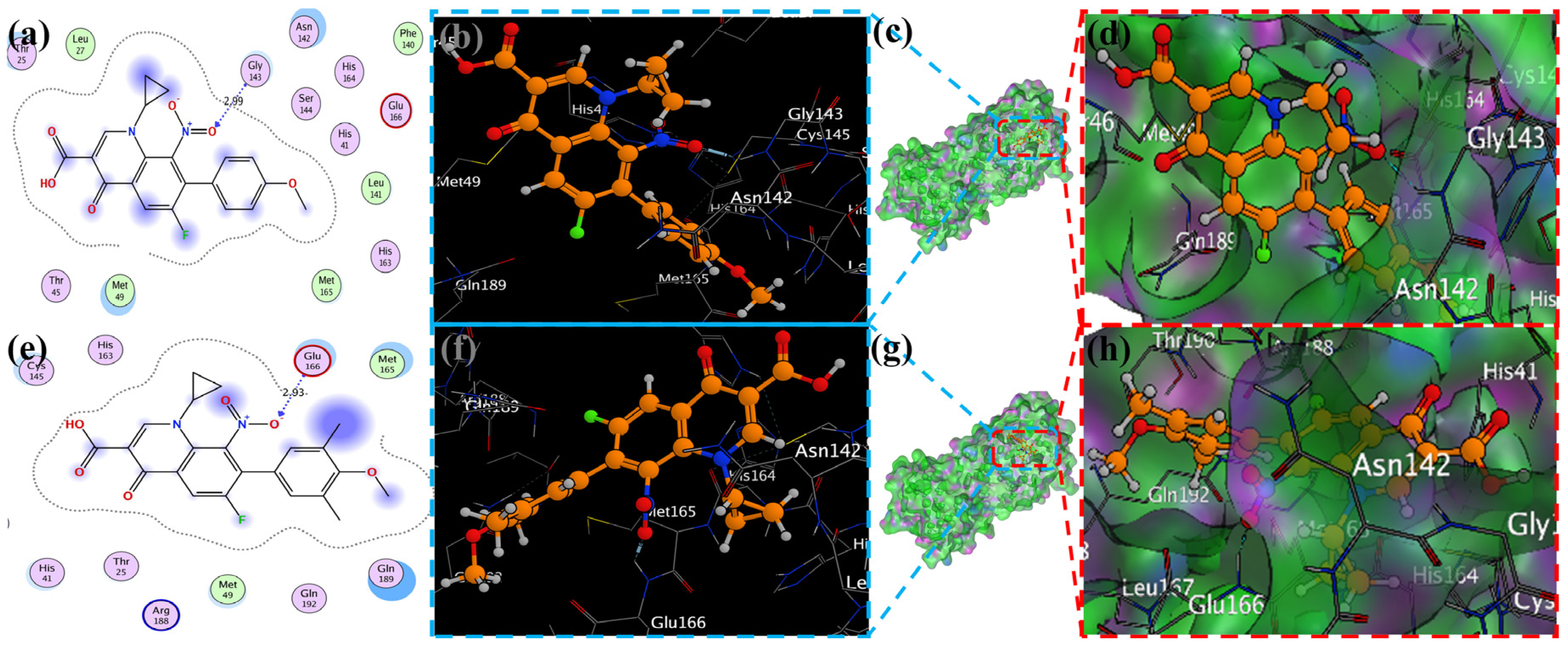
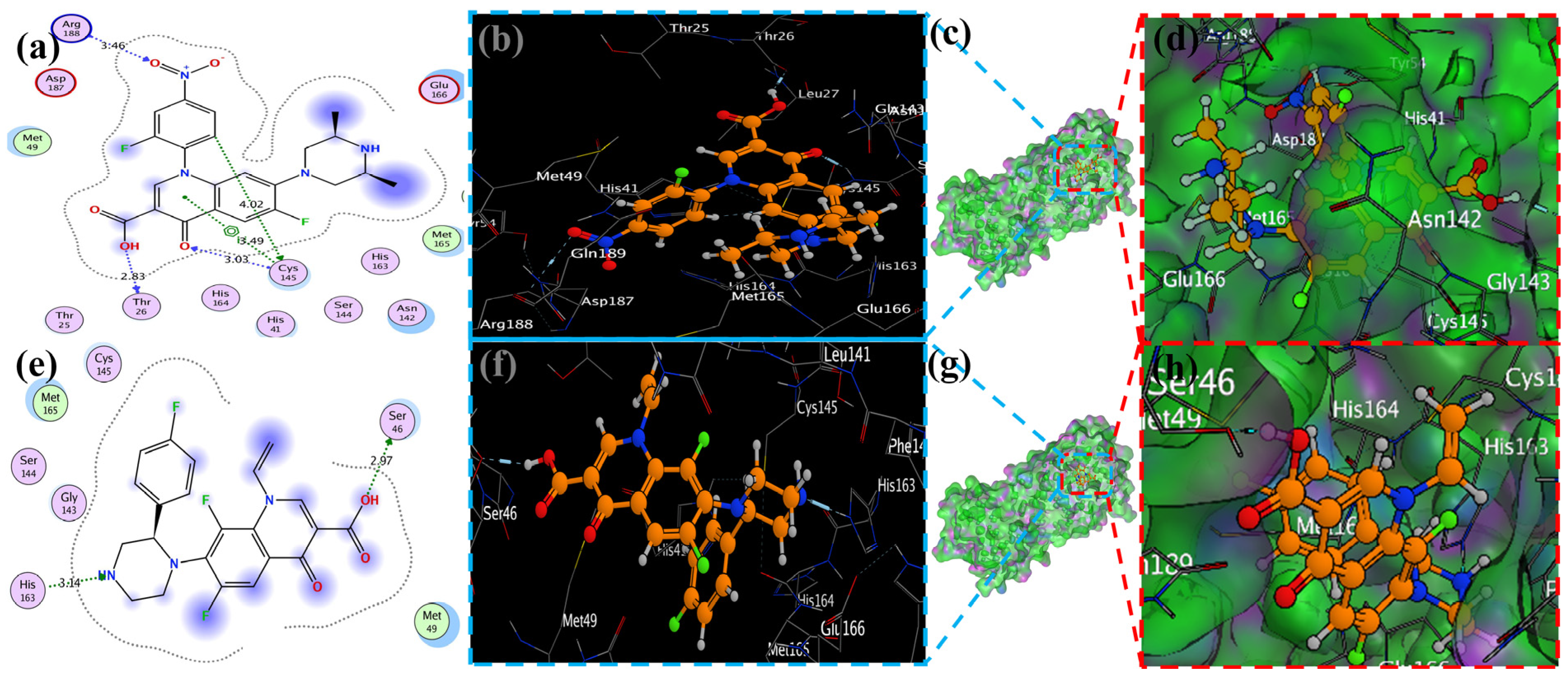
2.2. Molecular Docking Study
2.3. Validation of MOE Software through Inhibitory Mechanism of N3
2.4. Visualization of Docking Results
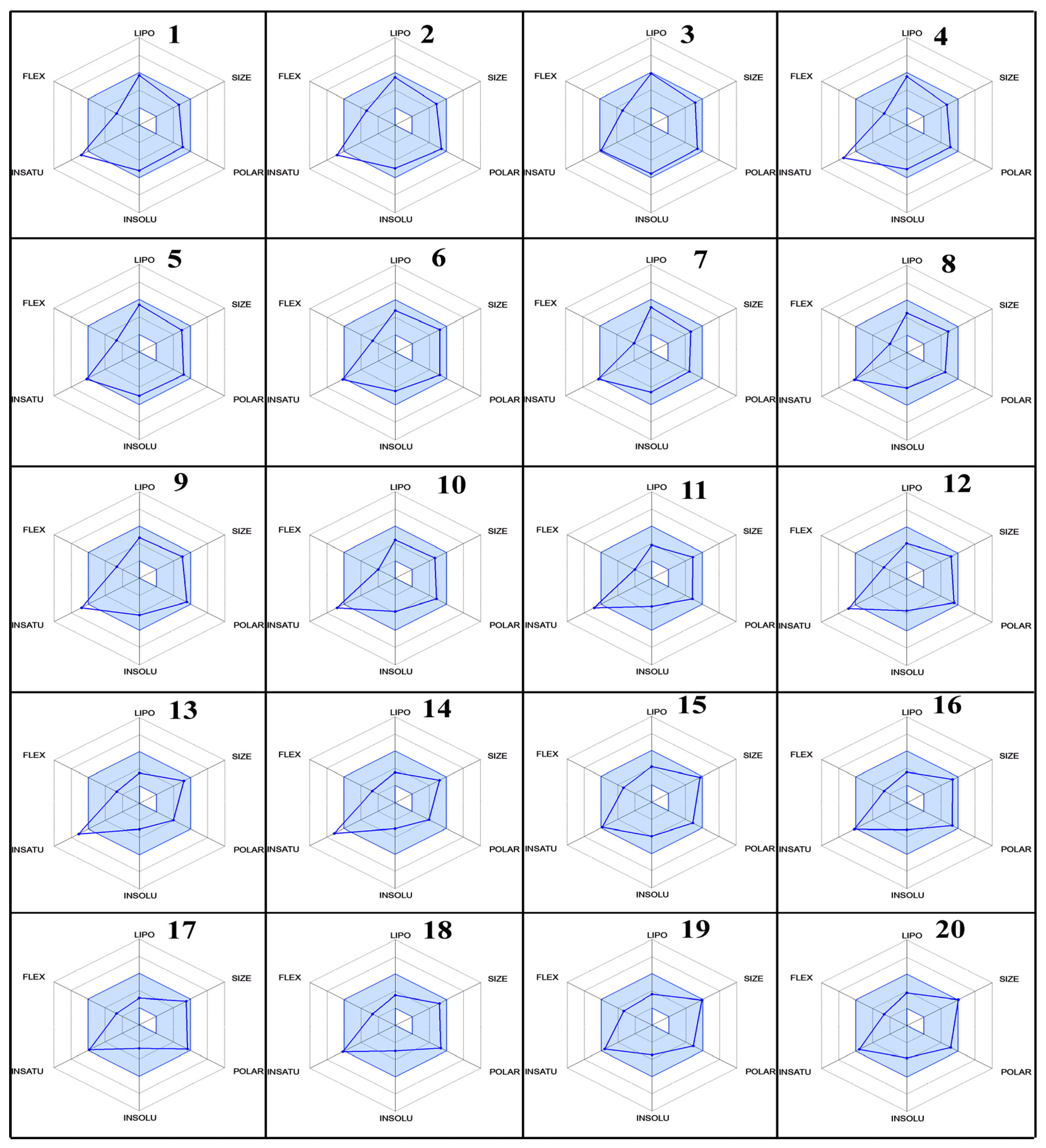
2.5. ADME Parameters, Pharmacokinetics, and Drug Likeness
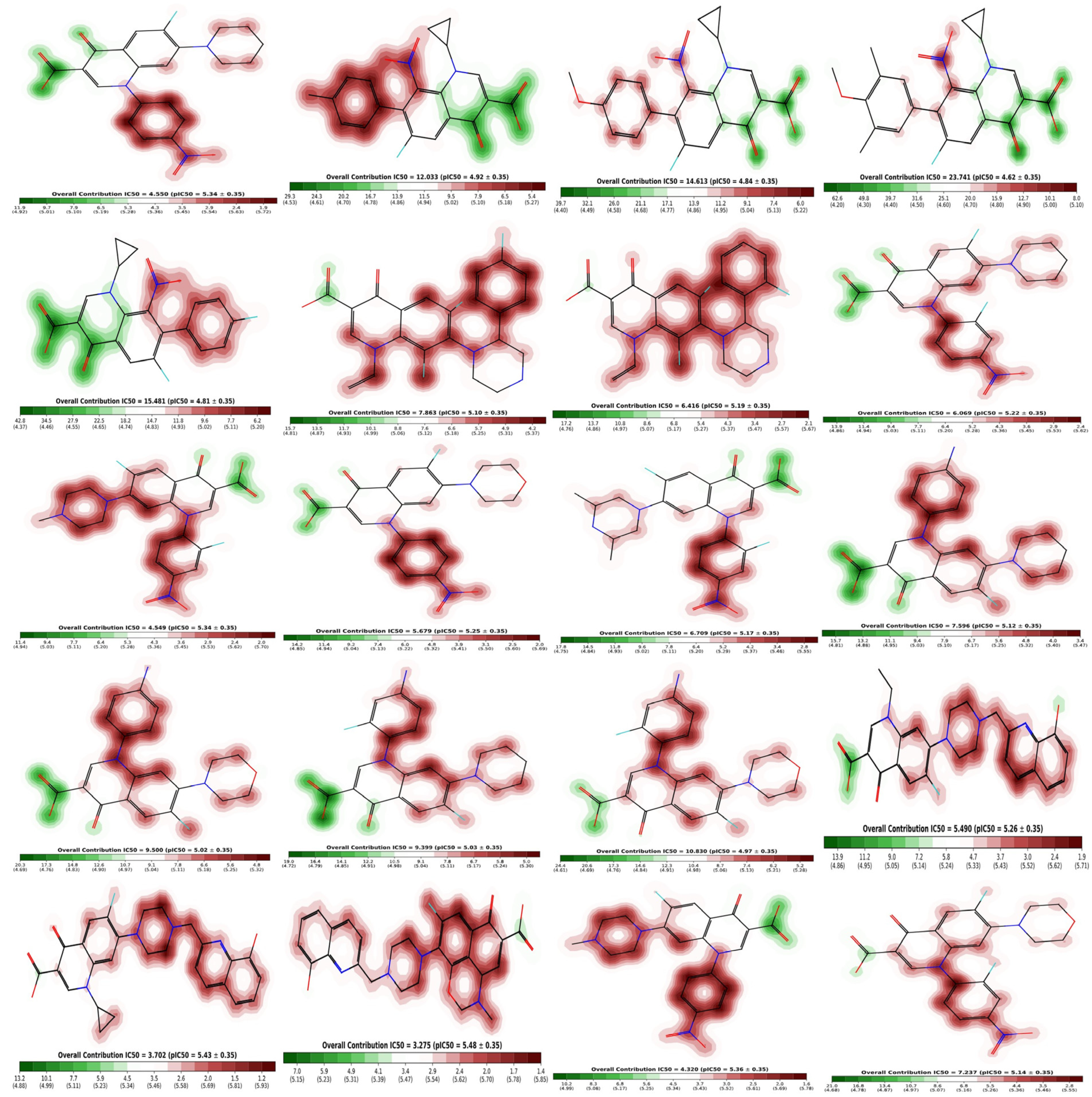
2.6. Cardiotoxicity of Compounds
3. Materials and Methods
3.1. Density Functional Theory (DFT) Study
3.2. Docking Study Using MOE (Molecular Operating Environment)
3.3. ADME Studies
3.4. Cardiotoxicity Studies of Compounds
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jebril, N.M.T. World Health Organization declared a pandemic public health menace: A systematic review of the coronavirus disease 2019 “COVID-19”. SSRN Electron. J. 3566298 2020. [Google Scholar] [CrossRef]
- Cucinotta, D.; Vanelli, M. WHO declares COVID-19 a pandemic. Acta. Biomed. 2020, 91, 157. [Google Scholar]
- Tsergouli, K.; Karampatakis, T.; Haidich, A.; Metallidis, S.; Papa, A. Nosocomial infections caused by Crimean–Congo haemorrhagic fever virus. J. Hosp. Infect. 2020, 105, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Moore, M.J.; Vasilieva, N.; Sui, J.; Wong, S.K.; Berne, M.A.; Somasundaran, M.; Sullivan, J.L.; Luzuriaga, K.; Greenough, T.C. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003, 426, 450–454. [Google Scholar] [CrossRef]
- Sun, M.; Yang, J.; Sun, Y.; Su, G. Inhibitors of RAS might be a good choice for the therapy of COVID-19 pneumonia. Chin. J. Tuberc. Respir. Dis. 2020, 43, E014. [Google Scholar]
- Batiha, G.E.-S.; Moubarak, M.; Shaheen, H.M.; Zakariya, A.M.; Usman, I.M.; Rauf, A.; Adhikari, A.; Dey, A.; Alexiou, A.; Hetta, H.F. Favipiravir in SARS-CoV-2 Infection: Is it Worth it? Comb. Chem. High Throughput Screen. 2022, 25, 2413–2428. [Google Scholar] [CrossRef]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef]
- Wang, M.; Cao, R.; Zhang, L.; Yang, X.; Liu, J.; Xu, M.; Shi, Z.; Hu, Z.; Zhong, W.; Xiao, G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020, 30, 269–271. [Google Scholar] [CrossRef]
- Yang, Y.; Islam, M.S.; Wang, J.; Li, Y.; Chen, X. Traditional Chinese medicine in the treatment of patients infected with 2019-new coronavirus (SARS-CoV-2): A review and perspective. Int. J. Biol. Sci. 2020, 16, 1708. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, Y. Traditional Chinese medicine treatment of COVID-19. Complement. Ther. Clin. Pract. 2020, 39, 101165. [Google Scholar] [CrossRef]
- Sampaio, F.; Sequeira, C.; Teixeira, L. Impact of COVID-19 outbreak on nurses’ mental health: A prospective cohort study. Environ. Res. 2021, 194, 110620. [Google Scholar] [CrossRef] [PubMed]
- Ling, C.-q. Traditional Chinese medicine is a resource for drug discovery against 2019 novel coronavirus (SARS-CoV-2). J. Integr. Med. 2020, 18, 87. [Google Scholar] [CrossRef] [PubMed]
- Hong-Zhi, D.; Xiao-Ying, H.; Yu-Huan, M.; Huang, B.-S.; Da-Hui, L. Traditional Chinese Medicine: An effective treatment for 2019 novel coronavirus pneumonia (NCP). Chin. J. Nat. Med. 2020, 18, 206–210. [Google Scholar]
- Feng, M.; Wang, X.; Chen, J.; Qu, R.; Sui, Y.; Cizmas, L.; Wang, Z.; Sharma, V.K. Degradation of fluoroquinolone antibiotics by ferrate (VI): Effects of water constituents and oxidized products. Water Res. 2016, 103, 48–57. [Google Scholar] [CrossRef]
- De Briyne, N.; Atkinson, J.; Borriello, S.; Pokludová, L. Antibiotics used most commonly to treat animals in Europe. Vet. Rec. 2014, 175, 325. [Google Scholar] [CrossRef]
- Daneman, N.; Chateau, D.; Dahl, M.; Zhang, J.; Fisher, A.; Sketris, I.S.; Quail, J.; Marra, F.; Ernst, P.; Bugden, S. Fluoroquinolone use for uncomplicated urinary tract infections in women: A retrospective cohort study. Clin. Microbiol. Infect. 2020, 26, 613–618. [Google Scholar] [CrossRef]
- Zhao, X.; Zhao, Y.; Ren, Z.; Li, Y. Combined QSAR/QSPR and molecular docking study on fluoroquinolones to reduce biological enrichment. Comput. Biol. Chem. 2019, 79, 177–184. [Google Scholar] [CrossRef]
- Kawczak, P.; Bober, L.; Bączek, T. Application of QSAR analysis and different quantum chemical calculation methods in activity evaluation of selected fluoroquinolones. Comb. Chem. High Throughput Screen. 2018, 21, 468–475. [Google Scholar] [CrossRef]
- Buglak, A.A.; Shanin, I.A.; Eremin, S.A.; Lei, H.-T.; Li, X.; Zherdev, A.V.; Dzantiev, B.B. Ciprofloxacin and clinafloxacin antibodies for an immunoassay of quinolones: Quantitative structure–activity analysis of cross-reactivities. Int. J. Mol. Sci. 2019, 20, 265. [Google Scholar] [CrossRef]
- Scientifico-Disciplinare, S.; Falabella, P.; Luca, F.; Scrano, L. Advanced Oxidation Processes (AOPs): Solutions for the degradation of emerging contaminants in liquid phases. Water 2023, 15, 1615. [Google Scholar]
- Appelbaum, P.; Hunter, P. The fluoroquinolone antibacterials: Past, present and future perspectives. Int. J. Antimicrob. Agents 2000, 16, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Dalhoff, A. Immunomodulatory activities of fluoroquinolones. Infection 2005, 33 (Suppl. 2), 55–70. [Google Scholar] [CrossRef]
- Kula, K.; Nagatsky, R.; Sadowski, M.; Siumka, Y.; Demchuk, O.M. Arylcyanomethylenequinone Oximes: An Overview of Synthesis, Chemical Transformations, and Biological Activity. Molecules 2023, 28, 5229. [Google Scholar] [CrossRef] [PubMed]
- Marciniec, K.; Beberok, A.; Pęcak, P.; Boryczka, S.; Wrześniok, D. Ciprofloxacin and moxifloxacin could interact with SARS-CoV-2 protease: Preliminary in silico analysis. Pharmacol. Rep. 2020, 72, 1553–1561. [Google Scholar] [CrossRef] [PubMed]
- Scroggs, S.L.; Offerdahl, D.K.; Flather, D.P.; Morris, C.N.; Kendall, B.L.; Broeckel, R.M.; Beare, P.A.; Bloom, M.E. Fluoroquinolone antibiotics exhibit low antiviral activity against SARS-CoV-2 and MERS-CoV. Viruses 2020, 13, 8. [Google Scholar] [CrossRef]
- Chaudhary, K.K.; Mishra, N.J.D. A review on molecular docking: Novel tool for drug discovery. JSM Chem. 2016, 3, 1029. [Google Scholar]
- Santos, E.S.; Silva, P.C.; Sousa, P.S.A.; Aquino, C.C.; Pacheco, G.; Teixeira, L.F.L.S.; Araujo, A.R.; Sousa, F.B.M.; Barros, R.O.; Ramos, R.M.; et al. Antiviral potential of diminazene aceturate against SARS-CoV-2 proteases using computational and in vitro approaches. Chem. Biol. Interact. 2022, 367, 110161. [Google Scholar] [CrossRef]
- Barciszewska, A.-M. Elucidating of oxidative distress in COVID-19 and methods of its prevention. Chem. Biol. Interact. 2021, 344, 109501. [Google Scholar] [CrossRef]
- Zhao, Y.-L.; Chen, Y.-L.; Sheu, J.-Y.; Chen, I.-L.; Wang, T.-C.; Tzeng, C.-C.; Chemistry, M. Synthesis and antimycobacterial evaluation of certain fluoroquinolone derivatives. Bioorg. Med. Chem. 2005, 13, 3921–3926. [Google Scholar] [CrossRef]
- Srinivasan, S.; Shafreen, R.M.B.; Nithyanand, P.; Manisankar, P.; Pandian, S.K. Synthesis and in vitro antimicrobial evaluation of novel fluoroquinolone derivatives. Eur. J. Med. Chem. 2010, 45, 6101–6105. [Google Scholar] [CrossRef]
- Al-Trawneh, S.A.; El-Abadelah, M.M.; Al-Abadleh, M.M.; Zani, F.; Incerti, M.; Vicini, P. A new efficient route to 7-aryl-6-fluoro-8-nitroquinolones as potent antibacterial agents. Eur. J. Med. Chem. 2014, 86, 364–367. [Google Scholar] [CrossRef] [PubMed]
- Chhetri, A.; Chettri, S.; Rai, P.; Sinha, B.; Brahman, D. Exploration of inhibitory action of Azo imidazole derivatives against COVID-19 main protease (Mpro): A computational study. J. Mol. Struct. 2021, 1224, 129178. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Jing, X.; Wang, J.; Zheng, Y.; Qiu, Y.; Ji, H.; Peng, L.; Jiang, S.; Wu, W.; Guo, D. Safety considerations of chloroquine in the treatment of patients with diabetes and COVID-19. Chem. Biol. Interact. 2022, 361, 109954. [Google Scholar] [CrossRef]
- Ye, N.; Yang, Z.; Liu, Y. Applications of density functional theory in COVID-19 drug modeling. Drug Discov. Today 2022, 27, 1411–1419. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Xie, W.; Xue, X.; Yang, K.; Ma, J.; Liang, W.; Zhao, Q.; Zhou, Z.; Pei, D.; Ziebuhr, J. Design of wide-spectrum inhibitors targeting coronavirus main proteases. PLoS Biol. 2005, 3, e324. [Google Scholar]
- Anand, K.; Ziebuhr, J.; Wadhwani, P.; Mesters, J.R.; Hilgenfeld, R. Coronavirus main proteinase (3CLpro) structure: Basis for design of anti-SARS drugs. Science 2003, 300, 1763–1767. [Google Scholar] [CrossRef]
- Jaramillo, P.; Domingo, L.R.; Chamorro, E.; Pérez, P. A further exploration of a nucleophilicity index based on the gas-phase ionization potentials. J. Mol. Struct. THEOCHEM 2008, 865, 68–72. [Google Scholar] [CrossRef]
- Tariq, S.; Mutahir, S.; Khan, M.A.; Mutahir, Z.; Hussain, S.; Ashraf, M.; Bao, X.; Zhou, B.; Stark, C.B.W.; Khan, I.U. Synthesis, in Vitro Cholinesterase Inhibition, Molecular Docking, DFT, and ADME Studies of Novel 1,3,4-Oxadiazole-2-Thiol Derivatives. Chem. Biodivers. 2022, 19, e202200157. [Google Scholar] [CrossRef]
- Celik, S. DFT investigations and molecular docking as potent inhibitors of SARS-CoV-2 main protease of 4-phenylpyrimidine. J. Mol. Struct. 2023, 1277, 134895. [Google Scholar] [CrossRef]
- Lakshminarayanan, S.; Jeyasingh, V.; Murugesan, K.; Selvapalam, N.; Dass, G. Molecular electrostatic potential (MEP) surface analysis of chemo sensors: An extra supporting hand for strength, selectivity & non-traditional interactions. J. Photochem. Photobiol. 2021, 6, 100022. [Google Scholar]
- Liu, K.; Lu, X.; Shi, H.; Xu, X.; Kong, R.; Chang, S. nCoVDock2: A docking server to predict the binding modes between COVID-19 targets and its potential ligands. Nucleic Acids Res. 2023, 51, W365–W371. [Google Scholar] [CrossRef] [PubMed]
- Alghamdi, A.; Abouzied, A.S.; Alamri, A.; Anwar, S.; Ansari, M.; Khadra, I.; Zaki, Y.H.; Gomha, S.M. Synthesis, molecular docking, and dynamic simulation targeting main protease (Mpro) of new, Thiazole clubbed pyridine scaffolds as potential COVID-19 inhibitors. Curr. Issues Mol. Biol. 2023, 45, 1422–1442. [Google Scholar] [CrossRef]
- Hatada, R.; Okuwaki, K.; Mochizuki, Y.; Handa, Y.; Fukuzawa, K.; Komeiji, Y.; Okiyama, Y.; Tanaka, S. Fragment molecular orbital based interaction analyses on COVID-19 main protease—Inhibitor N3 complex (PDB ID: 6LU7). J. Chem. Inf. Model. 2020, 60, 3593–3602. [Google Scholar] [CrossRef]
- Khan, M.A.; Mutahir, S.; Jabar, G.; Wenwei, Z.; Tariq, M.A.; Almehizia, A.A.A.; Mustafa, M. DFT, Molecular Docking, ADME, and Cardiotoxicity studies of Persuasive Thiazoles as Potential Inhibitors of the Main Protease of SARS-CoV-2. Chem. Biodivers. 2024, e202401775. [Google Scholar] [CrossRef] [PubMed]
- Hosseini, F.S.; Amanlou, M. Simeprevir, potential candidate to repurpose for coronavirus infection: Virtual screening and molecular docking study. Life Sci. 2020, 258, 118205. [Google Scholar] [CrossRef]
- Ma, S.-K.E.; Wong, W.C.; Leung, C.-W.R.; Lai, S.-T.T.; Lo, Y.-C.J.; Wong, K.-H.H.; Chan, M.-C.; Que, T.-L.; Chow, K.-W.M.; Yuen, M.-C.; et al. Review of vector-borne diseases in Hong Kong. Travel Med. Infect. Dis. 2011, 9, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Carvelli, J.; Demaria, O.; Vély, F.; Batista, L.; Benmansour, N.C.; Fares, J.; Carpentier, S.; Thibult, M.-L.; Morel, A.; André, P. Identification of immune checkpoints in COVID-19. 2020. [Google Scholar] [CrossRef]
- Liu, C.; Zhou, Q.; Li, Y.; Garner, L.V.; Watkins, S.P.; Carter, L.J.; Smoot, J.; Gregg, A.C.; Daniels, A.D.; Jervey, S. Research and development on therapeutic agents and vaccines for COVID-19 and related human coronavirus diseases. ACS Cent. Sci. 2020, 6, 315–331. [Google Scholar] [CrossRef]
- Sivasankarapillai, V.S.; Pillai, A.M.; Rahdar, A.; Sobha, A.P.; Das, S.S.; Mitropoulos, A.C.; Mokarrar, M.H.; Kyzas, G.Z.J.N. On facing the SARS-CoV-2 (COVID-19) with combination of nanomaterials and medicine: Possible strategies and first challenges. Nanomaterials 2020, 10, 852. [Google Scholar] [CrossRef] [PubMed]
- Owis, A.I.; El-Hawary, M.S.; El Amir, D.; Aly, O.M.; Abdelmohsen, U.R.; Kamel, M.S.J.R.A. Molecular docking reveals the potential of Salvadora persica flavonoids to inhibit COVID-19 virus main protease. RSC Adv. 2020, 10, 19570–19575. [Google Scholar] [CrossRef]
- Mutahir, S.; Khan, M.A.; Naglah, A.M.; Al-Omar, M.A.; Almehizia, A.A.; Huwaimel, B.; Abouzied, A.S.; Alharbi, A.S.; Refat, M.S. Structural Characterization and Molecular Docking Screening of Most Potent 1, 2, 4-Triazine Sulfonamide Derivatives as Anti-Cancer Agents. Crystals 2023, 13, 767. [Google Scholar] [CrossRef]
- Arshad, R.; Khan, M.A.; Mutahir, S.; Hussain, S.; Al-Hazmi, G.H.; Refat, M.S. DFT, Molecular Docking and ADME Studies of Thiazolidinones as Tyrosinase Inhibitors. Polycycl. Aromat. Compd. 2022, 43, 6750–6765. [Google Scholar] [CrossRef]
- Bahl, A.S.; Verma, V.K.; Bhatia, J.; Arya, D.S. Integrating in silico and in vivo approach for investigating the role of polyherbal oil in prevention and treatment of COVID-19 infection. Chem. Biol. Interact. 2022, 367, 110179. [Google Scholar] [CrossRef] [PubMed]
- Garg, S.; Roy, A. In silico analysis of selected alkaloids against main protease (Mpro) of SARS-CoV-2. Chem. Biol. Interact. 2020, 332, 109309. [Google Scholar] [CrossRef]
- Shao, H.P.; Wang, T.H.; Zhai, H.L.; Bi, K.X.; Zhao, B.Q. Discovery of inhibitors against SARS-CoV-2 main protease using fragment-based drug design. Chem. Biol. Interact. 2023, 371, 110352. [Google Scholar] [CrossRef]
- Nazeer, U.; Rasool, N.; Mujahid, A.; Mansha, A.; Zubair, M.; Kosar, N.; Mahmood, T.; Raza Shah, A.; Shah, S.A.A.; Zakaria, Z.A. Selective arylation of 2-bromo-4-chlorophenyl-2-bromobutanoate via a Pd-catalyzed Suzuki cross-coupling reaction and its electronic and non-linear optical (NLO) properties via DFT studies. Molecules 2020, 25, 3521. [Google Scholar] [CrossRef] [PubMed]
- Aliveisi, R.; Taherpour, A.; Yavari, I. A DFT Study of Electronic Structures and Relative Stabilities of Isomeric n, m-Diazaphenanthrenes. Polycycl. Aromat. Compd. 2019, 39, 462–469. [Google Scholar] [CrossRef]
- Mebi, C.A. DFT study on structure, electronic properties, and reactivity of cis-isomers of [(NC5H4S)2 Fe(CO)2]. J. Chem. Sci. 2011, 123, 727–731. [Google Scholar] [CrossRef]
- Orio, M.; Pantazis, D.A.; Neese, F. Density functional theory. Photosynth. Res. 2009, 102, 443–453. [Google Scholar] [CrossRef]
- Mutahir, S.; Khan, M.A.; Almehizia, A.A.; Abouzied, A.S.; Khalifa, N.E.; Naglah, A.M.; Deng, H.; Refat, M.S.; Khojali, W.M.; Huwaimel, B. Design, Synthesis, Characterization and Computational Studies of Mannich Bases Oxadiazole Derivatives as New Class of Jack Bean Urease Inhibitors. Chem. Biodivers. 2023, 20, e202300241. [Google Scholar] [CrossRef]
- Fadlalla, M.; Ahmed, M.; Ali, M.; Elshiekh, A.A.; Yousef, B.A. Molecular docking as a potential approach in repurposing drugs against COVID-19: A systematic review and novel pharmacophore models. Curr. Pharmacol. Rep. 2022, 8, 212–226. [Google Scholar] [CrossRef]
- Mutahir, S.; Khan, M.A.; Khan, I.U.; Yar, M.; Ashraf, M.; Tariq, S.; Ye, R.-L.; Zhou, B.-J. Organocatalyzed and mechanochemical solvent-free synthesis of novel and functionalized bis-biphenyl substituted thiazolidinones as potent tyrosinase inhibitors: SAR and molecular modeling studies. Eur. J. Med. Chem. 2017, 134, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V.J.S.r. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Bikiewicz, A.; Banach, M.; von Haehling, S.; Maciejewski, M.; Bielecka-Dabrowa, A.J.E.H.F. Adjuvant breast cancer treatments cardiotoxicity and modern methods of detection and prevention of cardiac complications. ESC Heart Fail. 2021, 8, 2397–2418. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comp | ELUMO (eV) | EHOMO (eV) | ∆E (HOMO-LUMO) (eV) | Ionization Potential (I) (eV) | Electron Affinity (A) (eV) | Chemical Hardness (η) (eV) | Chemical Softness (ζ) (eV) | ELECTRONEGATIVITY (χ) (eV) | Chemical Potential (μ) (eV) | Electrophilicity Index (ω) (eV) |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | −0.3103 | −0.0681 | 0.2422 | 0.3104 | 0.0681 | 0.1211 | 4.1281 | 0.1892 | −0.3440 | 0.0072 |
| 2 | −0.2973 | −0.0673 | 0.2300 | 0.2974 | 0.0674 | 0.1150 | 4.3474 | 0.1823 | −0.3310 | 0.0063 |
| 3 | −0.3069 | −0.0674 | 0.2395 | 0.3070 | 0.0675 | 0.1197 | 4.1755 | 0.1872 | −0.3400 | 0.0070 |
| 4 | −0.3147 | −0.0732 | 0.2415 | 0.3147 | 0.0733 | 0.1207 | 4.1411 | 0.1939 | −0.3510 | 0.0075 |
| 5 | −0.2933 | −0.0805 | 0.2127 | 0.2933 | 0.0806 | 0.1064 | 4.7006 | 0.1869 | −0.3330 | 0.0059 |
| 6 | −0.2932 | −0.0869 | 0.2063 | 0.2933 | 0.0870 | 0.1031 | 4.8475 | 0.1901 | −0.3360 | 0.0058 |
| 7 | −0.2745 | −0.0229 | 0.2516 | 0.2745 | 0.0229 | 0.1258 | 3.9739 | 0.1487 | −0.2850 | 0.0051 |
| 8 | −0.2698 | −0.0118 | 0.2581 | 0.2699 | 0.0118 | 0.1290 | 3.8751 | 0.1408 | −0.2750 | 0.0049 |
| 9 | −0.2904 | −0.0811 | 0.2093 | 0.2904 | 0.0811 | 0.1046 | 4.7785 | 0.1857 | −0.3300 | 0.0057 |
| 10 | −0.2826 | −0.0306 | 0.2520 | 0.2827 | 0.0307 | 0.1260 | 3.9683 | 0.1566 | −0.2970 | 0.0056 |
| 11 | −0.2752 | −0.0149 | 0.2604 | 0.2753 | 0.0149 | 0.1302 | 3.8408 | 0.1451 | −0.2820 | 0.0052 |
| 12 | −0.2892 | −0.0889 | 0.2003 | 0.2892 | 0.0889 | 0.1002 | 4.9925 | 0.1890 | −0.3330 | 0.0056 |
| 13 | −0.2930 | −0.0470 | 0.2466 | 0.2936 | 0.0470 | 0.1233 | 4.0556 | 0.1703 | −0.3170 | 0.0062 |
| 14 | −0.2834 | −0.0847 | 0.1988 | 0.2835 | 0.0847 | 0.0994 | 5.0312 | 0.1840 | −0.3250 | 0.0053 |
| 15 | −0.2775 | −0.0328 | 0.2447 | 0.2775 | 0.0328 | 0.1224 | 4.0865 | 0.1551 | −0.2930 | 0.0053 |
| 16 | −0.2891 | −0.0440 | 0.2450 | 0.2891 | 0.0441 | 0.1225 | 4.0810 | 0.1666 | −0.3110 | 0.0059 |
| 17 | −0.2040 | −0.0833 | 0.1207 | 0.2041 | 0.0834 | 0.0604 | 8.2850 | 0.1437 | −0.2450 | 0.0018 |
| 18 | −0.2950 | −0.0807 | 0.2144 | 0.2951 | 0.0807 | 0.1072 | 4.6650 | 0.1879 | −0.3350 | 0.0060 |
| 19 | −0.2752 | −0.0300 | 0.2451 | 0.2752 | 0.0301 | 0.1225 | 4.0793 | 0.1526 | −0.2900 | 0.0052 |
| 20 | −0.2668 | −0.0556 | 0.2111 | 0.2668 | 0.0557 | 0.1056 | 4.7366 | 0.1612 | −0.2940 | 0.0046 |
| Ligand | Docking Score kcal/mol | Predicted Inhibitory Constant (pKi) µM | ∆G Energy kcal/mol |
|---|---|---|---|
| 1 | −6.4577 | 2.3159 | −34.8948 |
| 2 | −6.1160 | 0.9924 | −32.3975 |
| 3 | −7.4168 | 0.8129 | −43.3306 |
| 4 | −5.8692 | 0.8136 | −31.0542 |
| 5 | −7.5820 | 1.7860 | −45.1559 |
| 6 | −7.2341 | 3.1690 | −40.8352 |
| 7 | −6.8824 | 2.7863 | −35.2465 |
| 8 | −7.2390 | 1.4395 | −37.6927 |
| 9 | −7.2094 | 1.4301 | −41.1037 |
| 10 | −6.8317 | 2.1947 | −41.7742 |
| 11 | −6.9915 | 1.3738 | −35.5844 |
| 12 | −7.1314 | 1.1245 | −34.9606 |
| 13 | −6.5379 | 3.4226 | −41.3863 |
| 14 | −7.3066 | 2.6392 | −41.6989 |
| 15 | −7.4876 | 0.8797 | −42.5053 |
| 16 | −7.2495 | 2.4672 | −41.5726 |
| 17 | −7.5876 | 2.2114 | −43.3508 |
| 18 | −7.8704 | 1.9804 | −48.5983 |
| 19 | −7.2012 | 2.6694 | −41.3393 |
| 20 | −7.2246 | 2.3682 | −41.6298 |
| Compounds (Drugs) | Binding Energy, ∆G (kcal/mol) |
|---|---|
| Remdesivir | −5.8 [45], −7.215 [46], −2.47 [47], −6–5 [48], −9.70 [49], −7.5 [45], −3.62 [47], −5.1 [48], −5.75 [50]. |
| Ligand 01 | −6.4577 (this work) |
| Ligand 03 | −7.4168 (this work) |
| Ligand 17 | −7.3066 (this work) |
| Ligand 15 | −7.4876 (this work) |
| Ligand 18 | −7.8704 (this work) |
| Title | Mol MW | Donor HB | Accept HB | QP logPo/w | QP logS | QPP Caco | Metab | Qplog Khsa | Human Oral Absorption | Percent Human Oral Absorption | Rule of Five | Rule of Three |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 382.347 | 0.00 | 4.50 | 3.511 | −4.453 | 43.305 | 2 | 0.174 | 3 | 76.795 | 0 | 0 |
| 2 | 398.347 | 0.00 | 5.25 | 3.206 | −3.906 | 43.379 | 2 | −0.041 | 3 | 75.024 | 0 | 0 |
| 3 | 426.4 | 0.00 | 5.25 | 3.863 | −4.918 | 46.489 | 4 | 0.278 | 3 | 79.405 | 0 | 0 |
| 4 | 386.311 | 0.00 | 4.50 | 3.436 | −4.245 | 43.157 | 1 | 0.052 | 3 | 76.329 | 0 | 0 |
| 5 | 411.389 | 0.00 | 5.50 | 3.482 | −5.576 | 12.778 | 1 | 0.314 | 2 | 67.138 | 0 | 1 |
| 6 | 429.379 | 0.00 | 5.50 | 3.647 | −5.757 | 13.127 | 1 | 0.344 | 2 | 68.313 | 0 | 2 |
| 7 | 381.406 | 1.50 | 5.50 | 3.482 | −5.586 | 26.62 | 1 | 0.381 | 2 | 72.839 | 0 | 0 |
| 8 | 399.396 | 1.50 | 5.50 | 3.654 | −5.813 | 27.321 | 1 | 0.411 | 2 | 74.053 | 0 | 1 |
| 9 | 413.361 | 0.00 | 7.20 | 2.38 | −4.211 | 12.778 | 2 | −0.266 | 2 | 60.682 | 0 | 1 |
| 10 | 383.378 | 1.50 | 7.20 | 2.551 | −4.609 | 26.62 | 2 | −0.03 | 2 | 67.393 | 0 | 0 |
| 11 | 401.369 | 1.50 | 7.20 | 2.725 | −4.84 | 27.321 | 2 | −0.001 | 2 | 68.612 | 0 | 0 |
| 12 | 431.352 | 0.00 | 7.20 | 2.547 | −4.4 | 13.127 | 2 | −0.234 | 2 | 61.873 | 0 | 1 |
| 13 | 429.398 | 1.00 | 6.00 | 1.6 | −5.138 | 13.313 | 1 | 0.393 | 2 | 56.434 | 0 | 1 |
| 14 | 429.398 | 1.00 | 6.00 | 1.465 | −4.885 | 13.318 | 2 | 0.373 | 2 | 55.645 | 0 | 1 |
| 15 | 476.506 | 1.00 | 8.25 | 1.966 | −5.959 | 10.746 | 4 | 0.413 | 2 | 56.913 | 0 | 2 |
| 16 | 444.394 | 0.00 | 7.50 | 0.503 | −4.617 | 2.887 | 2 | −0.074 | 1 | 38.134 | 0 | 1 |
| 17 | 458.421 | 1.00 | 7.00 | 1.032 | −5.617 | 2.219 | 1 | 0.422 | 1 | 39.183 | 0 | 1 |
| 18 | 426.403 | 0.00 | 7.50 | 0.327 | −4.373 | 2.814 | 2 | −0.108 | 1 | 36.903 | 0 | 1 |
| 19 | 488.517 | 1.00 | 8.25 | 2.037 | −6.573 | 7.478 | 3 | 0.535 | 1 | 54.514 | 0 | 2 |
| 20 | 504.517 | 1.00 | 10.5 | 0.638 | −3.949 | 2.767 | 5 | 0.053 | 1 | 25.635 | 1 | 1 |
| Compound | Prediction | Binary Reliability % | Multiclass Reliability % | Applicability Domain | IC50 Values µm | Reg. Prediction (plC50) |
|---|---|---|---|---|---|---|
| 1 | Non-blocker | 67.29 | 35.32 | Outside | 4.550 | 5.342 |
| 2 | Non-blocker | 87.45 | 41.2 | Outside | 12.033 | 4.92 |
| 3 | Non-blocker | 91.46 | 41.6 | Outside | 14.613 | 4.835 |
| 4 | Non-blocker | 95.48 | 46.4 | Outside | 23.741 | 4.625 |
| 5 | Non-blocker | 76.19 | 36.9 | Outside | 15.481 | 4.81 |
| 6 | Blocker | 64.25 | 34 | Outside | 7.863 | 5.104 |
| 7 | Non-blocker | 51.86 | 34.1 | Outside | 6.416 | 5.193 |
| 8 | Non-blocker | 95.01 | 31.16 | Outside | 6.069 | 5.217 |
| 9 | Non-blocker | 96.67 | 30.52 | Outside | 4.549 | 5.342 |
| 10 | Non-blocker | 90.21 | 34.1 | Outside | 5.679 | 5.246 |
| 11 | Non-blocker | 95.97 | 33.1 | Outside | 6.709 | 5.173 |
| 12 | Non-blocker | 77.55 | 33.44 | Outside | 7.596 | 5.119 |
| 13 | Non-blocker | 94.07 | 33.1 | Outside | 9.50 | 5.022 |
| 14 | Non-blocker | 99.37 | 32.28 | Outside | 9.399 | 5.027 |
| 15 | Non-blocker | 99.83 | 33.58 | Outside | 10.83 | 4.965 |
| 16 | Non-blocker | 80.06 | 30.4 | Outside | 5.490 | 5.26 |
| 17 | Non-blocker | 58.73 | 28.3 | Outside | 3.702 | 5.432 |
| 18 | Blocker | 56.83 | 30.3 | Outside | 3.275 | 5.485 |
| 19 | Non-blocker | 83.84 | 34.3 | Outside | 4.320 | 5.365 |
| 20 | Non-blocker | 98.51 | 33.41 | Outside | 7.237 | 5.14 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khan, M.A.; Mutahir, S.; Tariq, M.A.; Almehizia, A.A. Exploration of Specific Fluoroquinolone Interaction with SARS-CoV-2 Main Protease (Mpro) to Battle COVID-19: DFT, Molecular Docking, ADME and Cardiotoxicity Studies. Molecules 2024, 29, 4721. https://doi.org/10.3390/molecules29194721
Khan MA, Mutahir S, Tariq MA, Almehizia AA. Exploration of Specific Fluoroquinolone Interaction with SARS-CoV-2 Main Protease (Mpro) to Battle COVID-19: DFT, Molecular Docking, ADME and Cardiotoxicity Studies. Molecules. 2024; 29(19):4721. https://doi.org/10.3390/molecules29194721
Chicago/Turabian StyleKhan, Muhammad Asim, Sadaf Mutahir, Muhammad Atif Tariq, and Abdulrahman A. Almehizia. 2024. "Exploration of Specific Fluoroquinolone Interaction with SARS-CoV-2 Main Protease (Mpro) to Battle COVID-19: DFT, Molecular Docking, ADME and Cardiotoxicity Studies" Molecules 29, no. 19: 4721. https://doi.org/10.3390/molecules29194721
APA StyleKhan, M. A., Mutahir, S., Tariq, M. A., & Almehizia, A. A. (2024). Exploration of Specific Fluoroquinolone Interaction with SARS-CoV-2 Main Protease (Mpro) to Battle COVID-19: DFT, Molecular Docking, ADME and Cardiotoxicity Studies. Molecules, 29(19), 4721. https://doi.org/10.3390/molecules29194721